
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValence state engineering in multi-heteroatom-doped PAHs: a strategy for tunable photophysical properties and phototheranostic potentials†
Yao
Ma‡
a,
Dongxu
Li‡
a,
Xin
Chen‡
b,
Xinqiang
Hua
a,
Cheng-Shan
Yuan
a,
Jianguo
Wang
 *b,
Zitong
Liu
a,
Hao-Li
Zhang
a and
Xiangfeng
Shao
*a
*b,
Zitong
Liu
a,
Hao-Li
Zhang
a and
Xiangfeng
Shao
*a
aResearch Center for Free Radical Chemistry, State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Tianshui Southern Road 222, Lanzhou, Gansu Province, China. E-mail: shaoxf@lzu.edu.cn
bCollege of Chemistry and Chemical Engineering, College of Biomedical Sciences, Inner Mongolia Key Laboratory of Synthesis and Application of Organic Functional Molecules, Inner Mongolia University, Hohhot 010021, P. R. China. E-mail: wangjg@iccas.ac.cn
First published on 16th June 2025
Abstract
Precise modulation of the electronic structures in heteroatom-doped polycyclic aromatic hydrocarbons (hetero-PAHs) is essential for advancing organic optoelectronic materials. Herein, we report a facile synthetic strategy for hetero-PAHs co-doped with N, O, and S/Se, achieved via acid-catalyzed Pictet–Spengler reactions and thermally induced ipso-substitution. Systematic π-extension results in a large redshift in optical absorption/emission and enhances fluorescence quantum yield (ΦF) from 4.0% to 30.5%. Furthermore, a post-synthetic valence state engineering approach enables precise tuning of photophysical properties: (i) oxidation of thiophene units to thiophene S,S-dioxides increases ΦF to 42.3% and enhances singlet oxygen generation, and (ii) pyridinium-functionalized hetero-PAHs exhibit strong near-infrared absorption, leading to high photothermal conversion efficiency (up to 61%) at the molecular level. Notably, Se-doped derivatives outperform their S-doped counterparts, underscoring the heavy-atom effect in triplet-state modulation. This work provides a versatile platform for tailoring hetero-PAH electronic structures via valence state manipulation, offering potential applications for organic electronics and phototheranostics.
Introduction
Organic optoelectronic materials have garnered significant attention due to their potential in the development of flexible optoelectronic devices and their inherent biocompatibility, which facilitates applications in biological systems. Among these materials, polycyclic aromatic hydrocarbons (PAHs) have emerged as indispensable building blocks owing to their highly tunable electronic structures and versatile synthetic accessibility.1 Expanding the π-conjugated framework of PAHs effectively induces bathochromic shifts in both absorption and photoluminescence, particularly extending into the near-infrared (NIR) region, which is highly desirable for bioimaging and photothermal applications.2Beyond extending conjugation, the incorporation of heteroatoms into PAH scaffolds represents a powerful strategy to tailor their geometric and electronic structures, modulate charge transport and photophysical properties, and introduce novel functionalities, thereby broadening their applicability across various domains.3 Nitrogen is among the most widely utilized heteroatoms for PAH doping, yielding N-doped PAHs with outstanding optical and charge-transport properties.4 Meanwhile, chalcogen-doped PAHs (incorporating O, S, Se, or Te) have attracted considerable interest in crystal engineering and materials science.5,6 When multiple heteroatoms are simultaneously introduced into the frameworks of PAHs, their differences in electronegativity, valence states, and atomic radii further enrich the electronic properties, leading to enhanced light absorption, charge separation and transport, and energy conversion efficiencies.7 Notably, the incorporation of heavy atoms into organic dyes promotes intersystem crossing (ISC), thereby boosting singlet oxygen (1O2) generation and photothermal conversion efficiency, which are crucial for applications in photodynamic and photothermal therapies.8
In addition to heteroatom doping, modulating the valence states of heteroatom dopants provides an effective approach to fine-tune the optoelectronic properties of hetero-PAHs. For instance, in thiophene-containing hetero-PAHs, oxidation of the thiophene units to thiophene S,S-dioxides significantly enhances photoluminescence quantum yields9 and induces a transition from p-type to n-type charge transport characteristics.10 Similarly, nitrogen onium salts derived from N-doped PAHs exhibit intriguing optoelectronic properties and self-assembly behavior.11 A notable example is the N-methylated pyridinium salts of N-doped PAHs, which have demonstrated broad applicability in fluorescent dyes, cellular imaging, and phototheranostic applications.12
Given the growing demand for high-performance organic materials, rational molecular design strategies involving heteroatom doping and valence state manipulation of PAHs offer an exciting platform for tailoring optoelectronic properties. Herein, we report the design, synthesis, and optical properties of a series of multi-heteroatom-doped PAHs. Our molecular engineering approach integrates three key strategies: (i) multi-element heteroatom doping, (ii) π-conjugation extension, and (iii) main-group element valence state modulation.
We have synthesized a kind of hetero-PAHs co-doped with nitrogen and chalcogen,7c which display cyan green fluorescence with quantum yield (ΦF) of 4.0% (Scheme 1). Expanding the π-system plays a pivotal role in tuning absorption and emission properties. By fusing a benzopyran unit onto the periphery of a PAH scaffold, we introduce an additional oxygen dopant, achieving a bathochromic shift in absorption and emission with an enhanced ΦF of 11.7%. Further π-extension through bis-fusion results in red fluorescence with a significantly improved ΦF of 30.5%. Finally, the manipulation of valence states of heteroatom dopants is conducted, resulting distinct modulation on the electronic structures. Oxidation of thiophene rings to thiophene S,S-dioxides enhances the ΦF to 42.3% and enables efficient 1O2 generation. Meanwhile, pyridinium-functionalized hetero-PAHs exhibit strong NIR absorption, leading to high photothermal conversion efficiency (up to 61%) at the molecular level. Notably, selenium-doped derivatives outperform their sulfur analogs, highlighting the heavy-atom effect in triplet-state modulation. This work provides a facile synthetic approach for constructing multi-heteroatom-doped PAHs with tunable electronic structures and promising potential in phototheranostics.
 | ||
| Scheme 1 The molecular design principles underlying this work. | ||
Results and discussion
Synthesis of N, O, and S/Se co-doped hetero-PAHs
Developing efficient synthetic strategies for hetero-PAHs co-doped with nitrogen (N) and chalcogen atoms (O, S, and Se) is challenging. Herein, we report a scalable and systematic approach toward such kind of hetero-PAHs. To achieve the efficient synthesis of them, it is advantageous to begin with starting materials that are already doped with one of these heteroatoms. In this study, compounds 1a/1b, which feature sulfur (S) or selenium (Se) in their π-scaffolds, were selected as the precursors. These compounds are particularly suitable due to their scalable synthesis as reported in our previous work.7c Using a combination of acid-catalyzed Pictet–Spengler (P–S) reactions13 and thermally induced ipso-substitutions,7b a series of hetero-PAHs co-doped with N, O, and S/Se were synthesized efficiently (Scheme 2). | ||
Scheme 2 Synthesis of hetero-PAHs 5a/5b and 7a/7b. Reagents and conditions: (i) TBN (2.0 equiv.), CH2Cl2, RT, 4 h; (ii) Zn powder (7.0 equiv.), AcOH (2.0 equiv.), C2H5OH-THF (v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), 90 °C, 6 h; (iii) TFA, 120 °C, 12 h; (iv) 185 °C for 12 h under vacuum. :5), 90 °C, 6 h; (iii) TFA, 120 °C, 12 h; (iv) 185 °C for 12 h under vacuum. | ||
The synthesis began with nitration of 1a/1b using tert-butyl nitrite (TBN) at room temperature (RT), followed by reduction with Zn/AcOH to yield intermediates 2a/2b in overall yields of 80% and 72%, respectively. The subsequent P–S reaction with benzaldehyde, catalyzed by trifluoroacetic acid (TFA), afforded 3a/3b in yields of 71% and 73%, respectively. Similarly, reacting 2a/2b with salicylaldehyde provided 4a/4b in isolated yields of 60% and 75%. To construct the final framework of the desired hetero-PAHs, thermally induced intramolecular ipso-substitution of 4a/4b was performed by heating at 185 °C under vacuum for 12 hours, yielding 5a/5b in isolated yields of 74%/92%. Furthermore, the P–S reaction of 2a/2b with 2,5-dihydroxy-1,4-benzenedicarboxaldehyde enabled a two-fold ring-cyclization, giving 6a/6b in yields of 51%/66%. The intermediates 6a/6b underwent a two-fold thermally induced ipso-substitution to afford 7a/7b in yields of 76%/81%. As a result, the hetero-PAHs 5a/5b and 7a/7b, co-doped with N, O, and S/Se, were successfully synthesized. This method demonstrates a robust and scalable pathway for synthesizing hetero-PAHs with tailored doping profiles. Remarkably, the structure of 7a/7b features a novel subunit that can be conceptualized as a hetero-heptacene framework (painted with red background in Scheme 2), which is composed of alternating chalcogenole, pyran, and pyridine moieties. Needless to say, this is the first construction of such kind of hetero-acenes.
