
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of low-dimensional network polymers with thermoresponsive properties using MOF scaffolds†
Yuki
Kametani‡
 a,
Ami
Nishijima‡
b,
Shu
Hiramoto
c and
Takashi
Uemura
*ab
a,
Ami
Nishijima‡
b,
Shu
Hiramoto
c and
Takashi
Uemura
*ab
aInstitute of Engineering Innovation, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: uemurat@g.ecc.u-tokyo.ac.jp
bDepartment of Applied Chemistry Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
cDepartment of Advanced Materials Science Graduate School of Frontier Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 12th May 2025
Abstract
Materials with ultimately thin (2D) or narrow (1D) structures have gained significant attention due to their exceptional properties. However, decreasing the dimensionality of soft polymer materials has been a formidable challenge due to the lack of rational synthetic methodology. Here, we performed cross-linking polymerization inside metal–organic frameworks (MOFs) as nanoporous scaffolds to afford poly(N-isopropylacrylamide) (PNIPAm) with unprecedented 1D and 2D network topologies: double strands and monolayer sheets. Remarkably, these polymer networks exhibited unique thermoresponsive properties in water that were strongly correlated with their specific topologies. Note that the transition temperature of double-stranded PNIPAm is among the lowest of known PNIPAm materials. The monolayer PNIPAm sheets exhibit a markedly slow thermal response over a wide temperature range. The dimensional constraint imposed on cross-linking by MOF-templated polymerization enables precisely controlling the chain orientation and proximity, providing new insights into the mechanism of the PNIPAm phase transition.
Introduction
The study of low-dimensional materials with extremely thin (2D) or narrow (1D) structures at the molecular/atomic level is one of the most exciting areas of materials science and engineering owing to their structural anisotropy and quantum confinement effects.1 The distinct properties of the low-dimensional materials, such as remarkable electrical conductivity,2 mechanical strength,3 and flexibility,4 make them ideal for a wide range of applications.5 In this research field, major efforts have been devoted to investigating “hard” inorganic materials, including metals,6 metal oxides,7 and carbons,8 because recent advancements in synthesis techniques, such as chemical vapor deposition and exfoliation methods,9 have made them easier to produce and manipulate. However, reports on dimensional reduction of fully “soft” polymer-based materials have been rather rare. This is because soft polymers are materials that can deform easily under stress and are characterized by their flexibility and low stiffness. Therefore, preparation of 1D or 2D polymeric material usually faces challenges, requiring specific conditions that are not compatible with the conventional polymer processing.The spatial arrangement of polymer chains significantly influences the mechanical, thermal, optical, and electronic properties of polymeric materials.10 One of the most common approaches to improve the performance of polymeric materials is cross-linking that can immobilize linear chains to form network structures.11 The properties of cross-linked polymers can be quantitatively optimized by modulating the degree of cross-linking because the segmental motion of the polymer chains becomes more restricted with increasing the degree of cross-linking. However, elucidating the effect of chain orientation and proximity on the properties of network polymers has been impossible, as precisely controlling the cross-link locations in polymer matrices has been extremely challenging in polymer synthesis, and cross-linking usually provides a three-dimensional (3D) random network.12
Metal–organic frameworks (MOFs), crystalline porous materials comprising self-assembled metal ions and bridging organic ligands, offer uniformly sized inner cavities of molecular dimensions.13 In this research area, an increasing number of papers on controlled reactions within the nanopores have appeared, demonstrating their utility in catalysis and synthetic chemistry.14 MOFs have recently gained prominence as nanoreactors for preparing polymers that are otherwise unobtainable through conventional approaches.15 Indeed, cross-linking polymerization has been controllably performed in MOF nanochannels and has enabled the formation of particular network topologies rather than random network structures, dictated by channel connectivity and dimensionality.16,17
Poly(N-isopropylacrylamide) (PNIPAm), a representative thermoresponsive polymer, exhibits a coil-to-globule transition in an aqueous solution.18 Upon heating, PNIPAm chains undergo drastic conformational change through dehydration to aggregate into insoluble globular particles. As the lower critical solution temperature (LCST) of linear PNIPAm is 32 °C, PNIPAm has found application in bioengineering,19 drug delivery,20 and nanotechnology.21 The tunability of the PNIPAm thermoresponsivity has been extensively studied because the transition temperature of PNIPAm is sensitive to the polymer microstructures and morphology (e.g., the molecular weight,22 tacticity,23 end-groups,24 and branching25). However, in many cases, understanding how the PNIPAm chains interact at temperatures near the LCST remains rather elusive.26 Particularly, the pivotal mechanism underlying the phase transition of PNIPAm with cross-linked structures is yet to be elucidated because of the randomness of the 3D network topology.27 Therefore, a viable strategy for regulating the cross-linked structures of PNIPAm is required to understand the transition mechanism and modulate the thermoresponsive properties.
