
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvolutionary insights into the selectivity of sterol oxidising cytochrome P450 enzymes based on ancestral sequence reconstruction†
Daniel Z.
Doherty‡
 *a,
James J.
De Voss
b,
John B.
Bruning
c and
Stephen G.
Bell
*a
*a,
James J.
De Voss
b,
John B.
Bruning
c and
Stephen G.
Bell
*a
aDepartment of Chemistry, University of Adelaide, Adelaide, South Australia 5005, Australia. E-mail: daniel.z.doherty@adelaide.edu.au; stephen.bell@adelaide.edu.au
bSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Queensland 4072, Australia
cSchool of Biological Sciences, University of Adelaide, SA 5005, Australia
First published on 13th May 2025
Abstract
The cytochrome P450 (CYP) enzyme CYP125A1 is a crucial enzyme for the long-term survival and pathogenicity of Mycobacterium tuberculosis. CYP125 genes are found not only in pathogenic mycobacteria but are also widely dispersed within the Actinobacteria phylum, with many species possessing multiple copies of CYP125 encoding genes. Their primary function is the catalytic hydroxylation of the terminal methyl group of cholesterol and phytosterols. We have previously shown that CYP125 enzymes from distinct mycobacteria have substrate selectivity preferences for animal versus plant steroid oxidation. An evolutionary understanding of this selectivity is not known. Here, we use Ancestral Sequence Reconstruction (ASR), to support the hypothesis that some CYP125 enzymes evolved in a manner reflective of their adaptation to a pathogenic niche. We constructed a maximum-likelihood, most-recent common ancestor of the CYP125 clade (CYP125MRCA). We were then able to produce and characterise this enzyme both functionally and structurally. We found that CYP125MRCA was able to catalyse the terminal hydroxylation of cholesterol, phytosterols, and vitamin D3 (cholecalciferol); the latter was hydroxylated at both C-25 and C-26. This is the first example to date of vitamin D3 oxidation by a CYP125 enzyme, thereby demonstrating an increased substrate range of CYP125MRCA relative to its characterised extant relatives. The X-ray crystal structures of CYP125MRCA bound with sitosterol and vitamin D3 were determined, providing important insight into the changes that enable the expanded substrate range.
Introduction
The bacterial P450 enzyme family CYP125 is known to be involved in cholesterol catabolism via catalytic oxidation of the terminal methyl group of cholesterol (Scheme 1) and its 3-keto form cholest-4-en-3-one.1–3 The first CYP125 family member to be identified was found within a gene cluster of the soil-dwelling actinobacterial species Rhodococcus jostii RHA1 that was implicated in cholesterol catabolism.4,5 This enzyme was subsequently named CYP125A14.2 Genome mining for analogous cholesterol catabolism gene clusters within related Actinobacteria resulted in the discovery of a cholesterol catabolism pathway in the well-known and pathogenic Mycobacterium tuberculosis (Mtb).6,7 The discovery of this analogous gene cluster in Mtb was consistent with the observation that cholesterol catabolism was an important survival mechanism for this pathogen in vivo, particularly during the chronic stages of infection within macrophages.8 In an effort to utilise this important catabolic pathway as a potential route towards the discovery of novel antibiotics, individual genes within the pathway have been structurally and biochemically characterised. The CYP125A1 enzyme from Mtb, exhibited the same ability to catalyse the oxidation of the terminal methyl group of cholesterol and its hydrogenated form, cholest-4-en-3-one.1,3 | ||
| Scheme 1 Oxidation of the cholesterol side-chain by CYP125 enzymes. | ||
CYP125A1 is located within the igr operon,9 which is essential for Mtb growth on cholesterol as its sole carbon source. This demonstrated the importance of CYP125A1 for Mtb viability in vivo where host-cell cholesterol would be a major carbon source. Some strains of Mtb contain P450 enzyme families with back-up functionality to CYP125, namely CYP142 and possibly CYP124.9,10 For strains of mycobacteria that do not possess a copy of CYP142 (such as the CDC1551 strain), CYP125A1 is essential for growth on cholesterol.1
The catabolism of the cholesterol side-chain by Actinomycetota was first elucidated in 1968, through the isolation of various radio-labelled intermediates.11 The enzymes involved in this pathway, including the P450 enzymes mentioned above, have largely been characterised in the last few decades.12–19 The catabolism is initiated by activation of the cholesterol side-chain, firstly through conversion of the C-3 hydroxyl group to the ketone, forming cholest-4-en-3-one, followed by oxidation of one of the terminal methyl groups by CYP125 or CYP142, forming 3-oxo-cholest-4-en-26-oic acid. CYP125 can oxidise cholesterol without prior conversion to cholest-4-en-3-one1 and this the precise order of these steps in vivo is unclear. A point of difference between these CYP enzymes is that CYP125 produces the 25-S diastereomer, while CYP124/CYP142 mediated oxidation yields the 25-R product by oxidation at different diasterotopic methyl groups. Coenzyme A (CoA) is then ligated to the steroid, forming 3-oxo-cholest-4-en-26-oyl-CoA. At this point the sterol sidechain is activated for three rounds of β-oxidation in an analogous mechanism to that seen in fatty acid catabolism, releasing two units of propionyl-CoA and one unit of acetyl-CoA.20 Both propionyl-CoA and acetyl-CoA contribute to the central metabolism of Actinomycetota.20 In addition, in the case of mycobacteria, propionyl-CoA can be incorporated into the synthesis of phthiocerol dimycocerosate (PDIM) lipids which are important for the construction of the cell envelope, a critical virulence factor of Mtb.21 It is therefore evident that cholesterol-side chain breakdown has multiple benefits for mycobacterial survival. After side-chain degradation, the resulting androstenedione (AD) molecule is further degraded, yielding additional propionyl-CoA and acetyl-CoA moieties.
Genera from various Actinomycetota have been shown to grow on and metabolise not just cholesterol but also common plant sterols (phytosterols) such as β-sitosterol, stigmasterol and campesterol. This ability to use phytosterols as a carbon source has been demonstrated in industrially relevant strains of Mycobacterium neoaurum,22,23Mycobacterium smegmatis24 and various Nocardia species.25 The side-chain degrading capacity of these Actinomycetota can be combined with the mutation/knock out of various genes responsible for steroid ring degradation in order to yield the pharmaceutically valuable androstenedione (AD), 1,4-androstadiene-3,17-dione (ADD) and 9-hydroxyandrostenedione (9-OH-AD).26 The mechanism of sitosterol and campesterol oxidation by Actinomycetota has only been partially elucidated.27,28 Many steps, including the initial side-chain activation are assumed to be the same as that of cholesterol metabolism, however the presence of the additional C-24 carbon chain requires modifications to the overall pathway. The assumption that the CYP125 enzymes responsible for cholesterol oxidation in mycobacteria can also catalyse the oxidation of the phytosterol side-chains was only recently verified in vitro.29
The preference for cholesterol over sitosterol as a substrate for CYP125A1 (from Mtb), relative to other CYP125 enzymes from closely related mycobacteria, such as CYP125A7 from Mycobacterium ulcerans (Mulc), CYP125A6 from Mycobacterium marinum (Mmar) and from other more distantly related Actinomycetota, such as a CYP125 enzyme from Rhodococcus globerulus, has been examined.29 We hypothesised that this preference of CYP125A1 for cholesterol over phytosterols, when compared to bacteria found in non-pathogenic niches, is a result of the environment in which Mtb exists. In humans, cholesterol would be present in much greater abundance compared to phytosterols. A more general understanding of the evolution of steroid specificity within the CYP125 family remains unexplored. This family is classified as a blooming enzyme family within Actinomycetota, with many species containing multiple CYP125 copies, often with a large degree of sequence variability between them.30 This not only supports the notion that they are important for survival, but also that they could have a wider range of related steroidal substrates that are organism dependent. These interesting questions have led us to investigate the evolution of the CYP125 enzyme family within Actinomycetota using Ancestral Sequence Reconstruction (ASR), through which we have resurrected and characterised a most-probable, most-recent common ancestor of the CYP125 family (CYP125MRCA) and compared it to various extant Actinomycetota.