Manipulation of the valence states of heteroatom dopants
To further tailor the optoelectronic properties of the synthesized hetero-PAHs, we explored the valence state manipulation of the heteroatom dopants. Thiophene S,S-dioxides are well-documented for their excellent photoluminescence (PL) properties9 and n-type charge transport behavior,10a,d making their incorporation a promising strategy for enhancing material performance. To this end, the thiophene moieties in 5a and 7a were selectively oxidized to thiophene S,S-dioxides under optimized conditions (Scheme 3). The oxidation of 5a using hydrogen peroxide (H2O2) proceeded efficiently, yielding compound 8a with a 60% isolated yield.14 However, the oxidation of 7a with H2O2 resulted in complex mixtures that were hard to isolate by column chromatography. This limitation was overcome by employing meta-chloroperoxybenzoic acid (mCPBA) as the oxidant, which selectively oxidized the thiophene units in 7a to their corresponding S,S-dioxides, affording 9a in a yield of 40%.15 These results underscore the critical role of oxidant choice in controlling the reaction outcome. | ||
| Scheme 3 Synthesis of 8a and 9a. Reagents and conditions: (i) H2O2 (30% aqueous), CHCl3–AcOH (v/v = 1:1), 60 °C, 6 h; (ii) mCPBA (10.0 equiv.), CHCl3, 60 °C, 12 h. | ||
Additionally, the electronic structure of pyridine-containing hetero-PAHs will be altered by transforming the pyridine moieties into pyridinium salts.12 Hence, the pyridine moieties on 5a/5b and 7a/7b were transformed into the pyridinium salts as shown in Scheme 4. Methylation of the pyridine rings in 5a/5b and 7a/7b using methyl trifluoromethanesulfonate (MeOTf) at 120 °C afforded pyridinium salts 10a/10b (isolated yields, 62%/62%) and 11a/11b (isolated yields, 60%/66%). This transformation introduced a strong electron-withdrawing effect, significantly lowering the electron density across the PAH framework and further tuning their electronic properties. The integration of scalable starting materials, controlled doping, and post-synthetic valence state modulation demonstrates a versatile approach for tailoring the electronic structures and optoelectronic properties of PAHs.
 | ||
| Scheme 4 Synthesis of 10a/10b and 11a/11b. Reagents and conditions: (i) MeOTf (3.0 equiv.), toluene, 120 °C, 12 h. | ||
Molecular structures
To gain insight into the effects of π-system extension, heteroatom doping, and the valence state of dopants on molecular conformations, the structures of these hetero-PAHs were analyzed by theoretical calculations and single-crystal X-ray diffraction.The molecular structures of compounds 3a/3b, 5a/5b, and 7a/7b–11a/11b were optimized at the B3LYP/Def2-SVP level of theory, with n-butyl groups replaced by methyl groups, yielding 3a′/3b′, 5a′/5b′, and 7a′/7b′–11a′/11b′. The optimized structures of hetero-PAHs co-doped with N, O, and S are shown in Fig. 1, while those doped with N, O, and Se are presented in Fig. S39–S43.† Compound 3a′ adopts a bowl-shaped conformation with a bowl-depth of 0.32 Å (Fig. 1a). However, upon fusing a pyran moiety to the core structure, as in 5a′, the molecule has a nearly planar π-framework (Fig. 1b), suggesting that the introduction of the pyran moiety induces a flattening of the curved π-system. Oxidation of the thiophene moieties in 5a′ to thiophene S,S-dioxides further flattens the structure, resulting in a completely planar π-scaffold of 8a′ (Fig. 1c). In contrast, methylation of the pyridine ring in 5a′ leads to a shift from a planar structure to a bowl-shaped conformation in 10a′ (bowl-depth, 0.34 Å) as shown in Fig. 1d. Moreover, extension of the π-system from 5a′ to 7a′ changes the molecular conformation from planar to S-shaped, with bowl-depths of 0.41 Å and 0.31 Å for the two segments of 7a′ respectively (Fig. 1e). It is also found that altering the valence states of heteroatom dopants in 7a′ causes significant changes in molecular geometry. For instance, compound 9a′ takes a nearly planar conformation (Fig. 1f), while 11a′ possesses a boat-like conformation with bowl-depths of 0.43 Å and 0.36 Å for the two segments (Fig. 1g).
 | ||
| Fig. 1 Optimized molecular structures of (a) 3a′, (b) 5a′, (c) 8a′, (d) 10a′, (e) 7a′, (f) 9a′, and (g) 11a′. | ||
The results demonstrate that molecular conformation of these hetero-PAHs is highly influenced by the extension of the π-system, the nature of heteroatom doping, and the valence state of the dopants. The valence state modulation of heteroatoms in particular plays a crucial role in controlling molecular geometry, from curved to planar or even boat-like conformations. These insights are crucial for the rational design of molecular materials where control over the conjugation and structure can lead to tunable electronic properties.
Fortunately, we have obtained the single crystals of 9a, 10a, and 10b which were suitable for X-ray single-crystal diffraction analysis. Red needle-like crystals of 9a were grown by slow evaporation of a chlorobenzene solution at RT, while dark blue needle-like single crystals of 10a and 10b were harvested from a mixed solvent system of CH2Cl2 and CH3OH (v/v = 1:1). Herein, the crystal structures of 9a and 10a will be reported. The crystal structure of 10b is similar to that of 10a and is shown in Fig. S5.†
Compound 9a crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The crystal contains two distinct molecules (A and B), with molecule A and half of molecule B being crystallographically unique. Both molecules exhibit similar geometries, with the structure of molecule A depicted in Fig. 2a and b. The C
space group. The crystal contains two distinct molecules (A and B), with molecule A and half of molecule B being crystallographically unique. Both molecules exhibit similar geometries, with the structure of molecule A depicted in Fig. 2a and b. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths in the triphenylene moiety of A are consistent with those observed in trichalcogenasumanenes.6d However, the CC bonds within the pyran ring, highlighted in red in Fig. 2a, show notable bond length variation: bonds r1 (1.46 Å) and r2 (1.44 Å) are clearly longer than r3 (1.38 Å) and r4 (1.40 Å). The two C–O bonds (r5 and r6) in the pyran ring are nearly identical in length, measuring 1.37–1.38 Å. In contrast, the two C–N bonds in the pyridine ring (highlighted in blue) differ significantly, with r7 = 1.39 Å and r8 = 1.31 Å. Molecule A adopts a curved π-framework (Fig. 2b), with a dihedral angle of 13° between the mean planes of the two conjugated subunits (cyan dashed lines). In the crystal, molecules A and B aggregate in a columnar arrangement (Fig. 2c). Within the stacking columns, two A molecules and one B molecule form a trimer through multiple intermolecular C⋯C contacts (3.27–3.39 Å) between their π-frameworks, as indicated by green dashed lines. Additionally, C⋯O interactions (2.92–3.21 Å) are observed between the carbon π-framework and oxygen atoms on the thiophene S,S-dioxide units. Neighboring trimers are further connected by van der Waals interactions between their butoxyl chains.
C bond lengths in the triphenylene moiety of A are consistent with those observed in trichalcogenasumanenes.6d However, the CC bonds within the pyran ring, highlighted in red in Fig. 2a, show notable bond length variation: bonds r1 (1.46 Å) and r2 (1.44 Å) are clearly longer than r3 (1.38 Å) and r4 (1.40 Å). The two C–O bonds (r5 and r6) in the pyran ring are nearly identical in length, measuring 1.37–1.38 Å. In contrast, the two C–N bonds in the pyridine ring (highlighted in blue) differ significantly, with r7 = 1.39 Å and r8 = 1.31 Å. Molecule A adopts a curved π-framework (Fig. 2b), with a dihedral angle of 13° between the mean planes of the two conjugated subunits (cyan dashed lines). In the crystal, molecules A and B aggregate in a columnar arrangement (Fig. 2c). Within the stacking columns, two A molecules and one B molecule form a trimer through multiple intermolecular C⋯C contacts (3.27–3.39 Å) between their π-frameworks, as indicated by green dashed lines. Additionally, C⋯O interactions (2.92–3.21 Å) are observed between the carbon π-framework and oxygen atoms on the thiophene S,S-dioxide units. Neighboring trimers are further connected by van der Waals interactions between their butoxyl chains.