Here we performed the cross-linking polymerization of N-isopropylacrylamide (NIPAm) in the nanopores of different MOFs to yield PNIPAm networks with controlled chain arrangements. The use of low-dimensional MOF nanochannels as nanoporous templates successfully produced PNIPAm double strands (PNIPAm-1D) and monolayer sheets (PNIPAm-2D) with well-defined 1D and 2D network topologies, respectively (Fig. 1). These unprecedented topologies of PNIPAm significantly affect the thermoresponsive properties, which are clearly distinct from those of the conventional linear and 3D counterparts. This is the first demonstration that chain orientation and proximity in polymer matrices critically determine the thermal transition of PNIPAm.
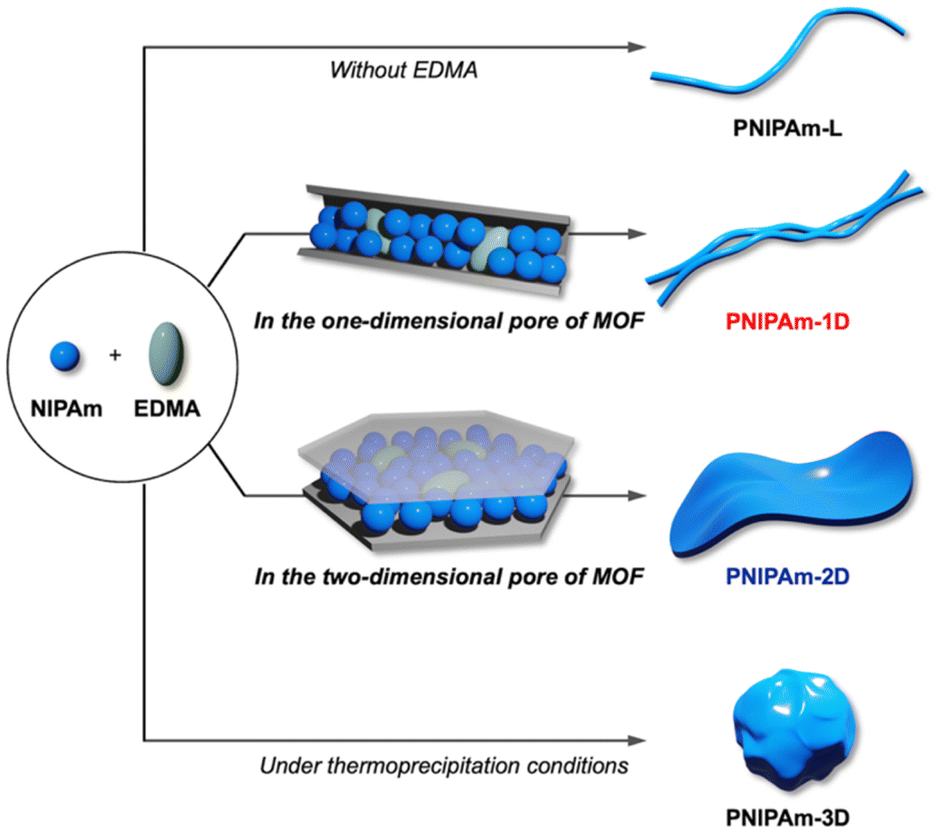 | ||
| Fig. 1 Schematic illustration of the synthesis of linear PNIPAm (PNIPAm-L) and PNIPAms with 1D, 2D, and 3D network topologies (PNIPAm-1D, PNIPAm-2D, and PNIPAm-3D, respectively). | ||
Results and discussions
Preparation of PNIPAm with regulated network topologies
To prepare PNIPAm-1D as double-stranded PNIPAm chains, we employed [In(OH)bdc]n (1, bdc: 1,4-benzenedicarboxylate, Fig. 2a), which has two types of 1D channels with diameters of 17 Å (hexagonal pores) and 5 Å (triangular pores).28 Although the smaller triangular pores cannot accommodate NIPAm monomers, the hexagonal pores are sufficiently large to align two PNIPAm chains exactly along the channels, as suggested by molecular dynamics simulations (Fig. S1†). Based on this observation, we infer that two spatially aligned PNIPAm chains within a pore would be cross-linked through EDMA during the polymerization process, resulting in a double-stranded structure within the nanochannel. | ||
| Fig. 2 (a) Crystal structure of 1; monomers (NIPAm) were inserted into the cavity by molecular modeling. (b) XRPD patterns of simulated 1, 1, and 1-PNIPAm composite. (c) Crystal structure of 2; the monomer was inserted into the cavity by molecular modeling. The pillar ligands (dpb) in 2 are shown as stick models for clarity. (d) XRPD patterns of simulated 2, 2, and 2-PNIPAm composite. Atoms: In (brown), Ni (purple), O (red), N (blue), C (gray), H (white). | ||
We then polymerized NIPAm within 1 in the presence of ethylene glycol dimethacrylate (EDMA) as a cross-linker. The monomer, cross-linker, and radical initiator were introduced into 1 by soaking the degassed host in a CH2Cl2 solution containing these reactants. Removing CH2Cl2 then yielded a composite that included only the reactants inside host 1 (Fig. S2†). The resulting composite was heated at 120 °C for 24 h to promote cross-linking polymerization in the nanochannels of 1 (conversions of 89% and 92% for NIPAm and EDMA, respectively). The X-ray powder diffraction (XRPD) profile of the composite remained unchanged after heating, indicating that the host crystal structure was maintained during polymerization (Fig. 2b).