Experimental
Ancestral sequence reconstruction
The most-probable, most-recent common ancestor (CYP125MRCA) of the CYP125 enzyme family was predicted using ASR via the FireProtASR software (see Fig. S1† for protein sequence).31 The FireProt software functions by exploring the evolutionary history of a given enzyme (search sequence; in this case CYP125A1) through a process of selection of 150 related sequences (between 30% and 90% global sequence identity to the search sequence). The search space is however much larger than 150 proteins, as commonly used protein databases are mined and clustered together into groups that contain greater than 90% global sequence identity for a total of 150 clusters. From each cluster a sequence is selected at random for further analysis. The 150 chosen sequences are then aligned to each other using Clustal Omega32 and this alignment is used to construct a maximum-likelihood phylogenetic tree using RAxML (Fig. S1†).33 Tree construction is followed by a maximum-likelihood ancestral reconstruction at each node of the tree using a modified version the Lazarus method.31,34 The posterior probability distribution at each position in the sequence alignment, at each ancestral node, were analysed and the amino acid with the highest posterior probability was chosen at each position in the alignment yielding the final reconstructed enzyme.To test the robustness of the reconstructed ancestral enzyme to uncertainty in the posterior probabilities at each animo acid position, an alternative ancestral sequence was created by substituting any amino acid with a less than 70% posterior probability and replacing it with the next most probable amino-acid. A similar approach has been used previously to assess different ancestral states.35 By applying this rule to every amino acid position in the sequence, an alternative ancestor CYP125MRCAAlt was reconstructed, which spans most of the total uncertainty in the ancestral sequence, and is a significantly less likely ancestral state than CYP125MRCA (see Fig. S1† for sequence).
CYP125MRCA and CYP125MRCAAlt protein expression and purification
The codon-optimised, predicted CYP125MRCA and CYP125MRCAAlt gene sequences with attached C-terminus 6 x His-tags and HindIII restriction sites were obtained in pET-29b(+) vectors from Twist Bioscience. For each enzyme, the resulting construct was transformed into competent BL21(DE3) E. coli cells. Viable colonies were selected by plating onto LBkan. Single colonies were used to inoculate 6 × 500 mL LBkan, and cultures were grown for ∼10 hours at 115 rpm and 37 °C. The temperature and rotation speed were then reduced to 16 °C and 90 rpm respectively. Ethanol (2% v/v) and benzyl alcohol (0.02% v/v) were added to the cooled cultures. Trace elements, enriched with an iron salt (0.3% v/v) were also added to aid heme synthesis. After 30 min, IPTG (100 μM) was added to induce protein production. After induction, the growth was carried out for 48 h at 16 °C and 90 rpm.Cells were harvested by centrifugation at 5000g and 4 °C for 10 min. The combined cell pellet was resuspended in 50 mM Tris (200 mL, pH 7.4) buffer. Cell lysis was achieved by sonication at 4 °C and 70% amplitude in cycles of 15 s on 45 s off for 40 min using a VC505 Ultrasonic Processor and an Autotune CV334 probe (Sonics & Materials, USA). The mixture was then centrifuged at 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and 4 °C for 20 min to remove cell debris. The supernatant was decanted and pooled for further purification.
000g and 4 °C for 20 min to remove cell debris. The supernatant was decanted and pooled for further purification.
The combined supernatant was loaded at 4 mL min−1 onto a 5 mL His-Trap column (Cytiva) that was pre-equilibrated in binding buffer (0.5 M NaCl, 20 mM sodium phosphate buffer, 20 mM imidazole, pH 7.4). Once loaded, the column was washed with 5 column volumes of binding buffer, followed by elution of the enzyme with elution buffer (0.5 M NaCl, 20 mM sodium phosphate buffer, 200 mM imidazole, pH 7.4). The red-coloured protein was collected in a volume of approximately 10 mL. Imidazole was removed by desalting at 4 °C using size-exclusion chromatography (medium grain Sephadex G-25 resin). The red-coloured fraction was collected, and imidazole removal was assessed by UV-Vis absorbance spectroscopy.
The protein was concentrated ∼10 mL by centrifugation before loading onto a 5 mL HiTrap Q HP anion exchange column (Cytiva) using an AKTA FPLC system. The sample was washed for 5 column volumes with 50 mM Tris pH 7.4 (low salt) before elution with a continuous low to high salt (400 mM KCl, 50 mM Tris pH 7.4) gradient (0–80% high salt) over 45 min at 2 mL min−1. Red-coloured fractions were collected in 2 mL intervals and the relative purity of each was quantified using UV-Vis absorbance spectroscopy via the Abs408nm/Abs280nm ratio. Fractions with Abs408nm/Abs280nm > 0.8 were pooled and concentrated to ∼10 mL. The sample was then desalted at 4 °C using size-exclusion chromatography (medium grain Sephadex G-25 resin). The desalted red fractions were pooled, concentrated to ∼7 mL, mixed with an equal volume of 80% glycerol, and stored at −20 °C until use.
CO binding and extinction coefficient determination
The extinction coefficients at the isosbestic point (408 nm) of CYP125MRCA and CYP125MRCAAlt were determined using the CO binding assay of the reduced form of the enzyme as previously described (Fig. S2†).36 All work involving CO was performed in a fume hood.Thermostability assays
Protein thermostability was estimated for CYP125MRCA and its extant relative, MulcCYP125A7 (ref. 37) using protein thermal shift assays (PTS). Assays were conducted on the purified enzymes (5 μM) in 50 mM Tris buffer (pH 7.4) combined with Protein Thermal Shift Dye™ (concentration of 1×, ThermoFisher Scientific) and Protein Thermal Shift Buffer™ (ThermoFisher Scientific) on a 20 μL scale. Reactions were conducted in triplicate. Thermal fluorescence curves were determined using a PCR instrument and established protocols.38 The resulting protein melting temperatures (Tm) were determined using Protein Thermal Shift™ Software (ThermoFisher Scientific).Substrate binding assays
To screen potential substrates for CYP125MRCA, the UV-Vis absorption spectrum (after buffer baseline subtraction) of the enzyme was recorded at 30 °C before and after the addition of substrate on a Agilent Cary 60 UV-Vis spectrophotometer (scan rate of 600 nm min−1 and spectral point intervals of 1 nm) between 250 and 700 nm. Substrate binding was detected by a shift in the wavelength of the UV-Vis absorbance of the Soret band from the low-spin, 6-coordinate and water bound heme iron centre at ∼418 nm to the high-spin, 5 coordinate and substrate bound heme iron centre at ∼390 nm. This is the initial step in the P450 catalytic cycle, where the binding of substrate triggers the dissociation of the 6th axial water ligand of the heme.39 Initially, the spectrum of CYP125MRCA or CYP125MRCAAlt (∼2 μM, 600 μL) was measured at intervals of 1 min until any changes in the UV-Vis absorbance spectrum ceased. This step is required because CYP125 enzymes exist with a mixture of LS and HS heme centres in their resting state. This ‘mixed’ resting state implies that the LS and HS heme energies of CYP125 enzymes are close in energy. The equilibrium between the states is therefore temperature dependent, and as a result equilibration of the sample inside the spectrophotometer (which is set to 30 °C) prior to substrate addition is required. Substrates (5 or 10 mM stocks in 40% (w/v) 2-hydroxypropyl-β-cyclodextrin (HPβCD) in 50 mM Tris pH 7.4, or ethanol) were then titrated with the enzyme 1 μL at a time, until no further changes in the UV-Vis absorbance spectrum were observed.Once binding of a compound was confirmed by an appropriate shift in the UV-Vis absorbance spectrum, binding constant (Kd) values were obtained by measuring the difference spectra (ΔA) induced by each consecutive addition of a known concentration of substrate to the enzyme (∼2 μM, 3 mL) after baseline subtraction of the absolute protein spectrum. Binding curves were modelled with a variety of models (see Table S1† for all models). All of the substrates tested exhibited binding behaviour consistent with the Hill equation (eqn (1)).40
 | (1) |
In vitro activity assays
Given that CYP125MRCA is a proposed ancestral enzyme, electron transfer partner enzymes are unknown. Therefore, the activity of CYP125MRCA for oxidation of those compounds that demonstrated spectral evidence of binding was measured using a reconstituted spinach ferredoxin/spinach ferredoxin reductase electron transfer system (Sigma-Aldrich/Merck), with NADPH used as the source of electrons. Reactions were conducted on a 600 μL scale in oxygenated 50 mM Tris buffer (pH 7.4). CYP125MRCA (2 μM) and substrate (100 μM) were allowed to equilibrate at room temperature for 3 min. Glucose 6-phosphate (5 mM), glucose 6-phosphate dehydrogenase (0.7 units per mL), spinach ferredoxin (4 μM), spinach ferredoxin reductase (0.2 units per mL) and bovine catalase (0.03 mg mL−1) were then added and allowed to equilibrate. Reactions were initiated by the addition of NADPH (1 mM) and were allowed to continue for 20 h at room temperature.After 20 h, reactions were quenched by the addition of 1 μL of concentrated HCl and internal standard (5 μL of 20 mM octanoic acid in ethanol). Substrate and products were extracted from the reaction mixture three times with ethyl acetate (1 × 400 μL, 2 × 300 μL) and the extract was dried with anhydrous MgSO4. Ethyl acetate was removed under N2, and the products were dissolved in anhydrous acetonitrile (150 μL). Samples were derivatised by the addition of 99:1 BSTFA:TMCS (15 μL) at 37 °C for 2 h.