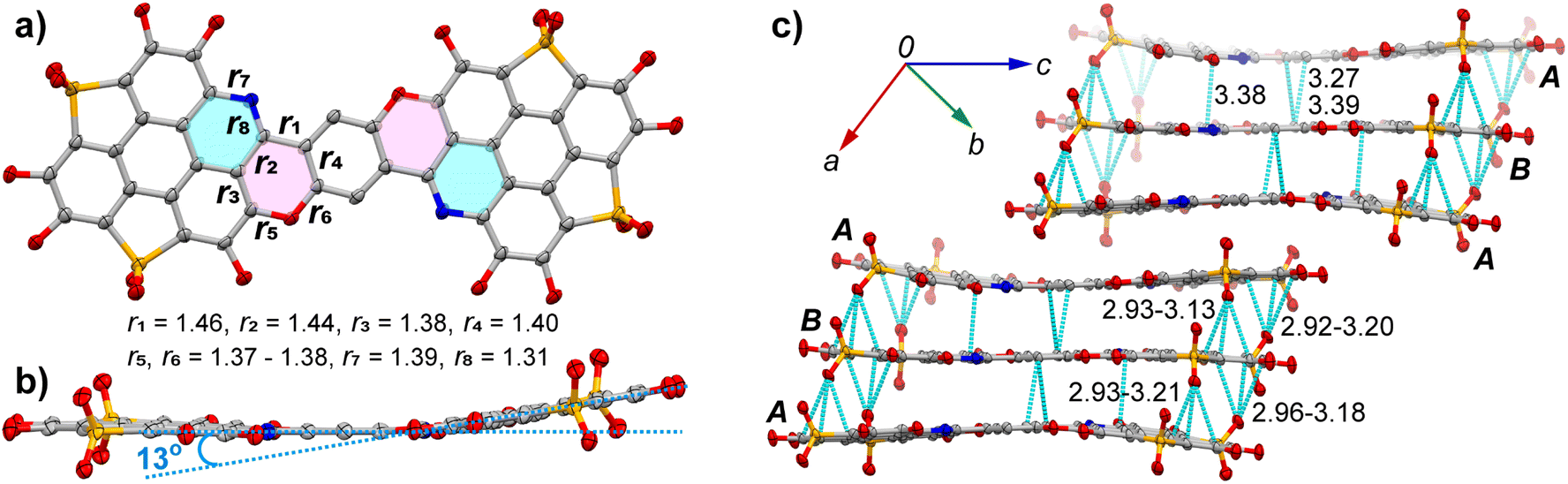 | ||
| Fig. 2 Crystal structure of 9a with n-butyl groups and hydrogen atoms omitted for clarity. (a) Top view of molecule A with selected bond lengths shown in unit of Å; (b) side view of A, showing the dihedral angle between the two planar subunits (cyan dashed lines); (c) packing structure with intermolecular short contacts shown in green dashed lines. The C, N, O, and S atoms are colored in grey, blue, red, and orange, respectively. | ||
Compound 10a adopts the P21/c space group with the asymmetric unit contains one molecule. The crystal structure of 10a is presented in Fig. 3. Similar to 9a, bond length variation is observed in the pyran ring, where r1 (1.45 Å) is longer than r2 (1.42 Å), r3 (1.41 Å), and r4 (1.41 Å) as shown in Fig. 3a. In comparison with 9a, the formation of a pyridinium salt in 10a gives rise to the elongation of the C–N bonds in the pyridine ring, with r7 = 1.43 Å and r8 = 1.36 Å. The π-conjugated skeleton of 10a is nearly planar, with an average deviation of 0.023 Å (Fig. 3b). However, steric hindrance from the methyl group on the pyridinium salt causes deviations from the π-conjugated plane, particularly for the nitrogen atom (0.245 Å), C24 (0.162 Å), and C25 (0.160 Å). Moreover, the methyl group on the pyridinium is distinctly displaced from the π-conjugated plane, with a torsion angle of 26° between the methyl group and the terminal benzene. In the crystal, 10a adopts a columnar arrangement (Fig. 3c), where 10a molecules are dimerized in a head-to-tail manner due to dipole–dipole interactions. Within a dimer, there are four C⋯C contacts (3.36–3.37 Å) between the π-frameworks of two molecules. Similar as that observed in 9a, the neighboring dimers of 10a are connected by van der Waals interactions between their butoxyl chains.
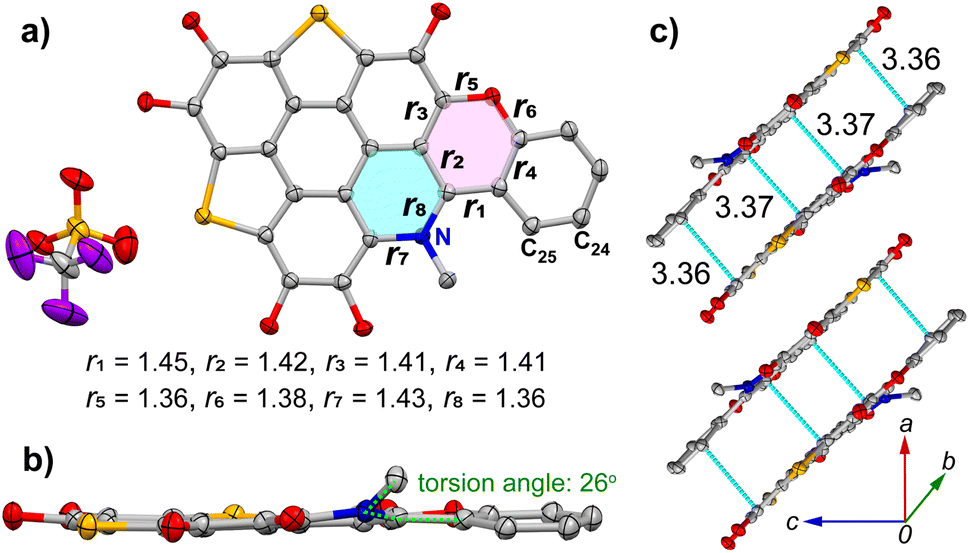 | ||
| Fig. 3 Crystal structure of 10a with n-butyl groups and hydrogen atoms omitted for clarity. (a) Top view of molecule with the selected bond lengths in unit of Å; (b) side view with torsion angle shown; (c) packing structure with intermolecular short contacts shown in green dashed lines. The C, N, O, F, and S atoms are colored in grey, blue, red, purple, and orange, respectively. | ||
It is worth of noting that the optimized molecular structures of 9a/10a are different from those observed in their crystal structures. The key reason is attributed to the packing effect on the molecular conformation as has been documented in the early reports.14b,16 For instance, the strong interactions between the carbon π-framework and oxygen atoms on the thiophene S,S-dioxide units in 9a result in the deformation of molecular conformation.
Electronic structures
The electronic structures of the hetero-PAHs under investigation were analyzed through density functional theory (DFT) calculations. The frontier molecular orbitals (FMOs) of the hetero-PAHs were computed at the B3LYP/Def2-TZVP level of theory, based on the optimized structures of 3a′/3b′, 5a′/5b′, and 7a′/7b′–11a′/11b′. The calculated HOMO, LUMO, and energy levels for compounds 3a′, 5a′, 7a′, 9a′, and 11a′ are presented in Fig. 4. The N, O, and Se co-doped hetero-PAHs exhibit similar features as presented in Fig. S39–S43.†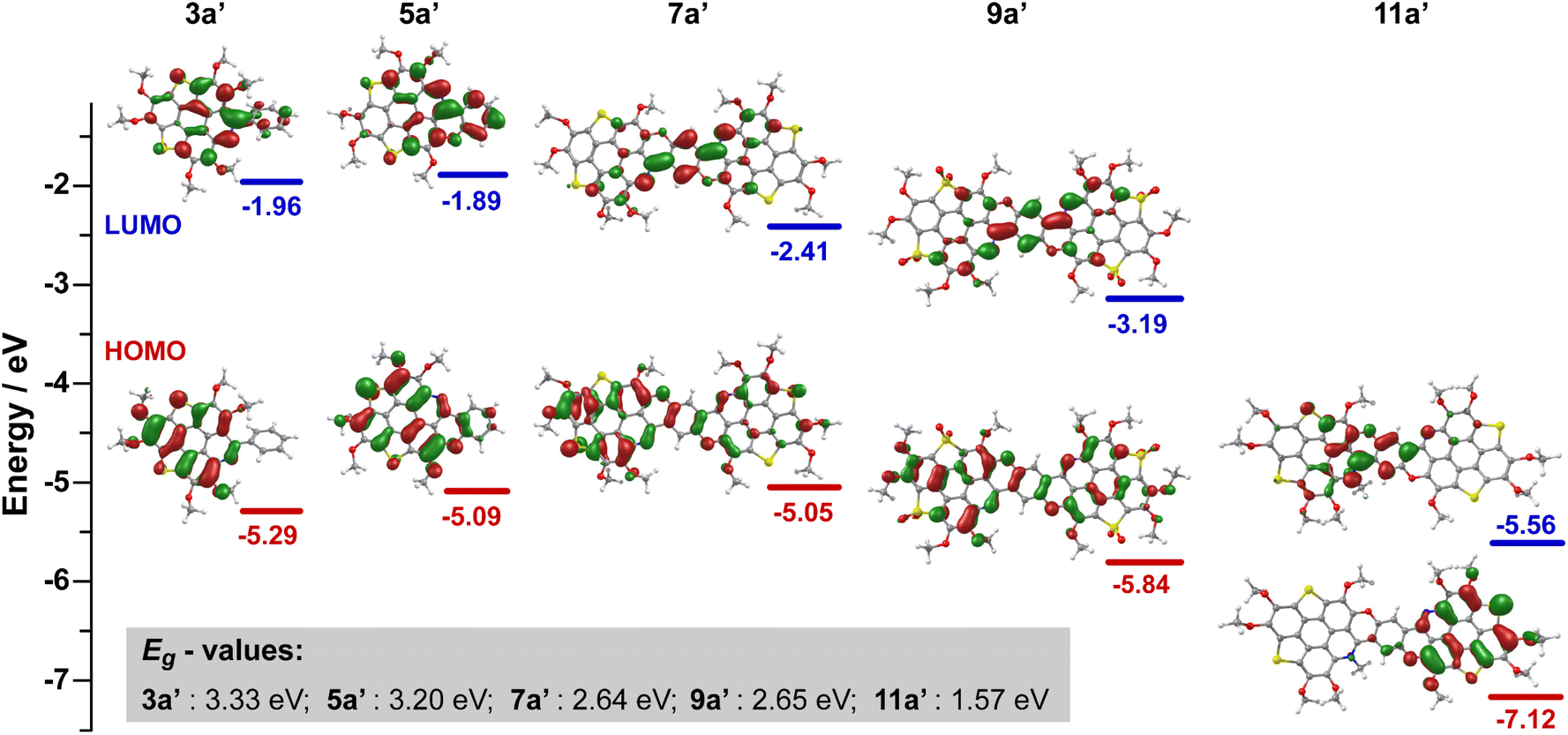 | ||
| Fig. 4 Calculated frontier molecular orbitals (FMOs) and energy levels of 3a′, 5a′, 7a′, 9a′, and 11a′. | ||
In the case of 3a′, the HOMO and LUMO are delocalized over the π-conjugated core, which consists of a triphenylene unit and two thiophene rings. Notably, the pyridine ring and terminal benzene ring contribute largely to the LUMO. For 5a′, the LUMO retains a similar distribution as in 3a′, but the HOMO extends across the π-conjugated core and the benzopyran moiety. As a result, the LUMO levels of 5a′ (−1.89 eV) and 3a′ (−1.96 eV) are comparable, but the HOMO level of 5a′ (−5.09 eV) is higher than that of 3a′ (−5.29 eV). This shift in energy of FMOs reflects the stabilization effect from the fusion of benzopyran moiety, which is further confirmed by the extension of π-framework from 5a′ to 7a′. The delocalization of π-electrons is markedly enhanced in 7a′, as evidenced by the HOMO and LUMO distributions. While the HOMO level of 7a′ (−5.05 eV) remains almost identical to that of 5a′ (−5.09 eV), the LUMO level of 7a′ (−2.41 eV) is distinctly lower than that of 5a′ (−1.89 eV). Consequently, the HOMO–LUMO gap (Eg) narrows in the order: 3a′ (3.33 eV) > 5a′ (3.20 eV) > 7a′ (2.64 eV).