Acid treatment of the MOF-polymer composite and purification yielded PNIPAm-1D as a white powder. Notably, the obtained polymer was highly soluble in many good solvents for PNIPAm, suggesting that the compartmentalization of the polymer chains inside the channels effectively suppressed unfavorable random cross-linking. This result was supported by the gel permeation chromatography (GPC) of PNIPAm-1D, demonstrating a unimodal curve (no shoulder peaks) with a number-average molecular weight (Mn) and polydispersity (Mw/Mn) of 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900, and 1.77, respectively (Fig. S3†). The 1H NMR spectrum of PNIPAm-1D exhibited peaks for the PNIPAm chains and EDMA cross-links (cross-linking ratio: 10 mol%) (Fig. 3a).
900, and 1.77, respectively (Fig. S3†). The 1H NMR spectrum of PNIPAm-1D exhibited peaks for the PNIPAm chains and EDMA cross-links (cross-linking ratio: 10 mol%) (Fig. 3a).
 | ||
| Fig. 3 1H NMR spectra of (a) PNIPAm-1D and (b) PNIPAm-2D (cross-linking ratios: 10 and 6 mol%, respectively). AFM images and height profiles of (c) PNIPAm-1D and (d) PNIPAm-2D. Samples were deposited on a mica substrate by spin-casting dilute CHCl3 solutions thereof. The height profiles were recorded along the yellow lines in the corresponding AFM images. | ||
PNIPAm-2D with a single-molecule-thick nanosheet structure was synthesized using [Ni(Hbtc)(dpb)]n (2, btc: 1,3,5-benz enetricarboxylate, dpb: 1,4-di(pyridin-4-yl)benzene) as a nanoporous template.17 The pillared-layer structure of this MOF, with an interlayer distance of 1.2 nm, can accommodate only single NIPAm and EDMA molecules between the 2D interstices (Fig. 2c and S1†). The narrow aperture (∼3 Å) of the [Ni(Hbtc)]n layers in 2, which does not allow interlayer exchange for NIPAm monomers and crosslinkers, enables the 2D-constrained growth of polymer networks. PNIPAm-2D was synthesized in 2 at 90 °C for 24 h (Fig. 2d and S2†). Note that cross-linking polymerization in 2 proceeded at a lower temperature than that in 1, likely because of the higher monomer mobility in the 2D nanospaces (conversions of 93% and 100% for NIPAm and EDMA, respectively). PNIPAm-2D was recovered from 2 by digesting the host framework in an aqueous chelate solution. Similar to PNIPAm-1D, isolated PNIPAm-2D was soluble in many solvents despite its cross-linked structure, enabling us to analyze its molecular weight using GPC (Fig. S3†). The calculated molecular weight (Mn = 20900, Mw/Mn = 2.28) could have been underestimated because of the 2D sheet morphology with compact chain assemblies.29 Formation of 2D PNIPAm network was also confirmed by 1H NMR spectroscopy of PNIPAm-2D, exhibiting the characteristic peaks of both EDMA and PNIPAm (cross-linking ratio: 6 mol%) (Fig. 3b).