Sample product distributions were then analysed using a Shimadzu QP2010S GC-MS coupled with an AOC-20S autosampler, an AOC-20i Plus auto-injector and a DB-5 MS fused silica column (30 m × 0.25 mm x 0.25 μm). The carrier gas was helium, and the column flow rate was 1.40 mL min−1. The injection temperature and interface temperature were both held at 280 °C. The sample was then injected (1 μL). The column temperature was set to 100 °C and held for 2 min. The temperature was increased at 9 °C min−1 to a final temperature of 280 °C (20 min). The column was then held at 280 °C for 10 min. Products were identified by their mass spectra and relative retention times.
X-ray crystallography
Before CYP125MRCA was crystallised, it was further purified by size-exclusion chromatography in 50 mM Tris (pH 7.4). High purity fractions (Abs408nm/Abs280nm > 0.9) were pooled and concentrated to 35 mg mL−1 by centrifugation. Substrate was then added to an approximately equimolar concentration. Crystal screening condition sets (Hampton Research) were used to find crystallisation conditions using the sitting-drop vapor diffusion method. Optimisation was conducted using the hanging-drop and vapor diffusion method. The protein mixture (1 μL) was mixed with crystallisation reagents (1 μL) under a reservoir of the same crystallisation reagent mixture (75 μL for screening and 500 μL for optimisation). Thin, rod-shaped crystals were obtained after approximately 1 week of growth at 16 °C in conditions using 0.1 M sodium citrate tribasic trihydrate, 10–25% isopropanol and 10–25% PEG 4000 (pH 5.2–5.6). Crystals were then mounted on nylon loops (Hampton Research) or micromounts (Mitagen), cryoprotected in Parabar 10312 (Paratone-N, Hampton Research) and flash frozen in liquid N2.
X-ray diffraction data were acquired on the MX2 beamline at the Australian Synchrotron.41 Diffraction data was indexed, integrated, scaled, and merged using the XDS package as part of the Australian Synchrotron's automated pipeline.42 R-free flag labelling (5% of the reflections, randomly selected) was performed using Phenix prior to molecular replacement.43 Phases were solved by molecular replacement, using the Phaser algorithm in the CCP4 program suite.40 The search model for molecular replacement was generated from AlphaFold's predicted protein-chain structure for CYP125MRCA, without including the heme, ligands, or solvent molecules.44 Refinement on the initial solution was carried out using WinCoot (by minimising Fo–Fc)45 and the model electron density was recalculated using Phenix.43 This process was continued iteratively, until Rfree was minimised. Unbiased FEM maps46 were generated to confirm the presence of ligand in the CYP125MRCA active site. Solvent-accessible channels were calculated using the Caver 3.0.1 PyMol plugin.47 Errors in atomic coordinates were calculated using an online diffraction precision index (DPI) calculator.48
Results and discussion
Reconstruction of CYP125MRCA
Using the workflow described in the experimental section, we created a P450 phylogenetic tree stretching back to a time that is estimated to precede the split between the related cholesterol degrading P450 enzyme families CYP125, CYP142 and CYP124 in Actinomycetota (Fig. S1†). Despite it being difficult to verify the precise phylogeny of bacteria, it has been shown that there is very little difference between ASR accuracy when phylogenetic uncertainty is incorporated or not.34 We defined the most-recent common ancestor of the CYP125 family (CYP125MRCA) to be the first ancestral node for which all descendent branches contained at least one CYP125 enzyme (Fig. 1). CYP125MRCA retained a >40% global sequence identity to all of its descendant nodes and leaves, implying that it is also a member of the CYP125 family according to current P450 nomenclature definitions.49 CYP125MRCA retained ∼54% overall sequence identity with CYP125A1 from Mtb and was 419 amino acids in length (∼98% the length of CYP125A1). We aligned CYP125MRCA with all extant leaves and ancestral nodes within the CYP125 clade (Fig. S3 and S4†). Whilst there were many differences at the amino acid level between CYP125MRCA and extant CYP125 enzymes, the alignment highlighted the overall conservation of key P450 heme and substrate binding motifs (Fig. S4†). The alignment highlighted that only short 2–3 amino acid insertions and deletions were observed when moving forward in evolutionary time. There was a 3-amino acid insertion after position 84 in CYP125MRCA. This insertion occurs between nodes at Anc292 and is only partially retained in certain extant descendants. It resides on the edge of the B′-helix, at the interface of the outer surface of the enzyme active-site cavity and the solvent (Fig. S4 and S6†). A deletion of positions 196–197 in CYP125MRCA occurs from Anc292 and this deletion is maintained in CYP125A1. These two residues also lie at the cap of the active-site, this time on the exterior of the G-helix (Fig. S6†). A two-point insertion also occurs between CYP125MRCA and Anc291/Anc292, at position 47 in CYP125MRCA (Fig. S4†). This region of the protein is in a flexible loop region close to the N terminus and A-helix.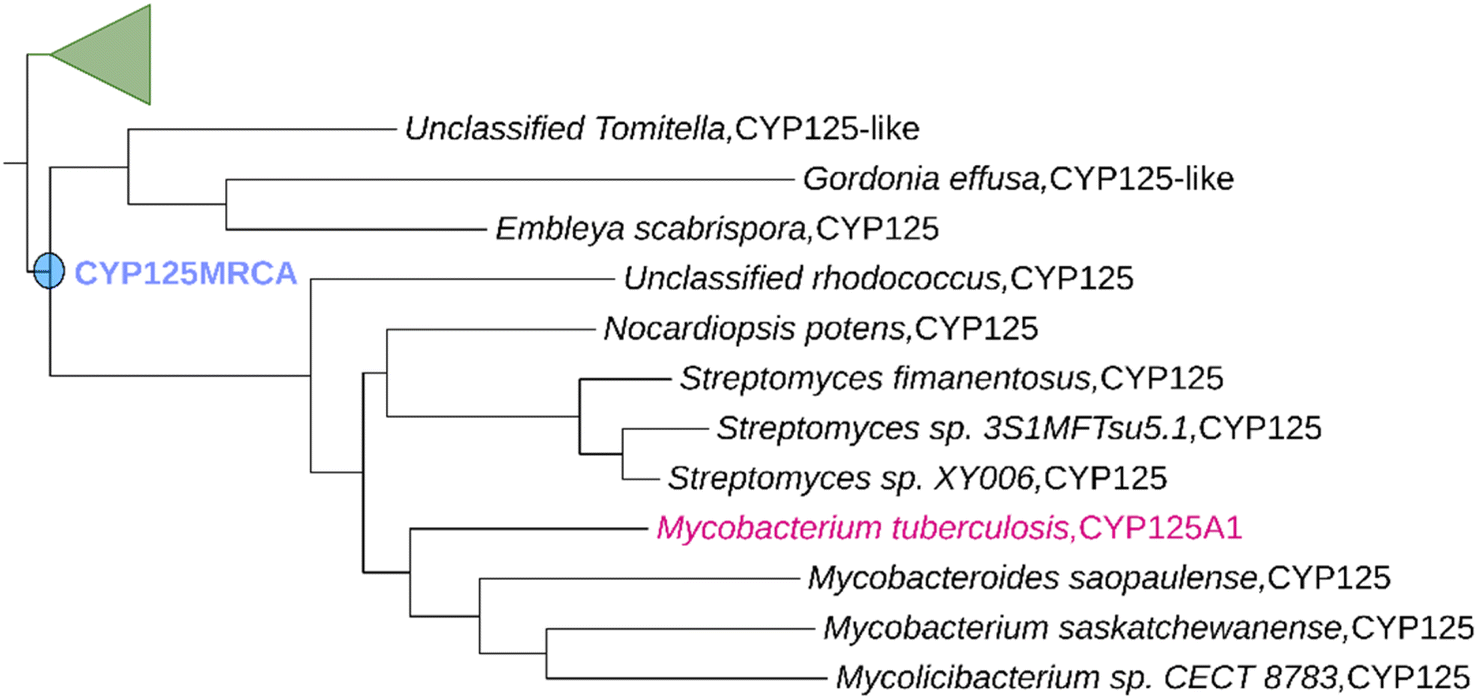 | ||
| Fig. 1 Ancestral Sequence Reconstruction of CYP125MRCA. Only a portion of the constructed phylogenetic tree is shown for clarity (see Fig. S1† for full tree). CYP125A1 was used as the search sequence (shown in magenta). The ancestrally reconstructed CYP125MRCA node is highlighted in blue. | ||
This region is unlikely to have a direct impact on catalytic activity. The CYP125 clade deduced from our ASR results in extant leaves of the tree that represent CYP125 enzymes from a variety of different genera within the Actinomycetota phylum, including Mycobacteria, Gordonia, Nocardia and Streptomyces, along with the recently differentiated Mycolicibacterium (Fig. 1).50,51 This is consistent with the current understanding of the evolution of Actinomycetota52 and supports the notion that all CYP125 enzymes originated from an ancient actinobacterial ancestor.