Upon oxidation of the thiophene rings in 7a′ to thiophene S,S-dioxides, as in 9a′, the thiophene S,S-dioxides contribute minimally to the HOMO coefficient, as observed in both 9a′ and 8a′ (Fig. S35–S36†). Both the HOMO (−3.19 eV) and LUMO (−5.84 eV) energy levels of 9a′ decrease markedly as compared to 7a′, which is attributed to the transformation of electron-rich thiophene rings into electron-deficient thiophene S,S-dioxides. Despite these shifts, the Eg-value of 9a′ (2.65 eV) is comparable to that of 7a′.
Finally, methylation of the pyridine ring in 7a′ results in a significant change in both the HOMO and LUMO coefficients and their respective energy levels. In 11a′, the HOMO and LUMO are localized on segments without and with the methylation, respectively. This spatial separation of HOMO and LUMO suggests the potential for intramolecular charge-transfer (ICT) transitions. Compared to 7a′ and 9a′, the HOMO (−5.56 eV) and LUMO (−7.12 eV) energy levels of 11a′ are significantly lowered, and the Eg-value (1.57 eV) of 11a′ is much narrower than those of 7a′ and 9a′.
The computational investigations reveal that the extension of the π-conjugation leads to enhanced electron delocalization and narrowing of HOMO–LUMO gap. Furthermore, the modifications on the valence states of dopants, such as oxidation and methylation, introduce significant changes to the electronic properties. This systematic control over the electronic structure highlights the tunability of these materials.
Photophysical properties
The photophysical properties of the present hetero-PAHs were systematically evaluated using UV-vis absorption and emission spectroscopy. As representative examples, Fig. 5 illustrates the UV-vis absorption and emission spectra for the N, O, and S co-doped PAHs co-doped 3a, 5a, 7a, 8a, and 9a in the CHCl3 solution (c = 1 × 10−5 mol L−1) measured at RT. | ||
| Fig. 5 Absorption and emission spectra of N, O, and S co-doped hetero-PAHs measured in CHCl3 (c = 1 × 10−5 mol L−1): (a) UV-vis absorption and (b) emission spectra for 3a, 5a, and 7a; (c) UV-vis absorption and (d) emission spectra of 5a, 7a, 8a, and 9a. | ||
Compound 3a exhibits three distinct absorption peaks at 299 nm, 328 nm, and 341 nm, alongside a broad absorption band spanning 365–470 nm (Fig. 5a). Upon extending the π-system by fusing a benzopyran moiety onto 3a, as seen in compound 5a, the absorption spectrum shifts bathochromically. Specifically, 5a displays a sharp absorption peak at 309 nm and a broader band at 390–500 nm, indicating the influence of π-system extension on the electronic structure. A further extension of π-system in 7a leads to a significantly broadened low-energy absorption band, which shifts to 430–600 nm. These trends are consistent with the calculated energy gaps (Eg) for 3a, 5a, and 7a.
Theoretical simulations using time-dependent DFT (TD-DFT) at the TD-ωB97X/Def2-SVP/IEFPCM(CHCl3) level (Fig. S54–S65 and Tables S7–S18†) provide further insight into these trends. For 3a, the lowest energy absorption band is attributed to the S0 → S2 transition with an excitation energy (ΔE) of 3.61 eV and an oscillator strength (f) of 0.2673. This excitation primarily involves the HOMO → LUMO transition (66.5% contribution). For 5a and 7a, the lowest energy absorption bands correspond to the S0 → S1 transition, with ΔE values of 3.28 eV (f = 0.3963) and 2.96 eV (f = 1.0927), respectively. The excitations in both cases predominantly arise from the HOMO → LUMO transition (>79.0% contribution).
In terms of emission properties, 3a exhibits green fluorescence with a maximum emission (λem) at 496 nm and ΦF of 4.0% (Fig. 5b). Upon modification on molecular structures, 5a displays a slightly red-shifted emission (λem = 503 nm) and an improved ΦF of 11.7%. In contrast, 7a shows a distinct red-shift in its fluorescence with λem at 585 nm and a higher ΦF of 30.5%.
The oxidation of the thiophene rings in 5a and 7a to thiophene S,S-dioxides, resulting in compounds 8a and 9a, induces further modulation of the photophysical properties. The absorption spectrum of 8a is slightly red-shifted compared to 5a (Fig. 5c), while 9a exhibits a slight blue-shift relative to 7a. TD-DFT simulations reveal that the lowest energy absorption bands of both 8a and 9a are attributed to the S0 → S1 transition, with ΔE values of 3.28 eV (f = 0.5221) and 2.97 eV (f = 1.1726), respectively (Fig. S60, S61 and Tables S13, S14†). These transitions predominantly arise from the HOMO → LUMO transition (>77.9% contribution). Notably, the oscillator strengths for these excitations are larger than those of 5a and 7a, resulting in stronger low energy absorption features for 8a and 9a. Fluorescence measurements show that 8a exhibits green emission (λem = 520 nm), red-shifted by approximately 17 nm compared to 5a (Fig. 5d), while 9a displays blue-shifted emission (λem = 565 nm) compared to 7a. The ΦF of 9a is much enhanced to 42.3%, surpassing that of 7a. In contrast, the ΦF of 8a is reduced to 3.9%, which is lower than that of 5a.
Further chemical modification through methylation of the pyridine rings on 5a and 7a to form compounds 10a and 11a leads to substantial changes in the absorption spectra. As shown in Fig. 6, 10a exhibits a broad absorption band between 410–670 nm, with the absorption maximum red-shifted by approximately 106 nm compared to 5a. For 11a, the low-energy absorption extends into the near-infrared (NIR) region, say, 600–860 nm. TD-DFT simulations indicate that the lowest energy absorption bands for 10a and 11a correspond to the S0 → S1 transition (Fig. S62–S64 and Tables S15–S17†). For 10a, this excitation is dominated by the HOMO → LUMO transition (91.0% contribution), with ΔE = 2.62 eV and f = 0.2791. For 11a, the S0 → S1 excitation arises from a combination of the HOMO → LUMO and HOMO-2 → LUMO transitions, with contributions of 58.3% and 27.2%, respectively. The calculated excitation energy for 11a is 2.40 eV, and the oscillator strength is 0.8097.