The morphologies of PNIPAm-1D and PNIPAm-2D were verified by atomic force microscopy (AFM) (Fig. 3c and d). Remarkably, the morphology of PNIPAm-1D was a linear chain structure. The height profile of PNIPAm-1D demonstrated that each chain had a thickness comparable to the diameter of a single PNIPAm chain (0.8 nm), indicating the side-by-side alignment of two PNIPAm chains on the substrate rather than vertical stacking.16 In contrast, the AFM image of PNIPAm-2D revealed uniformly thick 2D nanosheets, the thickness of which sheets corresponded to the cross-sectional diameter of a single PNIPAm chain. These results indicate that the topology and dimensionality of the cross-linked polymers can be successfully controlled using MOFs as nanoporous scaffolds.
To understand the effect of the cross-linking topology on the properties of PNIPAm, we prepared conventional linear PNIPAm without cross-links (PNIPAm-L) by solution polymerization (Fig. S3 and S4,†Mn = 44400, Mw/Mn = 1.63). A randomly cross-linked 3D network was also prepared by cross-linking polymerization under thermoprecipitation conditions, which yielded PNIPAm-3D as a microgel (cross-linking ratio: 8 mol%, similar to those of PNIPAm-1D and PNIPAm-2D) (Fig. S5†).30
Thermoresponsive properties of PNIPAm with regulated network topologies
The thermoresponsive behavior of the PNIPAm samples in diluted aqueous solutions (1 g L−1) was monitored by optical transmittance and dynamic light scattering (DLS) measurements.31 Upon heating, the transmittance of the PNIPAm-L solution changed abruptly through dehydration at the typical coil-to-globule transition temperature of 37 °C (Fig. 4), which was accompanied by a drastic increase in the size of the PNIPAm particles via chain agglomeration (Fig. S6†). This behavior is consistent with the well-known phenomenon for single PNIPAm chains as mentioned above.18 The phase transition of PNIPAm was strongly influenced by cross-linking, as the PNIPAm-3D solution apparently did not show such a clear change (Fig. 4). However, DLS analysis of the PNIPAm-3D solution revealed a marked decrease in hydrodynamic diameter (Dh) from 110 to 50 nm around 30 °C (Fig. 5 and S6†). This contrasting behavior of PNIPAm-3D against PNIPAm-L in terms of temperature-dependent size changes can be explained by the dehydration-induced contraction of microgels, known as volume phase transition.30 | ||
| Fig. 4 (a) Appearances of aqueous solutions of PNIPAm at different temperatures. (b) Temperature dependence of the optical transmittance of aqueous solutions of PNIPAm-L (black), PNIPAm-1D (red), PNIPAm-2D (blue), and PNIPAm-3D (green). | ||
 | ||
| Fig. 5 Hydrodynamic diameters (Dh) of PNIPAm-1D (red), PNIPAm-2D (blue), and PNIPAm-3D (green) in aqueous solutions, measured by DLS at different temperatures. The plotted particle sizes correspond to the peak with the highest volume fraction. The volume-based distributions are shown in Fig. S8.† | ||
The effect of the network topology on the phase transition was examined by investigating the thermoresponsive properties of PNIPAm-1D and PNIPAm-2D in water. Strikingly, the PNIPAm-1D solution remained transparent (transmittance > 99%) in the applied temperature range (Fig. 4). This is because the PNIPAm-1D particles had a very small hydrodynamic diameter (Dh = 20 nm), even after heating (Fig. 5). However, the transition of PNIPAm-1D was clearly detectable at 17.5 °C with DLS, and its transition temperature was much lower than those of PNIPAm-L and PNIPAm-3D (Fig. 5 and S6†). On the other hand, the aqueous PNIPAm-2D solution slowly became turbid upon heating (Fig. 4). The phase transition, which started at 20 °C with a progressive decrease in the transmittance over a wide temperature range, was ascribed to the gradual growth of PNIPAm aggregates, with Dh changing from 30 to 160 nm (Fig. 5 and S6†). Interestingly, the transition of PNIPAm-2D occurred at temperatures between those of PNIPAm-1D and PNIPAm-3D. The progressive thermoresponsive behavior of PNIPAm-2D was not kinetically directed because gradual transmittance changes were also observed during the cooling process (Fig. S9†) and even during a slower temperature sweep (Fig. S10†). We speculate that the planar, sheet-like topology of PNIPAm-2D allows the polymers to aggregate without forming irreversible entanglements, resulting in aggregates that adopt a thermodynamically favorable size at each temperature.