An alternative and less likely ancestor CYP125MRCAAlt (see Fig. S1† for sequence) was also reconstructed by applying the rule that any most-likely amino acid with a posterior probability <70% was to be substituted with the next most probable amino acid. CYP125MRCAAlt was also, by definition, 419 amino acids long. An alignment of CYP125MRCA and CYP125MRCAAlt (Fig. S7†) shows a large degree of sequence conservation, particularly in sections corresponding to core regions of the protein involved in heme recognition and in characteristic P450 helices. These include the I-helix, a helix close to the heme (on the distal side) responsible for oxygen binding and activation, and the F- and G-helices, which provide a cap at the entrance to the protein's active site and aid in substrate recognition (Fig. S7†). The sequence of the B′-helix, which forms the second part of the active-site envelope in the CYP125 family on the opposite side to the F/G-helices was also conserved (Fig. S7†). The overall % sequence identity between the two ancestors was ∼82%, with an additional 10% of residues being chemically similar in nature. Overall, the alignment of CYP125MRCAAlt and CYP125MRCA highlights that the amino-acid differences between the two sequences are scattered across the enzyme, and regions of difference are mostly 1–2 amino acids in length.
P450 enzymes are often screened for the binding of carbon monoxide (CO) to their reduced ferrous form as an indicator of a correctly folded and functional enzyme.53 Reduction of the CYP125MRCA and CYP125MRCAAlt enzymes with excess dithionite followed by saturation with CO resulted in a difference UV-Vis spectrum with a peak 450 nm and troughs at ∼390 nm and ∼418 nm representing the conversion of the LS (∼418 nm)and HS (∼390 nm) ferric forms of the heme centre to the 6-coordinate, CO-bound reduced form (Fig. S2†). Through the complete conversion of these enzymes to the CO-bound form, the extinction coefficient at the isosbestic point of the ferric form (408 nm) was determined to be 77 mM−1 cm−1 for each ancestor, using established methods.36
CYP125MRCA binds and oxidises both animal and phytosterols
The first experimental difference observed between CYP125MRCA and CYP125MRCAAlt was found in their resting state UV-Vis absorption spectra. The resting-state of the heme centre of CYP125MRCA is an equilibrium of the LS and HS forms, ranging between 40–70% HS, whilst in CYP125MRCAAlt, the heme is essentially completely HS in character (Fig. S8†). This equilibrium of LS and HS implies that the ancestor exists in an equilibrium of 6-coordinate, water bond heme (LS) and 5-coordinate heme (HS) with the 6th axial water ligand being absent. In most other P450 families, this 5-coordinate, HS species usually requires effective substrate binding in the active-site to be generated.54 The cause of this difference in resting state was not determined, however it must be derived from some part of the 20% difference in amino-acid sequence identity between the two enzymes. Importantly, this difference between the two resting states represents the extremes that have so far been observed for biochemically characterised CYP125 enzymes, with CYP125A1 having a resting state of >80% HS heme and MulcCYP125A7 having between 20 and 50>% HS.1,55 This result further evidences the fact that the energy of the LS and HS states of CYP125 enzymes are sufficiently close in energy that relatively small changes in their amino acid sequences at residues remote to the heme centre can alter the spin-state equilibrium.We tested plant and animal sterols or their derivatives as substrate for CYP125MRCA. We also tested if the substrate range of this ancestor would be broader, as has been reported for other classes of ancestral proteins.56 To test the substrate range of CYP125MRCA, we used UV-Vis absorbance spectroscopy to assess substrate-induced shifts in the Soret band resulting from the transition of the P450 heme centre from LS (∼418 nm) to HS (∼390 nm).57 We first tested CYP125MRCA for binding to the known substrates of CYP125A1, cholesterol and cholest-4-en-3-one (Fig. 4 and S9†).
CYP125MRCA enzyme was able to completely shift the Soret band to ∼390 nm upon binding of cholest-4-en-3-one and cholesterol which indicated a complete displacement of the axial aqua ligand of the 6-coordinate heme iron centre resulting in a decrease in the ∼416 nm low spin absorbance peak (Fig. S9†). This implies that these two steroids bound to the active-site in a likely catalytically productive way and was consistent with previously studied CYP125 enzymes from various mycobacteria and Rhodococcus species.1–3 Complete or near-complete spin-state shifts were also observed for cholesterol derivatives with changes to the steroid B ring including 7-ketocholesterol, 5,6α-epoxycholesterol, 7α-hydroxycholesterol, 7β-hydroxycholesterol and 7-dehydrocholesterol (Fig. S9†). Binding and oxidation of these cholesterol derivatives has been observed for MmarCYP125A6 and MmarCYP125A7.55
We also found that CYP125MRCA bound the three major plant sterols sitosterol, stigmasterol and campesterol, as well as stigmast-4-en-3-one (the plant equivalent to cholest-4-en-3-one) with a complete shift of the Soret band to the high spin peak at ∼391 nm (Fig. S9†). This contrasts the behaviour induced by the binding of phytosterols to extant CYP125A1, CYP125A6 and CYP125A7 enzymes from various mycobacteria, which only shifted their Soret bands partially (∼5% shift towards the high spin peak).29 Other C-27 alkyl steroids including desmosterol and ergosterol induced minimal perturbations of the heme centre.