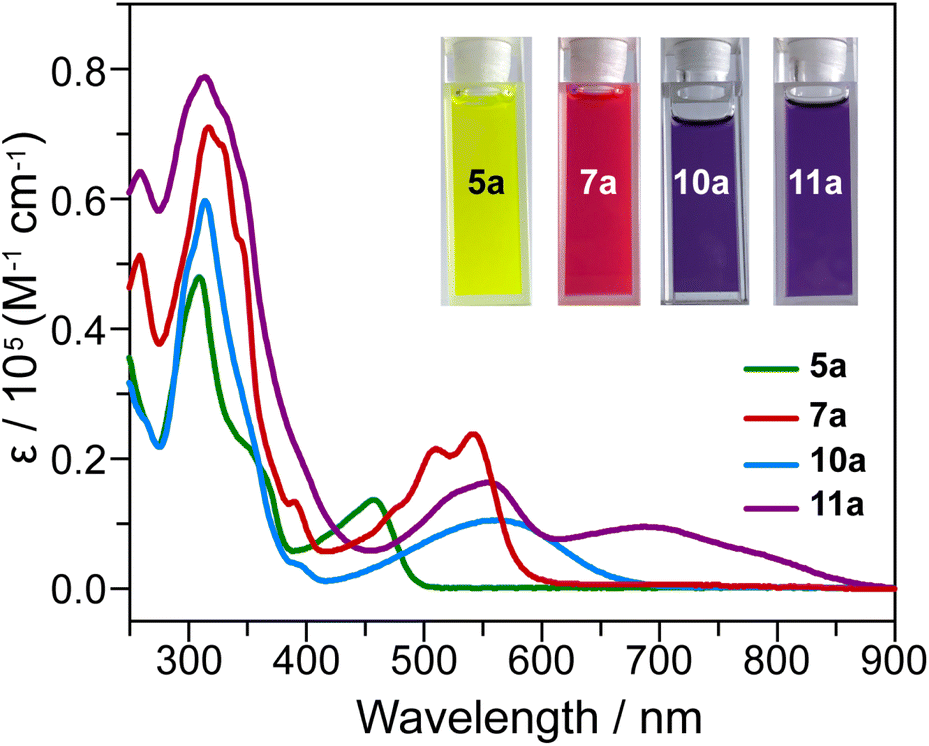 | ||
| Fig. 6 UV-vis-NIR absorption spectra of 5a, 7a, 10a and 11a in CHCl3 (c = 1 × 10−5 mol L−1). | ||
These results indicate that photophysical properties of hetero-PAHs can be effectively tuned through structural modifications, particularly the extension or alteration of the π-system and functional group modifications such as oxidation and methylation, which can significantly impact absorption, emission, and fluorescence quantum yield.
The optical properties of compounds 7a/7b and 11a/11b display notable tunability under acidic conditions. Representative examples are shown in Fig. 7, which illustrates the spectroscopic behavior of 7a and 11a upon treatment with trifluoroacetic acid (TFA), alongside theoretical simulation results. Compounds 7b and 11b exhibit similar trends, as shown in Fig. S11, S12 and S48–S51.†
 | ||
| Fig. 7 (a) UV-vis and (b) emission spectra of 7a upon titration with TFA; (c) UV-vis-NIR of 11a upon titration with TFA; (d) calculated frontier molecular orbitals (FMOs) and energy levels of 7a′, 7a′[H+], 7a′[H+]2, 11a′, 11a′[H+] and 7a′[CH3+]2. Measurement conditions: 20 °C, CHCl3 solution with c = 1 × 10−5 mol L−1. | ||
As shown in Fig. 7a, gradual addition of TFA to a CHCl3 solution of 7a (c = 1 × 10−5 mol L−1) leads to a decrease in the absorption band at 430–600 nm and the emergence of a new band spanning 450–850 nm, with a maximum at 663 nm. Concurrently, the solution color transitions from red to magenta (TFA, 100 eq.), then to blue (TFA, 1000 eq.), and finally to dark green (TFA, 3000 eq.). Notably, the UV-vis-NIR absorption spectrum of 7a treated with 200 equivalents of TFA closely resembles that of pure 11a in CHCl3, suggesting structural and electronic convergence upon protonation. In addition, the fluorescence of 7a is quenched upon TFA addition, as shown in Fig. 7b.
Upon titration of 11a with TFA (Fig. 7c), the original absorption band at 425–610 nm disappears, and a new band appears between 460–850 nm, with a λmax at 680 nm. Under conditions of excess TFA, both 7a and 11a converge to similar UV-vis-NIR absorption profiles, indicating stepwise protonation of the two pyridine rings in 7a.
Theoretical calculations support the notion that protonation of 7a is electronically analogous to methylation. As illustrated in Fig. 7d, the HOMO (−7.02 eV) and LUMO (−5.48 eV) energy levels, along with the energy gap (Eg = 1.54 eV) of the monoprotonated species 7a′[H+], closely match those of 11a′. Furthermore, the doubly protonated species 7a′[H+]2 displays electronic characteristics similar to the further protonated form of 11a′, with both showing decreased HOMO and LUMO energy levels and a slightly altered Eg. The doubly methylated 7a′[CH3+]2 exhibits HOMO (−9.54 eV), LUMO (−8.00 eV), and Eg (1.54 eV) values comparable to those of the doubly protonated 7a′[H+]2, further supporting the electronic analogy between methylation and protonation.
Generation of singlet oxygen (1O2)
Phototheranostics, composed of photodynamic therapy (PDT) and photothermal therapy (PTT), have garnered significant attention due to their noninvasive nature, high precision, and minimal side effects on healthy tissues.17 PDT is an effective tumor treatment strategy that utilizes photosensitizers (PSs) to generate reactive oxygen species (ROS), particularly 1O2, upon light irradiation, thereby inducing cellular apoptosis.18 The development of organic PDT materials has focused on optimizing photophysical properties, including extending absorption spectra to longer wavelength to enhance tissue penetration and increasing ΦF. A key strategy involves designing conjugated polymers or small organic molecules with large π-systems and incorporating heavy atoms to facilitate intersystem crossing (ISC) and improve ROS generation efficiency.8,19 Additionally, push–pull organic frameworks, featuring integrated electron-donating and electron-accepting groups, have demonstrated great potential for enhancing PDT performance by fine-tuning absorption properties and increasing 1O2 production.In this study, compounds 7a/7b, 9a, and 11a/11b exhibit potential as PSs for 1O2 generation, owing to their large π-systems and the presence of heavy chalcogen dopants (S, Se). The ability of these compounds to generate 1O2 was evaluated using 1,3-diphenylisobenzofuran (DPBF) as an oxidative probe, which undergoes a ring-opening reaction in the presence of 1O2 (Fig. 8a), leading to a decrease in its absorption at ∼415 nm.20 A control experiment showed that, in the absence of PSs, DPBF's absorption remained unchanged under light irradiation (660 nm) for 10 minutes (Fig. S19†). However, upon the addition of 7a/7b, 9a, and 11a/11b (10 μM), the DPBF absorption at 415 nm significantly decreased, confirming efficient 1O2 production. Time-dependent absorption spectra for DPBF in the presence of representative PSs (9a, 11a, and 11b) are shown in Fig. 8b–d, while those for 7a/7b are provided in Fig. S20 and S21.†
 | ||
| Fig. 8 (a) Chemical process for the ring-opening reaction of DPBF in the presence of 1O2 generated by PSs; time-dependent absorption spectra of DPBF (in CH2Cl2, c = 1 × 10−4 mol L−1) upon irradiation at 660 nm (2.5 W cm−2) in the presence of (b) 9a, (c) 11a, (d) 11b; (e) decay rate of DPBF absorbance at 415 nm with different PSs; (f) energy level diagram of singlet and triplet excited states of 11b. | ||
Using 7a as a PS, the DPBF absorbance at 415 nm declined to 15% of its original value after 15 minutes of irradiation (Fig. S20†). In contrast, 9a exhibited more efficient 1O2 production, completely quenching DPBF within 10 minutes (Fig. 8b). Similarly, 11a reduced the DPBF absorbance to 20% within 10 minutes (Fig. 8c). These results indicate that the manipulation of the valence states of heteroatom dopants shows significant influence on the efficiency of 1O2 generation. A notable approach to enhancing 1O2 generation is replacing sulfur with selenium, as demonstrated by the superior performance of 7b and 11b. The DPBF absorbance at 415 nm dropped rapidly, almost disappearing after 5 minutes with 7b as the PS (Fig. S21†). Remarkably, with 11b, complete degradation of DPBF was observed within just 2 minutes (Fig. 8d).
The decay rates of DPBF absorption (Fig. 8e) indicate the order of 1O2 generation efficiency: 7a < 11a < 9a < 7b < 11b. The superior performance of Se-doped PSs (7b and 11b) over S-doped analogs (7a, 9a, and 11a) is attributed to the heavy-atom effect, which enhances ISC and stabilizes the triplet state.21 Furthermore, the higher photosensitizing ability of 11b compared to 7b is primarily ascribed to its smaller singlet-triplet energy gap (ΔES1–T1). TD-DFT calculations reveal that ΔES1–T1 for 11b is 0.20 eV (Fig. 8f), significantly lower than that of 7b (0.52 eV), thereby facilitating more efficient 1O2 generation. Additionally, the higher fluorescence quantum yield (ΦF) of 9a (42.3%) compared to 7a (ΦF = 30.5%) further accounts for its enhanced PDT performance.22
Photothermal performance
PTT employs light-absorbing materials to convert light energy into heat, effectively inducing tumor cell apoptosis by localized hyperthermia.23 Organic PSs with strong near-infrared (NIR) absorption (600–1000 nm) are highly promising candidates for PTT applications.2g,24 In this study, compounds 11a/11b exhibit broad absorption bands spanning 600–860 nm, making them well-suited for PTT. Their photothermal characteristics were thoroughly investigated under 660 nm laser irradiation.The photothermal properties of 11a/11b were evaluated by monitoring temperature changes in their o-dichlorobenzene solutions using an infrared thermal camera. As shown in Fig. 9a, the temperature of 11a solution steadily increases upon irradiation, reaching a plateau within 6 minutes. The maximum stable temperature (Tmax) increases with power density and reaches 100 °C at 0.5 W cm−2. Tmax also exhibits concentration dependence, rising from 42 °C to 79 °C as the concentration of 11a increases from 10 μM to 70 μM (Fig. 9b). According to the reported method,25 the photothermal conversion efficiency (η) of 11a was calculated to be 51% based on a heating-cooling cycle experiment (Fig. 9c).
 | ||
| Fig. 9 Photothermal properties of 11a/11b in o-dichlorobenzene solution under 660 nm laser irradiation: (a) power-dependent (70 μM) and (b) concentration-dependent (0.3 W cm−2) temperature, and (c) temperature increasing/decreasing curve (70 μM, 0.3 W cm−2) and calculation of η (θ: temperature driving force) of 11a; (d) power-dependent (70 μM) and (e) concentration-dependent (0.3 W cm−2), and (f) temperature increasing/decreasing curve (70 μM, 0.3 W cm−2) and calculation of η of 11b; (g) photothermal stabilities of 11a/11b during 5 cycles of heating-cooling (70 μM, 0.3 W cm−2); (h) photothermal images of 11a (irradiation for 60 s, 0.5 W cm−2). | ||
Compared to 11a, 11b demonstrated similar power- and concentration-dependent trends but exhibited superior photothermal performance. Under identical irradiation conditions, the Tmax of 11b reached 118 °C at 0.5 W cm−2 (Fig. 9d). As the concentration of 11b increases from 10 μM to 70 μM, Tmax ascends from 45 °C to 88 °C (Fig. 9e). The calculated η for 11b is 61%, surpassing most reported organic PTT materials (Fig. S31†).