To further investigate the effect of cross-linking, we synthesized PNIPAms with different cross-linking ratios and monitored the coil-to-globule transition process (Fig. S11 and S12†). The transition temperatures of the cross-linked PNIPAms decreased with the increasing cross-linking ratio. In addition to the effect of the cross-linking ratios, the effect of the cross-linking location on the PNIPAm phase transition is disclosed here for the first time. Even with similar cross-linking ratios, the transition temperatures of the cross-linked PNIPAms decreased in the order of the dimensional constraint (PNIPAm-1D < PNIPAm-2D < PNIPAm-3D). Notably, the transition temperature of PNIPAm-1D is among the lowest of all known PNIPAm materials.23,32,33 The remarkable properties of PNIPAm-1D are likely the result of the 1D network topology with its parallel arrangement of double PNIPAm chains. The alignment of adjacent hydrophobic isopropyl groups could maximize the interactions between the two chains, facilitating the dehydration of PNIPAm at lower temperatures and favoring aggregation within the double strand, which reduces the number of chains participating in single aggregates.33,34 The resulting spherical particles (only several tens of nanometers in size) remained stable in aqueous solution for a few weeks, even at room temperature (above the transition temperature) (Fig. S13†). For PNIPAm-2D, cross-linking polymerization in 2 yielded uniformly thick nanosheets with a 2D network topology. Immobilizing the polymer chains in the same plane can facilitate hydrophobic interactions within the monolayers, thereby lowering the transition temperature. However, because the possible multiple in-plane orientations of its chains prevent proximal chain arrangement, its transition temperature is higher than that of PNIPAm-1D. In contrast with the other PNIPAm samples, PNIPAm-2D showed a gradual transition due to its 2D network geometry that topologically disables entanglement among the polymer networks. Above the transition temperature, dehydration triggers the stacking of the 2D sheets rather than chain entanglement, initially forming morphologically anisotropic aggregates. The heterogeneity of the primary particles in terms of size and morphology plausibly leads to the gradual thermoresponse of PNIPAm-2D in water over a wide temperature range. By contrast, the other PNIPAms form spherical particles with small surface areas via chain entanglement, which prevents further merging.35 The plausible aggregation behavior of low-dimensionally crosslinked PNIPAm were illustrated in Fig. S14.†
Conclusions
In conclusion, the cross-linking polymerization of NIPAm in the 1D and 2D nanochannels of MOFs formed PNIPAm chains with double-stranded and unimolecularly thick nanosheet structures with unprecedented network topologies. These well-defined network topologies significantly affected the thermoresponsive behavior of PNIPAm in water by critically determining the temperature and aggregation during the coil-to-globule transition. The parallel alignment of the double PNIPAm chains significantly lowered the transition temperature, whereas monolayer sheets significantly delayed the transition of the aggregates. Imposing dimensional constraints on cross-linking rationally affords network topologies that are inaccessible by solution and bulk polymerization; additionally, it offers new insights into polymeric properties, thereby advancing the development of networked polymers.Experimental
Synthesis of PNIPAm-L
NIPAm (2.00 g) and AIBN (8.6 mg) were dissolved in 2-propanol (8 mL) and degassed by bubbling N2 for 5 min. Heating the mixture at 70 °C for 2 h, followed by reprecipitation into n-hexane three times and drying under reduced pressure, afforded PNIPAm-L as a linear reference (973.3 mg). Mn and Mw/Mn were measured using GPC (Fig. S3,†Mn = 44400, Mw/Mn = 1.63).