CYP125MRCAAlt was also screened for binding to these potential substrate steroids to examine whether the substrate range was robust to statistical uncertainty in the ancestral state. Given that the resting state of CYP125MRCAAlt was >95% HS, it was first treated with imidazole (∼1.3 mM), a known P450 ligand that coordinates to the heme centre, to induce a partial Type II shift in the Soret band to ∼424 nm. This resulted in a ∼50% LS CYP125MRCAAlt state. The imidazole treatment allowed for the binding of substrates to be detected, via a shift back to the HS peak at ∼390 nm. Of the substrates tested, CYP125MRCAAlt was found to bind cholesterol, cholest-4-en-3-one, vitamin D3, sitosterol and stigmast-4-en-3-one (Fig. S10†). These substrate binding assays clearly demonstrated that the differences in sequence between CYP125MRCA and CYP125MRCAAlt did not result in significant differences in substrate range. So even though the positions in the ancestral sequence with greatest uncertainty did alter the electronics of the heme resting state they did not significantly change substrate recognition.
Through titration of these substrates to CYP125MRCA and monitoring the UV-Vis absorbance difference spectrum, their Kd values were determined (Fig. S11†). The Kd values presented in Table 1 are derived by fitting the data to the Hill binding model, where n is the Hill coefficient (see Table S1† for model comparisons).40
| Substrate | K d (μM) (n*) |
|---|---|
| a *n is the Hill coefficient as shown in the Hill equation (eqn (1)). | |
| Cholest-4-en-3-one | 0.85 ± 0.02 (2.15) |
| Cholesterol | 2.00 ± 0.05 (4.45) |
| Sitosterol | 1.48 ± 0.03 (3.74) |
| Stigmast-4-en-3-one | 1.61 ± 0.11 (1.98) |
| Stigmasterol | 2.04 ± 0.12 (2.36) |
| Campesterol | 4.19 ± 0.05 (3.68) |
| Cholecalciferol (VD3) | 1.60 ± 0.22 (1.84) |
A comparison of the binding of cholesterol and cholest-4-en-3-one to CYP125MRCA with data published for CYP125A1 (an extant relative) highlights similar affinities for cholest-4-en-3-one (Kd CYP125A1 = 1.18 ± 0.11 μM) and cholesterol (Kd CYP125A1 = 0.11 ± 0.06 μM).1 No cooperativity was reported in the binding of these substrates with CYP125A1 (Fig. S11†).1 MmarCYP125A7 also bound cholest-4-en-3-one with a similar strength as CYP125MRCA, with a Kd of 0.70 ± 0.4 μM.37
We cannot directly compare the binding of phytosterols to CYP125MRCA and extant CYP125 enzymes, as we have not been able to calculate the Kd of phytosterol binding to the latter, due to the lack of significant perturbations of the UV-Vis absorbance spectrum.29
We next conducted in vitro oxidation assays of CYP125MRCA for the substrates that were demonstrated to bind to this enzyme, using a reconstituted electron transfer system in which NADPH was the source of electrons, and spinach ferredoxin/spinach ferredoxin reductase were used as the redox partners. We found that CYP125MRCA was able to oxidise cholest-4-en-3-one and cholesterol to their respective 26-hydroxy oxidation products (Fig. 2, S12 and S13†).
 | ||
| Fig. 2 CYP125MRCA can bind and oxidise both animal and phytosterols. (a) Terminal methyl oxidation of both animal and phytosterols in their 3-keto or 3-hydroxy forms by CYP125MRCA. (b) Bar-chart demonstrating the binding constants (Kd) of various animal and phytosterols to CYP125MRCA. * Indicates that the substrate bound but was not oxidised. (c) GC chromatogram of the in vitro oxidation of cholest-4-en-3-one (left) and sitosterol (right) by CYP125MRCA using a reconstituted NADPH and spinach ferredoxin/ferredoxin reductase electron transfer system. * Indicates substrate/product peaks arising from a campesterol impurity in the sitosterol sample. The enzymatic reactions are shown in red and the no P450 substrate control reactions are shown in black. (d) Crystal structure of CYP125MRCA in complex with sitosterol. Left: Feature-enhanced map depicting the electron density (σ = 1.0, carve radius = 2.0) of the sitosterol substrate within the CYP125MRCA active-site (sitosterol in green, protein in yellow). Right: The CYP125MRCA structure overlaid with cholest-4-en-3-one (magenta) bound CYP125A1 (blue, PDB ID: 2X5W). Residues shown are within 5 Å of the substrates. | ||
Additionally, CYP125MRCA was able to effectively oxidise the major phytosterols whose sidechains were saturated, namely sitosterol and campesterol (Fig. 2, S12 and S14†). One difference between the oxidation of these phytosterols and cholesterol is the ability of CYP125MRCA to readily oxidise the former to their carboxylic acid equivalents, suggesting effective turnover of not just the phytosterol, but also the 26-hydroxy intermediates (Fig. 2). Furthermore, this activity is markedly different from the results observed for CYP125A1 and MulcCYP125A7 mediated sitosterol oxidation,29 in which no 26-sitostenoic acid product was observed. Even when compared to CYP125A6, which did produce a small amount of 26-sitostenoic acid, the acid:alcohol ratio for sitosterol products observed for CYP125MRCA was much higher.
We measured the thermal stability of CYP125MRCA relative to an extant descendant, in this case MulcCYP125A7 (see Fig. S15† for melting curves). CYP125MRCA had a melting temperature (Tm) of 64.4 ± 0.8 °C, over 10 °C higher than that of MulcCYP125A7 (51.2 ± 0.8 °C). This increase in enzyme stability when using ASR has been observed for various other P450 families.58,59
To assess the tolerance of activity of the ancestral enzyme to uncertainties in the posterior probability distribution, CYP125MRCAAlt was also tested for oxidation activity with cholesterol. This alternative ancestor was also able to oxidise cholesterol, albeit with a lower efficiency (Fig. S16†). Furthermore, differences in the product distribution between the two ancestral forms were evident. Whilst CYP125MRCA preferred to oxidise cholesterol at C-26, with 26-cholestenoic acid being the major product (Fig. S16†), CYP125MRCAAlt produced no acid metabolite, with 26-hydroxycholesterol being the major product. Two minor products were also produced by CYP125MRCAAlt, from desaturation of cholesterol and hydroxylation at C-25 (Fig. S16†).
To investigate the structure of CYP125MRCA and the binding mode of sitosterol, the crystal structure of this enzyme–substrate complex was solved (Fig. 3d). The resolution of the structure was 2.06 Å (see Table S2† for full data collection and refinement statistics). The majority (96%) of the 419 residues were modelled, with the first seven and last four residues not modelled due to a lack of discernible electron density. The surface loop region close to the A-helix and the N-terminus, spanning residues 44–49, was not modelled for the same reason. The presence of the sitosterol within the active-site channel was confirmed by the construction of feature-enhanced maps (FEM) which clearly demonstrated the electron density associated with the substrate (Fig. 2d). The overall fold of the protein was consistent with the canonical P450 fold with key structural components for substrate interactions being the I-helix located parallel to the heme centre and the F-G-helices/B′-helix, which together form the active-site envelope in a concordant fashion to CYP125A1 (Fig. S6†).3 The hydrophobic residues deep in the binding pocket allow for favourable interactions with the hydrophobic steroidal ring system and side-chain, while the hydrophilic E197/K92 residue pair caps the active-site (Fig. 2d).