The photothermal stability of 11a/11b was further examined through multiple heating-cooling cycles (Fig. 9g). Both compounds maintain consistent temperature elevations after five cycles, confirming excellent photostability. The photothermal effect of 11a/11b was also visualized through IR thermal imaging (Fig. 9h), demonstrating a rapid photothermal response. These results highlight the excellent photothermal conversion efficiency and stability of 11a/11b, underscoring their strong potential for PTT applications.
Conclusion
In summary, we have developed a versatile strategy for the synthesis and post-synthetic modulation of hetero-PAHs co-doped with N, O, and S/Se. By integrating acid-catalyzed P–S reactions with thermally induced ipso-substitution, we successfully synthesized hetero-PAHs (3a/3b, 5a/5b, and 7a/7b) featuring chalcogenole, pyran, and pyridine-fused architectures. The systematic expansion of π-conjugation from 3a/3b to 5a/5b and 7a/7b induces a pronounced redshift in both absorption and emission spectra, accompanied by a remarkable enhancement in fluorescence quantum yield (ΦF) from 4.0% (3a) to 30.5% (7a).Beyond structural design, we demonstrate a post-synthetic valence state engineering strategy to precisely modulate photophysical properties. The oxidation of thiophene units into thiophene S,S-dioxides (8a and 9a) and the methylation of pyridine into pyridinium salts (11a and 11b) significantly alter electronic structures and intersystem crossing efficiencies. Notably, the introduction of thiophene S,S-dioxides in 9a boosts ΦF to 42.3%, leading to enhanced singlet oxygen (1O2) generation. Meanwhile, pyridinium-based hetero-PAHs (11a and 11b) show strong near-infrared absorption, accordingly they exhibit excellent photothermal performance, achieving a photothermal conversion efficiency of up to 61%. Furthermore, the superior performance of Se-doped analogs (7b and 11b) over their S-doped counterparts (7a, 9a, and 11a) in 1O2 generation and/or photothermal conversion underscores the critical role of the heavy-atom effect in optimizing triplet-state dynamics.
This work establishes a facile synthetic blueprint for designing multi-heteroatom-doped PAHs with tunable electronic structures. By leveraging valence state modulation, we unlock new opportunities for tailoring optoelectronic properties, paving the way for applications in organic electronics and phototheranostics.
Data availability
All experimental procedures and characterisation data can be found in the article or in the ESI.†Author contributions
X. Shao designed the research. Y. Ma and D. Li conducted the synthesis, crystal structure analysis, photophysical measurements; X. Chen and J. Wang carried out the photothermal analysis; X. Hua finished the DFT calculation; C.-S. Yuan, Z. Liu, and H.-L. Zhang provided suggestions for the structural characterizations, optical measurements, and singlet oxygen generation. All authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (22175080, 22371107, 22161034, 22371151) and The Science and Technology Major Program of Gansu Province of China (22ZD6FA006, 23ZDFA015, 24ZD13FA017).Notes and references
- (a) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed; (b) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC; (c) T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed; (d) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed.
- (a) S. Zhang, W. Guo, J. Wei, C. Li, X.-J. Liang and M. Yin, ACS Nano, 2017, 11, 3797–3805 CrossRef CAS PubMed; (b) Y. Cai, Z. Wei, C. Song, C. Tang, X. Huang, Q. Hu, X. Dong and W. Han, Chem. Commun., 2019, 55, 8967–8970 RSC; (c) C. Ji, W. Cheng, Q. Yuan, K. Müllen and M. Yin, Acc. Chem. Res., 2019, 52, 2266–2277 CrossRef CAS PubMed; (d) C. Liu, C. Ji, Z. Fan, R. Ma and M. Yin, Chem. Commun., 2021, 57, 13126–13129 RSC; (e) Q. Wang, X. Niu, L. Yang, J. Liu, J. Wang, X. Xu, W. Tang, W. Huang and Q. Fan, Mater. Chem. Front., 2021, 5, 5689–5697 RSC; (f) W. Jia, F. Huang, Q. Zhang, L. Zhao, C. Li and Y. Lu, Chem. Commun., 2022, 58, 6340–6343 RSC; (g) K. Liu, Z. Jiang, R. A. Lalancette, X. Tang and F. Jäkle, J. Am. Chem. Soc., 2022, 144, 18908–18917 CrossRef CAS PubMed; (h) C. Li, G. Jiang, J. Yu, W. Ji, L. Liu, P. Zhang, J. Du, C. Zhan, J. Wang and B. Z. Tang, Adv. Mater., 2023, 35, 2208229 CrossRef CAS PubMed; (i) M. Li, Z. Lu, J. Zhang, L. Chen, X. Tang, Q. Jiang, Q. Hu, L. Li, J. Liu and W. Huang, Adv. Mater., 2023, 35, 2209647 CrossRef CAS PubMed; (j) Z.-H. Wu, M. Peng, C. Ji, P. Kardasis, I. Tzourtzouklis, M. Baumgarten, H. Wu, T. Basché, G. Floudas, M. Yin and K. Müllen, J. Am. Chem. Soc., 2023, 145, 26487–26493 CrossRef CAS PubMed; (k) M. Liang, L. Liu, Y. Sun, J. Li, L. e. Zhang, X. Jiang and W. Wu, Aggregate, 2024, 5, e458 CrossRef CAS; (l) X. Liu, B. Chen, M. Wang, X. Gu and L. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202416160 CrossRef CAS PubMed.
- (a) W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC; (b) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (c) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed; (d) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed; (e) A. Winter and U. S. Schubert, Mater. Chem. Front., 2019, 3, 2308–2325 RSC; (f) W. Wang and X. Shao, Org. Biomol. Chem., 2021, 19, 101–122 RSC.
- (a) J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem., Int. Ed., 2010, 49, 8209–8213 CrossRef CAS PubMed; (b) Z. Liang, Q. Tang, R. Mao, D. Liu, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 5514–5518 CrossRef CAS PubMed; (c) Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed; (d) U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed; (e) S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed; (f) Q. Tan, H. Chen, H. Xia, B. Liu and B. Xu, Chem. Commun., 2015, 52, 537–540 RSC; (g) H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed; (h) Y. S. Park, D. J. Dibble, J. Kim, R. C. Lopez, E. Vargas and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2016, 55, 3352–3355 CrossRef CAS PubMed; (i) Y. Zhao, Q. Zhang, K. Chen, H. Gao, H. Qi, X. Shi, Y. Han, J. Wei and C. Zhang, J. Mater. Chem. C, 2017, 5, 4293–4301 RSC; (j) A. Hirono, H. Sakai, S. Kochi, T. Sato and T. Hasobe, J. Phys. Chem. B, 2020, 124, 9921–9930 CrossRef PubMed; (k) R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, J. Am. Chem. Soc., 2021, 143, 6593–6600 CrossRef CAS PubMed; (l) M. Nishimoto, Y. Uetake, Y. Yakiyama and H. Sakurai, Chem.–Asian J., 2023, 18, e202201103 CrossRef CAS PubMed; (m) X.-L. Chen, S.-Q. Yu, J.-Q. Liang, X. Huang and H.-Y. Gong, Org. Chem. Front., 2025, 12, 1340–1354 RSC.