Synthesis of PNIPAm-1D
1 (2.12 g) was evacuated at 120 °C overnight to remove residual guest molecules from the pores. CH2Cl2 (2 mL) was initially added to the activated 1. Then, a CH2Cl2 solution (6 mL) of NIPAm, (740 mg), EDMA (150 mg), and benzoyl peroxide (20.0 mg) was added to 1 in a round-bottom flask and left for 30 min to encapsulate the monomers in the channels of 1. The solvent was slowly removed under reduced pressure (40 kPa) and then vacuumed at 0.1 kPa for 1 h. Subsequently, radical polymerization was performed at 120 °C for 24 h under a nitrogen atmosphere, yielding a composite of 1 with PNIPAm-1D in the channels. The conversions were determined from the 1H NMR spectra of the MOF composites digested in DMSO-d6/D2O (35% DCl) (v/v = 9:1). The integral ratio of peaks corresponding to the vinyl protons was compared before and after polymerization, using the peaks of bdcH2 as an internal standard (89% and 92% for NIPAm and EDMA, respectively). The composite was added to DMF-aqueous HCl (9/1) solution to decompose the host framework, and the obtained polymer was purified by dialysis in DMF and methanol twice each cycle. After dissolving the polymer in CHCl3, a MOF-derived residue was removed by passing through the Celite pad. Finally, the residual DMF was removed using a preparative GPC. The polymer solution was poured into hexane to obtain the powder sample of PNIPAm-1D (378 mg). The degree of cross-linking was calculated from the 1H NMR spectrum (Fig. 3a, cross-linking ratio = 10 mol%). Mn and Mw/Mn were measured using GPC (Fig. S3,†Mn = 74900, Mw/Mn = 1.77, polystyrene standard).
Synthesis of PNIPAm-2D
A CH2Cl2 solution of NIPAm (180 mg), EDMA (15.8 mg), and AIBN (5 mg) was added to 2 (2 g) in a round-bottom flask and left for 30 min to allow the monomers to diffuse into 2. The solvent was slowly removed under reduced pressure and then vacuumed at 3 kPa for 30 min. Subsequently, radical polymerization was performed at 90 °C for 24 h under a nitrogen atmosphere, yielding a composite of 2 with PNIPAm-2D in the nanospaces. The conversions were determined from the 1H NMR spectra of the MOF composites digested in DMSO-d6/D2SO4 (4% D2O) (v/v = 9:1) (93% and 100% for NIPAm and EDMA, respectively; dpb as a standard). The composite was added to methanol-0.1 M aqueous Na4EDTA solution to decompose the host framework, and the polymer was purified by dialysis in methanol/H2O (v/v = 1:1) and methanol twice each cycle. After removing the solvent, the obtained polymer was dissolved in CH2Cl2, and the solution was poured into hexane to obtain the powder product of PNIPAm-2D (133 mg). The degree of cross-linking was calculated from the 1H NMR spectrum (Fig. 3b, cross-linking ratio = 6 mol%). Mn and Mw/Mn were measured using GPC (Fig. S3,†Mn = 20900, Mw/Mn = 2.28, polystyrene standard).
Synthesis of PNIPAm-3D
PNIPAm-3D was synthesized according to the literature with slight modifications.30 NIPAm (501.0 mg), EDMA (76.8 mg), AIBN (11.9 mg), and sodium dodecyl sulfate (25.0 mg) were dissolved in water (25 mL) and stirred for 3 h. After degassing by N2 bubbling for 5 min, the solution was heated at 70 °C for 1 h. After purification by dialysis in water, the polymer was dissolved in CHCl3 and poured into hexane to obtain the powder product of PNIPAm-3D (502.9 mg).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. K.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, validation, visualization, writing – original draft, A. S.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, validation, visualization, writing – original draft, S. H.: data curation, formal analysis, methodology, T. U.: conceptualization, funding acquisition, project administration, resources, supervision, validation, visualization, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (23K13788 and 23K13789 for Y. K. and A. N., respectively). T. U. acknowledges CREST program (JPMJCR20T3) of Japan Science and Technology Agency (JST) and Data Creation and Utilization-Type Material Research and Development Project (JPMXP1122714694) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan.Notes and references
- (a) S. N. V. T. Kuchibhatla, A. S. Karakoti, D. Bera and S. Seal, Prog. Mater. Sci., 2007, 52, 699 CrossRef CAS; (b) C. Tian, W. Miao, L. Zhao and J. Wang, Rev. Phys., 2023, 10, 100082 CrossRef; (c) C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225 CrossRef CAS.