 | ||
| Fig. 3 CYP125MRCA is capable of oxidising vitamin D3 (a) Proposed oxidation of vitamin D3 (cholecalciferol) and isomer by CYP125MRCA. The blue arrows indicate the pathway in which isomerism occurs before hydroxylation by CYP125MRCA, while the magenta arrows indicate the opposite pathway, in which isomerism occurs after hydroxylation. While isotachysterol is shown here as the alternative isomer leading to hydroxylation, due to acidification of the sample prior to GC-MS, hydroxylation of the other isomers cannot be ruled out. (b) The shift of the Soret band of CYP125MRCA in the UV-Vis region that is induced by the binding of vitamin D3 in the active-site. (c) GC chromatogram of the in vitro oxidation of vitamin D3 by CYP125MRCA using a reconstituted NADPH and spinach ferredoxin/ferredoxin reductase electron transfer system. * Indicates the two substrate peaks. * (blue) are products arising from hydroxylation and * (green) is a partially underivatised hydroxylation product. The enzymatic reaction is shown in red and the no P450 substrate control reaction in black. Left: Crystal structure of CYP125MRCA in complex with vitamin D3 (vitamin D3 in orange, protein in purple). The feature-enhanced map depicting the electron density of the substrate (σ = 1.0, carve radius = 2.0) within the CYP125MRCA active-site Right: The CYP125MRCA-vitamin D3-bound structure overlaid with the CYP125MRCA sitosterol-bound structure, with the sitosterol-bound structure residues shown in yellow and the sitosterol ligand in green. Residues shown are within 5 Å of the substrates. | ||
We then analysed differences in phytosterol versus mammalian sterol binding by superimposing the structure of sitosterol bound CYP125MRCA with that of cholest-4-en-3-one bound CYP125A1 (PDB ID: 2X5W3). The overall active-site topology between the two enzymes was similar, with many conserved hydrophobic residues close to the heme centre (Fig. 2d). Notable differences include the F400/W414 and M243/F260 residue changes. The M243/F260 difference appears to result in a re-positioning of the steroid ring system of cholest-4-en-3-one in CYP125A1 relative to the sitosterol ring-system in CYP125MRCA, to avoid steric clashes. This shifting of the ring system results in a movement of the position of the C25–C27 carbon atoms of sitosterol above the heme centre.
CYP125MRCA binds and oxidises vitamin D3 at both C-25 and C-26
Addition of vitamin D3 (cholecalciferol), a breakdown product of 7-dehydrocholesterol, to CYP125MRCA, generated a complete shift in the Soret band to HS, thereby indicating that this enzyme can bind substrates that have their steroid ring nucleus partially opened (Fig. 3). This is a significant difference from CYP125A1, CYP125A6 and CYP125A7, enzymes for which it has already been established that vitamin D3 does not induce a spectral shift in the Soret band.29,60 The binding affinity (Kd) of vitamin D3 was comparable to sitosterol (Table 1).By carrying out in vitro oxidation assays, it was found that vitamin D3 was oxidised by CYP125MRCA into four hydroxylation products and one partially underivatised alcohol product (Fig. 3 and S17†). Rather than being four different oxidation products arising from reactions at different C–H bonds of the substrate, we assigned this product distribution to a combination of the oxidation of different isomers of vitamin D3 that may result from the derivatisation process and/or GC-MS analysis conditions (Fig. 3a).61 This is supported by the presence of two substrate peaks in the GC-MS chromatogram (Fig. 3a).
The mass spectra of the product peaks (Fig. S17†) suggest that the first hydroxylation peak at ∼20.6 min is a result of C-25 hydroxylation, with a high intensity mass peak at 131 m/z.62 The presence of 25-hydroxyvitamin D3 was confirmed by the use of an authentic standard control (Fig. S18†). The second hydroxylation peak at ∼21.3 min produces a mass fragmentation patterns consistent with the C-26 hydroxylation observed for cholesterol.63 These two metabolites were formed in roughly equal amounts. The partially underivatised alcohol product most likely arises from difficulties adding the trimethylsilyl group to the sterically hindered 25-hydroxy metabolite.
The two smaller peaks, at retention times of ∼22.3 and 23.1 min respectively, result in mass fragmentation patterns that are indicative of 25-OH and 26-OH products, likely arising from the second, higher retention time isomer of the vitamin D3 (Fig. 3). The ability of this enzyme to oxidise vitamin D3 is to our knowledge the first evidence of a CYP125 enzyme that is able to do so. CYP125A1 does not oxidise vitamin D3 (ref. 60) and neither does MulcCYP125A7 (Fig. S19†).
This result when combined with those described in the previous section demonstrates an increased substrate range for CYP125MRCA relative to its studied extant descendants. The loss of activity for vitamin D3 in CYP125A1 and MulcCYP125A7 may reflect an increased specialisation of the CYP125A subfamily towards solely steroids with an intact ring nucleus, with the oxidation of vitamin D3 being primarily undertaken by the CYP124 family64 or other CYP125 subfamilies. This may allow for a greater selectivity of gene activity in response to different host immune responses.60
The level of vitamin D3 catalysed oxidation was much lower for CYP125MRCAAlt, implying that the activity is sensitive to the uncertainty in the posterior probability distribution of the ancestor. No 25- or 26-hydroxylation activity was found with this alternative enzyme, although trace amounts of desaturation metabolites were observed (Fig. S18†).
Structural rationale for increased substrate range and altered product distribution
To understand the structural determinants that permit CYP125MRCA to oxidise vitamin D3, the crystal structure of vitamin D3 bound CYP125MRCA was solved (Fig. 3d). This structure was solved to 1.78 Å and 96% of the residues were modelled (see Table S2† for full data collection and refinement statistics). The first seven residues and the loop region of amino acids 44–50 were not modelled due to a lack of defined electron density, suggesting a high level of flexibility in this loop. By overlaying the structure of vitamin D3 bound CYP125MRCA with that of the sitosterol bound enzyme, key differences were highlighted in the former's binding mode compared to sitosterol (Fig. 3d). The breaking of the steroid B ring in vitamin D3 relative to sitosterol results in a movement of A ring towards a pocket facing M243 (Fig. 3d). The shape, size and flexibility of this residue are critical for providing space for vitamin D3 to bind in this manner.When comparing this to the CYP125A1-cholest-4-en-3-one structure, the replacement of M243 by F260 removes a side-channel in CYP125A1, that is present in CYP125MRCA, to the active-site (Fig. S20†). This side-channel is necessary for the binding of the A ring of vitamin D3 without steric clashes. We therefore have a structural rationale for the difference in both binding and activity of CYP125MRCA and CYP125A1 for vitamin D3. This could be investigated via mutagenesis studies or further examination of more extant CYP125 enzymes from other bacteria.
Changes in key ancestral active-site residues and activity has been described in previous ancestral reconstructions of other P450 families. For example, the study of various ancestors of the CYP116B family revealed that the change of two adjacent active-site residues (A205T and F206W) in various ancestral lineages resulted in greater selectivity for subterminal fatty acid hydroxylation, as opposed to the mid-chain oxidation of the extant CYP116B46 enzyme.65 The ancestral reconstruction of CYP125MRCA highlights that ASR is also capable of altering the regioselectivity of oxidation as well as the substrate range.
Aside from active-site differences, CYP125A1 and CYP125MRCA also differ greatly in their solvent-accessible channels (Fig. S21 and S22†). While CYP125MRCA has multiple channels surrounding the F′ and G′ helices and their associated loop regions, CYP125A1 has markedly less space in this region and instead has a channel that extends over the I-helix. These differences in solvent channels may also explain the variation in the resting spin-state equilibria observed between CYP125MRCA and CYP125A1, due to the differences in the proportion of enzymes within the solution that would have water molecules available for heme coordination.
In both the vitamin D3-CYP125MRCA and sitosterol-CYP125MRCA structures, the C-25 carbon atoms of the substrates are significantly closer to the heme than the C-25 carbon in the cholest-4-en-3-one-bound CYP125A1 structure (Table 2 and Fig. S23†). This would enhance the possibility of the C-25 hydroxylation that is observed in the analysis of vitamin D3 (Fig. 3). The pro-R C-26 carbon of vitamin D3 is significantly closer to the heme than either C-25 or the pro-S C-26.
| Fe–C distance (Å) | |||
|---|---|---|---|
| C# | CYP125 | CYP125 | CYP125A1 |
| MRCA | MRCA | Cholest-4- | |
| Sitosterol | Vitamin D3 | en-3-one | |
| 25 | 4.5 ± 0.17 | 4.5 ± 0.12 | 5.1 ± 0.07 |
| Pro-S C-26 | 4.8 ± 0.18 | 4.9 ± 0.12 | 4.3 ± 0.08 |
| Pro-R C-26 | 4.6 ± 0.17 | 4.0 ± 0.12 | 4.8 ± 0.08 |
The physiological significance of hydroxylation of vitamin D3 at C-25 by CYP125MRCA is unclear, as hydroxylation at this position would not allow for the standard mycobacterial degradation of the steroid sidechain that results from C-26 hydroxylation. This activity for vitamin D3 hydroxylation may be a result of an evolutionary vestige of the older common ancestor of both the CYP125 and CYP124 families, given that CYP124A1 from Mtb is known to oxidise vitamin D3 at C-26. It also may arise from having a larger active site to accommodate the larger alkyl side chain of plant sterols. In the environment these would be a source of different vitamin D analogues with branched side chains, for example, ergocalciferol and these may be oxidised more selectively.60
When combined, this structural data provides a rationale for the substrate range difference that we observe between CYP125MRCA and CYP125A1. Despite this, the similarities in overall fold and active-site topology strengthen the evidence that this ancestrally reconstructed CYP125 enzyme is indeed a member of that family and potential progenitor of it. Therefore, we hypothesise that the two terminal hydroxylation products observed via GC-MS would give the 25-(R) diastereomer. Contrastingly, the uncertainties in the coordinates of the C-26 and C-27 in the CYP125MRCA-sitosterol structure make it more challenging to predict which C–H bond is most likely to be oxidised.