- O-doped PAHs: (a) D. Stassen, N. Demitri and D. Bonifazi, Angew. Chem., Int. Ed., 2016, 55, 5947–5951 CrossRef CAS PubMed; (b) T. Miletić, A. Fermi, I. Orfanos, A. Avramopoulos, F. De Leo, N. Demitri, G. Bergamini, P. Ceroni, M. G. Papadopoulos, S. Couris and D. Bonifazi, Chem.–Eur. J., 2017, 23, 2363–2378 CrossRef PubMed; (c) A. Berezin, N. Biot, T. Battisti and D. Bonifazi, Angew. Chem., Int. Ed., 2018, 57, 8942–8946 CrossRef CAS PubMed; (d) A. Sciutto, A. Fermi, A. Folli, T. Battisti, J. M. Beames, D. M. Murphy and D. Bonifazi, Chem.–Eur. J., 2018, 24, 4382–4389 CrossRef CAS PubMed; (e) S. Dong, T. Y. Gopalakrishna, Y. Han, H. Phan, T. Tao, Y. Ni, G. Liu and C. Chi, J. Am. Chem. Soc., 2019, 141, 62–66 CrossRef CAS PubMed; (f) Y. Wang, S. Qiu, S. Xie, L. Zhou, Y. Hong, J. Chang, J. Wu and Z. Zeng, J. Am. Chem. Soc., 2019, 141, 2169–2176 CrossRef CAS PubMed; (g) C. M. Wehrmann, R. T. Charlton and M. S. Chen, J. Am. Chem. Soc., 2019, 141, 3240–3248 CrossRef CAS PubMed; (h) L. Đorđević, C. Valentini, N. Demitri, C. Mézière, M. Allain, M. Sallé, A. Folli, D. Murphy, S. Mañas-Valero, E. Coronado and D. Bonifazi, Angew. Chem., Int. Ed., 2020, 59, 4106–4114 CrossRef PubMed; (i) M. Nishimoto, Y. Uetake, Y. Yakiyama, F. Ishiwari, A. Saeki and H. Sakurai, J. Org. Chem., 2022, 87, 2508–2519 CrossRef CAS PubMed; (j) K. Zhang, J. Guo, H. Liu, X. Wang, Y. Yao, K. Yang and Z. Zeng, Chem. Commun., 2023, 59, 4947–4950 RSC.
- S, Se, or Te doped PAHs: (a) H. Ebata, E. Miyazaki, T. Yamamoto and K. Takimiya, Org. Lett., 2007, 9, 4499–4502 CrossRef CAS PubMed; (b) C. Mitsui, T. Okamoto, H. Matsui, M. Yamagishi, T. Matsushita, J. Soeda, K. Miwa, H. Sato, A. Yamano, T. Uemura and J. Takeya, Chem. Mater., 2013, 25, 3952–3956 CrossRef CAS; (c) T. Okamoto, C. Mitsui, M. Yamagishi, K. Nakahara, J. Soeda, Y. Hirose, K. Miwa, H. Sato, A. Yamano, T. Matsushita, T. Uemura and J. Takeya, Adv. Mater., 2013, 25, 6392–6397 CrossRef CAS PubMed; (d) X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535–538 CrossRef CAS PubMed; (e) C. Wang, M. Abbas, G. Wantz, K. Kawabata and K. Takimiya, J. Mater. Chem. C, 2020, 8, 15119–15127 RSC; (f) T. Guo, Z. Li, L. Bi, L. Fan and P. Zhang, Tetrahedron, 2022, 112, 132752 CrossRef CAS; (g) S. Liu, D. Xia, J. Wang, C. Ge, S. Hao, J. Zhang, P. Wang, K. Lin, S. Dong, E. Tretyakov, Y. Yang and X. Gao, J. Mater. Chem. C, 2023, 11, 6089–6094 RSC; (h) Z. Liu, W. Han, J. Lan, L. Sun, J. Tang, C. Zhang and J. You, Angew. Chem., Int. Ed., 2023, 62, e202211412 CrossRef CAS PubMed.
- (a) D. Wu, W. Pisula, M. C. Haberecht, X. Feng and K. Müllen, Org. Lett., 2009, 11, 5686–5689 CrossRef CAS PubMed; (b) S. M. Elbert, M. Reinschmidt, K. Baumgärtner, F. Rominger and M. Mastalerz, Eur. J. Org Chem., 2018, 2018, 532–536 CrossRef CAS; (c) R. Geng, X. Hou, Y. Sun, C. Yan, Y. Wu, H.-L. Zhang and X. Shao, Mater. Chem. Front., 2018, 2, 1456–1461 RSC; (d) B. Liu, D. Shi, Y. Yang, D. Liu, M. Li, E. Liu, X. Wang, Q. Zhang, M. Yang, J. Li, X. Shi, W. Wang and J. Wei, Eur. J. Org Chem., 2018, 2018, 869–873 CrossRef CAS; (e) A. Goujon, L. Rocard, H. Melville, T. Cauchy, C. Cabanetos, S. Dabos-Seignon and P. Hudhomme, J. Mater. Chem. C, 2022, 10, 14939–14945 RSC; (f) C. Valentini, D. Gowland, C. G. Bezzu, D. Romito, N. Demitri, N. Bonini and D. Bonifazi, Chem. Sci., 2022, 13, 6335–6347 RSC; (g) A. H. G. David, D. Shymon, H. Melville, L.-A. Accou, A. Gapin, M. Allain, O. Alévêque, M. Force, A. Grosjean, P. Hudhomme, L. L. Bras and A. Goujon, J. Mater. Chem. C, 2023, 11, 14631–14640 RSC; (h) V. B. S., M. Vadivel, D. P. Singh, V. A. Raghunathan, A. Roy and S. Kumar, Chem.–Eur. J., 2023, 29, e202300227 CrossRef CAS PubMed; (i) W. Jiang, H. Zhang, X. Hua, Y. Ma, Y. Feng, C. Yuan, Z. Liu, H.-L. Zhang and X. Shao, Angew. Chem., Int. Ed., 2025, e202500391 CAS.
- (a) A. Rodriguez-Serrano, V. Rai-Constapel, M. C. Daza, M. Doerr and C. M. Marian, Phys. Chem. Chem. Phys., 2015, 17, 11350–11358 RSC; (b) G. C. Hoover and D. S. Seferos, Chem. Sci., 2019, 10, 9182–9188 RSC; (c) M. Santra, M. Owens, G. Birch and M. Bradley, ACS Appl. Bio Mater., 2021, 4, 8503–8508 CrossRef CAS PubMed; (d) X. Xia, R. Wang, Y. Hu, Q. Yao, S. Long, W. Sun, J. Fan and X. Peng, Sci. China:Chem., 2022, 65, 821–828 CrossRef CAS; (e) X. Chen, X. Ma, G. Yang, G. Huang, H. Dai, N. Liu and J. Yu, Nanophotonics, 2023, 12, 3645–3652 CrossRef CAS PubMed.
- (a) L. Antolini, E. Tedesco, G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, D. Casarini, G. Gigli and R. Cingolani, J. Am. Chem. Soc., 2000, 122, 9006–9013 CrossRef CAS; (b) G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, A. Bongini, C. Arbizzani, M. Mastragostino, M. Anni, G. Gigli and R. Cingolani, J. Am. Chem. Soc., 2000, 122, 11971–11978 CrossRef CAS; (c) C.-H. Tsai, D. N. Chirdon, A. B. Maurer, S. Bernhard and K. J. T. Noonan, Org. Lett., 2013, 15, 5230–5233 CrossRef CAS PubMed; (d) B. Chen, H. Zhang, W. Luo, H. Nie, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 960–968 RSC; (e) M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef CAS PubMed.
- (a) J. E. Cochran, E. Amir, K. Sivanandan, S.-Y. Ku, J. H. Seo, B. A. Collins, J. R. Tumbleston, M. F. Toney, H. Ade, C. J. Hawker and M. L. Chabinyc, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 48–56 CrossRef CAS; (b) S. Wei, J. Xia, E. J. Dell, Y. Jiang, R. Song, H. Lee, P. Rodenbough, A. L. Briseno and L. M. Campos, Angew. Chem., Int. Ed., 2014, 53, 1832–1836 CrossRef CAS PubMed; (c) E. J. Dell, B. Capozzi, J. Xia, L. Venkataraman and L. M. Campos, Nat. Chem., 2015, 7, 209–214 CrossRef CAS PubMed; (d) Y. Deng, B. Sun, Y. He, J. Quinn, C. Guo and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 3459–3462 CrossRef CAS PubMed; (e) C. Wang, Z. Zhang, S. Pejić, R. Li, M. Fukuto, L. Zhu and G. Sauvé, Macromolecules, 2018, 51, 9368–9381 CrossRef CAS; (f) M. Marinelli, A. Candini, F. Monti, A. Boschi, M. Zangoli, E. Salatelli, F. Pierini, M. Lanzi, A. Zanelli, M. Gazzano and F. D. Maria, J. Mater. Chem. C, 2021, 9, 11216–11228 RSC; (g) S. Jiang, Y. Yu, D. Li, Z. Chen, Y. He, M. Li, G.-X. Yang, W. Qiu, Z. Yang, Y. Gan, J. Lin, Y. Ma and S.-J. Su, Angew. Chem., Int. Ed., 2023, 62, e202218892 CrossRef CAS PubMed.