- (a) S. Gong and W. Cheng, Adv. Electron. Mater., 2017, 3, 1600314 CrossRef; (b) A. K. Katiyar, A. T. Hoang, D. Xu, J. Hong, B. J. Kim, S. Ji and J.-H. Ahn, Chem. Rev., 2024, 124, 318 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- M. N. Esfahani and B. E. Alaca, Adv. Eng. Mater., 2019, 21, 1900192 CrossRef.
- P. Kumbhakar, J. S. Jayan, A. S. Madhavikutty, P. R. Sreeram, A. Saritha, T. Ito and C. S. Tiwary, iScience, 2023, 26, 106671 CrossRef CAS PubMed.
- A. G. N. Sofiah, M. Samykano, K. Kadirgama, R. V. Mohan and N. A. C. Lah, Appl. Mater. Today, 2018, 11, 320 CrossRef.
- P. Kumbhakar, C. C. Gowda, P. L. Mahapatra, M. Mukherjee, K. D. Malviya, M. Chaker, A. Chandra, B. Lahiri, P. M. Ajayan, D. Jariwala, A. Singh and C. S. Tiwary, Mater. Today, 2021, 45, 142 CrossRef CAS.
- Y. Gu, Z. Qiu and K. Müllen, J. Am. Chem. Soc., 2022, 144, 11499 CrossRef CAS PubMed.
- (a) Z. Cai, B. Liu, X. Zou and H.-M. Cheng, Chem. Rev., 2018, 118, 6091 CrossRef CAS; (b) M. A. Islam, P. Serles, B. Kumral, P. G. Demingos, T. Qureshi, A. Meiyazhagan, A. B. Puthirath, M. S. B. Abdullah, S. R. Faysal, P. M. Ajayan, D. Panesar, C. V. Singh and T. Filleter, Appl. Phys. Rev., 2022, 9, 041301 CrossRef CAS.
- (a) R. Shrestha, P. Li, B. Chatterjee, T. Zheng, X. Wu, Z. Liu, T. Luo, S. Choi, K. Hippalgaonkar, M. de Boer and S. Shen, Nat. Commun., 2018, 9, 1664 CrossRef PubMed; (b) H. Ko, D. Kang, Y. Choi, Y. Wi, S. Kim, H. Pham, K. Lee, N. Godman, M. Mcconney and K. Jeong, J. Am. Chem. Soc., 2024, 146, 4393 CrossRef CAS; (c) Y. Jiang, L. Ning, C. Liu, Y. Sun, J. Li, Z. Liu, Y. Yi, D. Qiu, C. He, Y. Guo, W. Hu and Y. Liu, Chem, 2021, 7, 1258 CrossRef CAS; (d) G. Distefano, H. Suzuki, M. Tsujimoto, S. Isoda, S. Bracco, A. Comotti, P. Sozzani, T. Uemura and S. Kitagawa, Nat. Chem., 2013, 5, 335 CrossRef CAS PubMed.
- (a) C. K. Varnava and C. S. Patrickios, Polymer, 2021, 215, 123322 CrossRef CAS; (b) G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181 CrossRef CAS PubMed.
- (a) Y. S. Gao, D. Z. Zhou, J. Lyu, A. Sigen, Q. Xu, B. Newland, K. Matyjaszewski, H. Y. Tai and W. X. Wang, Nat. Rev. Chem., 2020, 4, 194 CrossRef CAS PubMed; (b) Y. W. Gu, J. L. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022 CrossRef CAS PubMed.
- (a) N. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- (a) L. Jiao, Y. Wang, H. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef; (b) M. Dinca, A. Iliescu, J. Oppenheim and C. Sun, Chem. Rev., 2023, 123, 6197 CrossRef.