CYP125MRCA prefers phytosterols over animal sterols
After establishing the substrate range of CY125MRCA, we decided to investigate its preference for phytosterols or animal sterols through competitive oxidation assays. We monitored the oxidation of a 1:1 mixture of plant and animal steroid substrates under the same conditions used for the oxidation of individual substrates. We first examined the preference of CYP125MRCA for cholest-4-en-3-one versus stigmast-4-en-3-one. There was a significant preference, 3.2-fold, for stigmast-4-en-3-one (Fig. 4). This differs to extant Mtb CYP125A1, which had a 1.8-fold preference for cholesterol over sitosterol.29 Other extant CYP125 enzymes have close to 1:1 ratio.29
 | ||
| Fig. 4 GC-MS chromatograms of the competitive oxidations of 1:1 mixtures of cholest-4-en-3-one versus stigmast-4-en-3-one (top), and sitosterol versus stigmast-4-en-3-one (bottom). The substrate control is shown in black and the enzyme catalysed oxidation reaction in red. * indicates a product peak arising from a campesterol impurity in the sitosterol stock. The top chromatogram shows a greater amount of the plant sterol stigmast-4-en-3-one 26-hydroxylated product than that of the animal sterol cholest-4-en-3-one (ratio of 3.2:1 versus a ratio of 1.5:1 for an equimolar mixture of the substrates). The bottom chromatogram demonstrates a slightly greater amount of hydroxylated product for sitosterol over the 3-keto form stigmast-4-en-3-one (a ratio of 1.9:1 versus a ratio of 1.2:1 for an equimolar mixture of the substrates). | ||
In contrast, only a small preference for sitosterol over stigmast-4-en-3-one oxidation was observed (Fig. 4). These results suggest that the phytosterols are the preferred substrates for these enzymes over the animal steroid cholest-4-en-3-one.
The preference of extant Mtb CYP125A1 for cholesterol hydroxylation is not unexpected, given that its host organism, Mycobacterium tuberculosis, is found in a cholesterol-rich environment when it infects a human host.8,29 The preference for plant sterols in a CYP125 ancestor aligns with the assumption that the bacterial ancestor to which CYP125MRCA belonged to a soil-dwelling Actinomycetota, and therefore lived in an environment in which plant sterols would have been more common.52
Conclusions
In summary, ASR has been used to produce and characterise a maximum-likelihood most-recent common ancestor of the CYP125 family. Binding data, in vitro oxidation assays and crystallographic structures provide valuable information about the evolution of the steroid-oxidising CYP125 enzyme family towards a crucial role in pathogenicity within the Mtb genome. These results are consistent with the notion that the CYP125 enzymes evolved from being generalists that were able to oxidise plant sterols, animal sterols and closely related molecules, to being increasingly specific to their environmental niche, whether as pathogens or environmental organisms. Additionally, the discovery that CYP125MRCA can oxidise vitamin D3 at C-25 provides an example of the potential of ASR to construct stable enzymes that can produce medicinally and pharmaceutically relevant compounds. A logical continuation of this work could be conducted on two fronts. Characterisation of other nodes of the phylogenetic tree and using alternative methods of ancestral gene construction would enable a more sophisticated understanding of CYP125 evolution. More specifically, it would allow for an estimate of the point at which and the preference for cholesterol/cholest-4-en-3-one over vitamin D3 was enhanced. Secondly, rational design could be employed to enhance the C-25 vitamin D3 activity of CYP125MRCA and other potential substrates such as plant-based triterpenoids could be investigated.Given the close relationship between the CYP125, CYP142 and CYP124 enzyme families, using an ASR approach to investigate a robust evolutionary history of the latter two families would be of interest, as well as the root ancestor of these three families (Fig. S1†). Such an investigation may explain the loss of phytosterol oxidation activity by CYP142 enzymes and the preference of CYP124 enzymes for fatty acids.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for X-ray crystal structures has been deposited at the PBD under PDB ID: 8VXI and 8VXG and can be obtained from https://www.rcsb.org/.Author contributions
Daniel Z. Doherty was responsible for conceptualization, experimentation, data analysis and writing of manuscript. Stephen G. Bell was responsible for conceptualization, funding acquisition, manuscript drafting, experimental advice and data analysis. John B, Bruning contributed to crystallographic data analysis. James J. De Voss provided ideas for the project and was involved in manuscript drafting.Conflicts of interest
There are no conflicts to declare.Abbreviations
| ASR: | Ancestral sequence reconstruction |
| CYP: | Cytochrome P450 |
| LS: | Low-spin |
| HPβCD: | 2-Hydroxypropyl-β-cyclodextrin |
| HS: | High-spin |
| MRCA: | Most-recent common ancestor |
| UV-Vis: | Ultraviolet-visible spectroscopy |
| (VD3): | vitamin D3, cholecalciferol |
Acknowledgements
We would like to acknowledge Kate Wegener for methodological advice regarding the protein melting temperature experiments. This research was supported by the Australian Research Council grant DP210103970, a grant awarded to S. G. B and J. D. V. This research was also supported by an AINSE Ltd Postgraduate Research Award (PGRA), awarded to D. Z. D. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.66Notes and references
- H. Ouellet, S. Guan, J. B. Johnston, E. D. Chow, P. M. Kells, A. L. Burlingame, J. S. Cox, L. M. Podust and P. R. de Montellano, Mol. Microbiol., 2010, 77, 730–742 CrossRef CAS PubMed.
- K. Z. Rosloniec, M. H. Wilbrink, J. K. Capyk, W. W. Mohn, M. Ostendorf, R. van der Geize, L. Dijkhuizen and L. D. Eltis, Mol. Microbiol., 2009, 74, 1031–1043 CrossRef PubMed.
- K. J. McLean, P. Lafite, C. Levy, M. R. Cheesman, N. Mast, I. A. Pikuleva, D. Leys and A. W. Munro, J. Biol. Chem., 2009, 284, 35524–35533 CrossRef CAS PubMed.
- M. P. McLeod, R. L. Warren, W. W. Hsiao, N. Araki, M. Myhre, C. Fernandes, D. Miyazawa, W. Wong, A. L. Lillquist, D. Wang, M. Dosanjh, H. Hara, A. Petrescu, R. D. Morin, G. Yang, J. M. Stott, J. E. Schein, H. Shin, D. Smailus, A. S. Siddiqui, M. A. Marra, S. J. Jones, R. Holt, F. S. Brinkman, K. Miyauchi, M. Fukuda, J. E. Davies, W. W. Mohn and L. D. Eltis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15582–15587 CrossRef PubMed.
- U. Haussmann, D. A. Wolters, B. Franzel, L. D. Eltis and A. Poetsch, J. Proteome Res., 2013, 12, 1188–1198 CrossRef CAS PubMed.
- R. Van der Geize, K. Yam, T. Heuser, M. H. Wilbrink, H. Hara, M. C. Anderton, E. Sim, L. Dijkhuizen, J. E. Davies, W. W. Mohn and L. D. Eltis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1947–1952 CrossRef CAS.