- (a) D. Wu, L. Zhi, G. J. Bodwell, G. Cui, N. Tsao and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5417–5420 CrossRef CAS PubMed; (b) J. Fortage, C. Peltier, F. Nastasi, F. Puntoriero, F. Tuyèras, S. Griveau, F. Bedioui, C. Adamo, I. Ciofini, S. Campagna and P. P. Lainé, J. Am. Chem. Soc., 2010, 132, 16700–16713 CrossRef CAS PubMed; (c) J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed; (d) K. Xu, Y. Fu, Y. Zhou, F. Hennersdorf, P. Machata, I. Vincon, J. J. Weigand, A. A. Popov, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 15876–15881 CrossRef CAS PubMed; (e) R. D. Mule, A. C. Shaikh, A. B. Gade and N. T. Patil, Chem. Commun., 2018, 54, 11909–11912 RSC; (f) N. Toriumi, N. Asano, K. Miyamoto, A. Muranaka and M. Uchiyama, J. Am. Chem. Soc., 2018, 140, 3858–3862 CrossRef CAS PubMed; (g) P. Karak, S. S. Rana and J. Choudhury, Chem. Commun., 2021, 58, 133–154 RSC; (h) Q.-Q. Li, Y. Hamamoto, C. C. H. Tan, H. Sato and S. Ito, Org. Chem. Front., 2022, 9, 4128–4134 RSC; (i) Y. Ohno, S. Ando, D. Furusho, R. Hifumi, Y. Nagata, I. Tomita and S. Inagi, Org. Lett., 2023, 25, 3951–3955 CrossRef CAS PubMed; (j) L. Huang, Q. Wang, P. Fu, Y. Sun, J. Xu, D. L. Browne and J. Huang, JACS Au, 2024, 4, 1623–1631 CrossRef CAS PubMed; (k) L. Liu, J. Gong, G. Jiang and J. Wang, Chem.–Eur. J., 2024, 30, e202400378 CrossRef CAS PubMed; (l) S. S. Rana, S. Manna and J. Choudhury, Chem. Commun., 2024, 60, 10942–10945 RSC.
- (a) W. Jiang, Y. Li, W. Yue, Y. Zhen, J. Qu and Z. Wang, Org. Lett., 2010, 12, 228–231 CrossRef CAS PubMed; (b) M. T. Gabr and F. C. Pigge, RSC Adv., 2015, 5, 90226–90234 RSC; (c) J.-W. Chen and C.-C. Chang, ACS Appl. Mater. Interfaces, 2016, 8, 29883–29892 CrossRef CAS PubMed; (d) X. Gu, E. Zhao, T. Zhao, M. Kang, C. Gui, J. W. Y. Lam, S. Du, M. M. T. Loy and B. Z. Tang, Adv. Mater., 2016, 28, 5064–5071 CrossRef CAS PubMed; (e) S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453–493 RSC; (f) Y. Gao, X. Wang, X. He, Z. He, X. Yang, S. Tian, F. Meng, D. Ding, L. Luo and B. Z. Tang, Adv. Funct. Mater., 2019, 29, 1902673 CrossRef; (g) M. Imran, C. M. Wehrmann and M. S. Chen, J. Am. Chem. Soc., 2020, 142, 38–43 CrossRef CAS PubMed; (h) Q. Wan, R. Zhang, Z. Zhuang, Y. Li, Y. Huang, Z. Wang, W. Zhang, J. Hou and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 2002057 CrossRef CAS; (i) C. M. Wehrmann, M. Imran, C. Pointer, L. A. Fredin, E. R. Young and M. S. Chen, Chem. Sci., 2020, 11, 10212–10219 RSC; (j) Z. Liu, Q. Wang, W. Qiu, Y. Lyu, Z. Zhu, X. Zhao and W.-H. Zhu, Chem. Sci., 2022, 13, 3599–3608 RSC; (k) L. Wang, L. Qi, Q. Zhang, B. Xue, Z. Zheng, P. Yin, Y. Xue, W. Yang and Y. Li, Chem. Sci., 2023, 14, 4612–4619 RSC; (l) Q.-L. Zhang, Q.-T. Fan, Y. Zhou, J. Zhang and F.-L. Zhang, Org. Chem. Front., 2024, 11, 2884–2890 RSC.
- (a) E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797–1842 CrossRef CAS; (b) A. K. Mandadapu, M. Saifuddin, P. K. Agarwal and B. Kundu, Org. Biomol. Chem., 2009, 7, 2796 RSC.
- (a) X. Hou, Y. Zhu, Y. Qin, L. Chen, X. Li, H.-L. Zhang, W. Xu, D. Zhu and X. Shao, Chem. Commun., 2017, 53, 1546–1549 RSC; (b) W. Wang, L. Feng, X. Hua, C. Yuan and X. Shao, Chin. J. Chem., 2021, 39, 3413–3420 CrossRef CAS.
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef CAS PubMed.
- (a) S. Liu, Y. Li, H. Zhang, Z. Zhao, X. Lu, J. W. Y. Lam and B. Z. Tang, ACS Mater. Lett., 2019, 1, 425–431 CrossRef CAS; (b) S. Furukawa, J. Wu, M. Koyama, K. Hayashi, N. Hoshino, T. Takeda, Y. Suzuki, J. Kawamata, M. Saito and T. Akutagawa, Nat. Commun., 2021, 12, 768 CrossRef CAS PubMed; (c) Y. Li, X. Fan, Y. Li, S. Liu, C. Chuah, Y. Tang, R. T. K. Kwok, J. W. Y. Lam, X. Lu, J. Qian and B. Z. Tang, ACS Nano, 2022, 16, 3323–3331 CrossRef CAS PubMed.
- (a) W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed; (b) G. Feng, G.-Q. Zhang and D. Ding, Chem. Soc. Rev., 2020, 49, 8179–8234 RSC; (c) Q. Xie, J. Tang, S. Guo, Q. Zhao and S. Li, Molecules, 2023, 28, 6038 CrossRef CAS PubMed; (d) D. Ma, H. Bian, M. Gu, L. Wang, X. Chen and X. Peng, Coord. Chem. Rev., 2024, 505, 215677 CrossRef CAS.
- (a) S. Liu, G. Feng, B. Z. Tang and B. Liu, Chem. Sci., 2021, 12, 6488–6506 RSC; (b) W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC; (c) X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- (a) K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. Peng, Q. Shi, X. Cai and H. Huang, Adv. Mater., 2022, 34, 2108146 CrossRef CAS PubMed; (b) S. Yao, Y. Chen, W. Ding, F. Xu, Z. Liu, Y. Li, Y. Wu, S. Li, W. He and Z. Guo, Chem. Sci., 2023, 14, 1234–1243 RSC.
- (a) K. C. Park, J. Cho and C. Y. Lee, RSC Adv., 2016, 6, 75478–75481 RSC; (b) F. Xu, H. Li, Q. Yao, H. Ge, J. Fan, W. Sun, J. Wang and X. Peng, Chem. Sci., 2019, 10, 10586–10594 RSC; (c) Q. Xu, Y. Gao, X. Wu, H. Hang, H. Li, Y. Chen, W. Wang and H. Tong, New J. Chem., 2019, 43, 16385–16390 RSC; (d) H. Bian, D. Ma, F. Pan, X. Zhang, K. Xin, X. Zhang, Y. Yang, X. Peng and Y. Xiao, J. Am. Chem. Soc., 2022, 144, 22562–22573 CrossRef CAS PubMed; (e) W. Cao, Y. Zhu, F. Wu, Y. Tian, Z. Chen, W. Xu, S. Liu, T. Liu and H. Xiong, Small, 2022, 18, 2204851 CrossRef CAS PubMed; (f) W.-X. Wang, J.-J. Chao, Z.-Q. Wang, T. Liu, G.-J. Mao, B. Yang and C.-Y. Li, Adv. Healthcare Mater., 2023, 12, 2301230 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- F.-Z. Xu, L. Zhu, H.-H. Han, J.-W. Zou, Y. Zang, J. Li, T. D. James, X.-P. He and C.-Y. Wang, Chem. Sci., 2022, 13, 9373–9380 RSC.
- (a) R.-L. Ge, P.-N. Yan, Y. Liu, Z.-S. Li, S.-Q. Shen and Y. Yu, Adv. Funct. Mater., 2023, 33, 2301138 CrossRef CAS; (b) H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC; (c) B. Lu, Y. Huang, Z. Zhang, H. Quan and Y. Yao, Mater. Chem. Front., 2022, 6, 2968–2993 RSC; (d) Z. Zhao, C. Chen, W. Wu, F. Wang, L. Du, X. Zhang, Y. Xiong, X. He, Y. Cai, R. T. K. Kwok, J. W. Y. Lam, X. Gao, P. Sun, D. L. Phillips, D. Ding and B. Z. Tang, Nat. Commun., 2019, 10, 768 CrossRef CAS PubMed.
- (a) S. Li, Q. Deng, Y. Zhang, X. Li, G. Wen, X. Cui, Y. Wan, Y. Huang, J. Chen, Z. Liu, L. Wang and C.-S. Lee, Adv. Mater., 2020, 32, 2001146 CrossRef CAS PubMed; (b) X. Chen, X. Ma, G. Yang, G. Huang, H. Dai, J. Yu and N. Liu, ACS Appl. Mater. Interfaces, 2024, 16, 12332–12338 CrossRef CAS PubMed; (c) M. Li, Z. Lu, J. Zhang, L. Chen, X. Tang, Q. Jiang, Q. Hu, L. Li, J. Liu and W. Huang, Adv. Mater., 2023, 35, 2209647 CrossRef CAS PubMed; (d) L. Feng, C. Li, L. Liu, X. Chen, G. Jiang, J. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202212673 CrossRef CAS PubMed; (e) H. Gu, W. Liu, W. Sun, J. Du, J. Fan and X. Peng, Chem. Sci., 2022, 13, 9719–9726 RSC.
- (a) D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636–3641 CrossRef CAS PubMed; (b) R. Marin, A. Skripka, L. V. Besteiro, A. Benayas, Z. Wang, A. O. Govorov, P. Canton and F. Vetrone, Small, 2018, 14, 1803282 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2423790–2423792. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02061a |
| ‡ These authors contributed equally: Yao Ma, Dongxu Li, Xin Chen. |
| This journal is © The Royal Society of Chemistry 2025 |