- (a) A. Nishijima, Y. Kametani and T. Uemura, Coord. Chem. Rev., 2022, 466, 214601 CrossRef CAS; (b) M. Kalaj, K. C. Bentz, S. Ayala, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, Chem. Rev., 2020, 120, 8267 CrossRef CAS; (c) B. Schmidt, Macromol. Rapid Commun., 2020, 41, 1900333 CrossRef CAS.
- M. Abe, Y. Kametani and T. Uemura, J. Am. Chem. Soc., 2023, 145, 2448 CrossRef CAS.
- N. Hosono, S. Mochizuki, Y. Hayashi and T. Uemura, Nat. Commun., 2020, 11, 3573 CrossRef CAS.
- (a) H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS; (b) A. Halperin, M. Kroger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342 CrossRef CAS PubMed.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607 RSC.
- H. Wei, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Prog. Polym. Sci., 2009, 34, 893 CrossRef CAS.
- J. Liu, L. Jiang, S. R. He, J. Zhang and W. Shao, Chem. Eng. J., 2022, 433, 133496 CrossRef CAS.
- Y. Xia, X. Yin, N. Burke and H. Stover, Macromolecules, 2005, 38, 5937 CrossRef CAS.
- B. Ray, Y. Okamoto, M. Kamigaito, M. Sawamoto, K. Seno, S. Kanaoka and S. Aoshima, Polym. J., 2005, 37, 234 CrossRef CAS.
- P. De, S. Gondi and B. Sumerlin, Biomacromolecules, 2008, 9, 1064 CrossRef CAS PubMed.
- H. Kojima, Y. Imamura, Y. Lu, S. Yamago and T. Koga, Macromolecules, 2022, 55, 7932 CrossRef CAS.
- (a) T. Kawaguchi, K. Kobayashi, M. Osa and T. Yoshizaki, J. Phys. Chem. B, 2009, 113, 5440 CrossRef CAS PubMed; (b) C. Wang and T. Hashimoto, Macromolecules, 2023, 56, 6354 CrossRef CAS.
- (a) M. I. Din, R. Khalid, F. Akbar, G. Ahmad, J. Najeeb and Z. U. N. Hussain, Soft Mater., 2018, 16, 228 CrossRef CAS; (b) B. Sierra-Martin, J. R. Retama, M. Laurenti, A. F. Barbero and E. L. Cabarcos, Adv. Colloid Interface Sci., 2014, 205, 113 CrossRef CAS; (c) R. Acciaro, T. Gilanyi and I. Varga, Langmuir, 2011, 27, 7917 CrossRef CAS.
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Ferey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892 CrossRef CAS PubMed.
- A. Nishijima, Y. Hayashi, K. Mayumi, N. Hosono and T. Uemura, Macromolecules, 2023, 56, 3141 CrossRef CAS.
- C. Obeso-Vera, J. Cornejo-Bravo, A. Serrano-Medina and A. Licea-Claverie, Polym. Bull., 2013, 70, 653 CrossRef CAS.
- A. V. Gorelov, A. DuChesne and K. A. Dawson, Phys. Stat. Mech. Appl., 1997, 240, 443 CrossRef CAS.
- (a) A. P. Vogt and B. S. Sumerlin, Macromolecules, 2008, 41, 7368 CrossRef CAS; (b) Q. Duan, A. Narumi, Y. Miura, X. Shen, S. Sato, T. Satoh and T. Kakuchi, Polym. J., 2006, 38, 306 CrossRef CAS.
- J. Ye, J. Xu, J. M. Hu, X. F. Wang, G. Z. Zhang, S. Y. Liu and C. Wu, Macromolecules, 2008, 41, 4416 CrossRef CAS.
- (a) M. X. Cao, Y. T. Liu, X. W. Zhang, F. Li and M. J. Zhong, Chem, 2022, 8, 1460 CrossRef CAS; (b) K. Terao, A. Masahiro, M. Nagase, S. Takeshima, S. Ida and S. Kanaoka, Macromolecules, 2023, 56, 5635 CrossRef CAS.
- J. Chuang, A. Y. Grosberg and T. Tanaka, J. Chem. Phys., 2000, 112, 6434 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02118a |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2025 |