- J. E. Griffin, J. D. Gawronski, M. A. Dejesus, T. R. Ioerger, B. J. Akerley and C. M. Sassetti, PLoS Pathog., 2011, 7, e1002251 CrossRef CAS.
- A. K. Pandey and C. M. Sassetti, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4376–4380 CrossRef CAS PubMed.
- M. D. Driscoll, K. J. McLean, C. Levy, N. Mast, I. A. Pikuleva, P. Lafite, S. E. Rigby, D. Leys and A. W. Munro, J. Biol. Chem., 2010, 285, 38270–38282 CrossRef CAS PubMed.
- J. B. Johnston, P. M. Kells, L. M. Podust and P. R. Ortiz de Montellano, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20687–20692 CrossRef CAS PubMed.
- C. J. Sih, H. H. Tai, Y. Y. Tsong, S. S. Lee and R. G. Coombe, Biochemistry, 1968, 7, 808–818 CrossRef CAS PubMed.
- M. H. Wilbrink, M. Petrusma, L. Dijkhuizen and R. van der Geize, Appl. Environ. Microbiol., 2011, 77, 4455–4464 CrossRef CAS PubMed.
- N. Wronska, A. Brzostek, R. Szewczyk, A. Sobon, J. Dziadek and K. Lisowska, Molecules, 2016, 21, 598 CrossRef PubMed.
- M. Yang, R. Lu, K. E. Guja, M. F. Wipperman, J. R. St Clair, A. C. Bonds, M. Garcia-Diaz and N. S. Sampson, ACS Infect. Dis., 2015, 1, 110–125 CrossRef CAS.
- C. M. Schaefer, R. Lu, N. M. Nesbitt, J. Schiebel, N. S. Sampson and C. Kisker, Structure, 2015, 23, 21–33 CrossRef CAS PubMed.
- T. Yuan, J. M. Werman, X. Yin, M. Yang, M. Garcia-Diaz and N. S. Sampson, ACS Infect. Dis., 2021, 7, 1739–1751 CrossRef CAS PubMed.
- S. T. Thomas and N. S. Sampson, Biochemistry, 2013, 52, 2895–2904 CrossRef CAS PubMed.
- M. Yang, K. E. Guja, S. T. Thomas, M. Garcia-Diaz and N. S. Sampson, ACS Chem. Biol., 2014, 9, 2632–2645 CrossRef CAS PubMed.
- T. Yuan, M. Yang, K. Gehring and N. S. Sampson, Biochemistry, 2019, 58, 4224–4235 CrossRef CAS PubMed.
- K. M. Wilburn, R. A. Fieweger and B. C. VanderVen, Pathog. Dis., 2018, 76, Fty021 Search PubMed.
- M. B. Goren, O. Brokl and W. B. Schaefer, Infect. Immun., 1974, 9, 150–158 CrossRef CAS PubMed.
- J. K. Loraine and M. C. M. Smith, Methods Mol. Biol., 2017, 1645, 93–108 CrossRef CAS PubMed.
- L. B. Xiong, H. H. Liu, M. Zhao, Y. J. Liu, L. Song, Z. Y. Xie, Y. X. Xu, F. Q. Wang and D. Z. Wei, Microb. Cell Fact., 2020, 19, 80 CrossRef CAS PubMed.
- B. Galan, I. Uhia, E. Garcia-Fernandez, I. Martinez, E. Bahillo, J. L. de la Fuente, J. L. Barredo, L. Fernandez-Cabezon and J. L. Garcia, Microb. Biotechnol., 2017, 10, 138–150 CrossRef CAS.
- P. Sharma, P. S. Slathia, P. Somal and P. Mehta, Ann. Microbiol., 2012, 62, 1651–1659 CrossRef CAS.
- K. Kong, M. Zhang, H. Zhang, C. Zhang, C. Wang and X. Yan, Syst. Microbiol. Biomanuf., 2022, 4, 1–19 Search PubMed.
- Y. Fujimoto, C. S. Chen, Z. Szeleczky, D. Ditullio and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 4718–4720 CrossRef CAS.
- Y. Fujimoto, C. S. Chen, A. S. Gopalan and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 4720–4722 CrossRef CAS.
- D. Z. Doherty, A. Ghith, A. Ho, J. J. De Voss and S. G. Bell, Chem. Commun., 2023, 59, 9392–9395 RSC.
- M. Parvez, L. B. Qhanya, N. T. Mthakathi, I. K. Kgosiemang, H. D. Bamal, N. S. Pagadala, T. Xie, H. Yang, H. Chen, C. W. Theron, R. Monyaki, S. C. Raselemane, V. Salewe, B. L. Mongale, R. G. Matowane, S. M. Abdalla, W. I. Booi, M. van Wyk, D. Olivier, C. E. Boucher, D. R. Nelson, J. A. Tuszynski, J. M. Blackburn, J. H. Yu, S. S. Mashele, W. Chen and K. Syed, Sci. Rep., 2016, 6, 33099 CrossRef CAS PubMed.
- M. Musil, R. T. Khan, A. Beier, J. Stourac, H. Konegger, J. Damborsky and D. Bednar, Briefings Bioinf., 2021, 22, bbaa337 CrossRef.
- J. D. Thompson, T. J. Gibson and D. G. Higgins, Curr. Protoc. Bioinf., 2003, 2–3, 2.3.1–2.3.22 Search PubMed.
- A. Stamatakis, Bioinformatics, 2014, 30, 1312–1313 CrossRef CAS PubMed.
- V. Hanson-Smith, B. Kolaczkowski and J. W. Thornton, Mol. Biol. Evol., 2010, 27, 1988–1999 CrossRef CAS PubMed.
- G. N. Eick, J. T. Bridgham, D. P. Anderson, M. J. Harms and J. W. Thornton, Mol. Biol. Evol., 2016, 34, 247–261 Search PubMed.
- F. P. Guengerich, M. V. Martin, C. D. Sohl and Q. Cheng, Nat. Protoc., 2009, 4, 1245–1251 CrossRef CAS PubMed.
- S. A. Child, A. Ghith, J. B. Bruning and S. G. Bell, J. Inorg. Biochem., 2020, 209, 111116 CrossRef CAS.
- K. Huynh and C. L. Partch, Curr. Protoc. Protein Sci., 2015, 79, 28.9.1–28.9.14 Search PubMed.
- E. M. Isin and F. P. Guengerich, Anal. Bioanal. Chem., 2008, 392, 1019–1030 CrossRef CAS PubMed.
- A. V. Hill, J. Physiol., 1910, 40, i–vii Search PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef.
- W. Kabsch, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- P. V. Afonine, N. W. Moriarty, M. Mustyakimov, O. V. Sobolev, T. C. Terwilliger, D. Turk, A. Urzhumtsev and P. D. Adams, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2015, 71, 646–666 CrossRef CAS PubMed.
- E. Chovancova, A. Pavelka, P. Benes, O. Strnad, J. Brezovsky, B. Kozlikova, A. Gora, V. Sustr, M. Klvana, P. Medek, L. Biedermannova, J. Sochor and J. Damborsky, PLoS Comput. Biol., 2012, 8, 1–12 CrossRef PubMed.
- K. S. D. Kumar, M. Gurusaran, S. N. Satheesh, P. Radha, S. Pavithra, K. P. S. Thulaa Tharshan, J. R. Helliwell and K. Sekar, J. Appl. Crystallogr., 2015, 48, 939–942 CrossRef CAS.
- D. R. Nelson, Methods Mol. Biol., 2006, 320, 1–10 CAS.
- R. S. Gupta, B. Lo and J. Son, Front. Microbiol., 2018, 9, 67 CrossRef PubMed.
- N. L. Bachmann, R. Salamzade, A. L. Manson, R. Whittington, V. Sintchenko, A. M. Earl and B. J. Marais, Front. Microbiol., 2019, 10, 3019 CrossRef PubMed.
- M. Ventura, C. Canchaya, A. Tauch, G. Chandra, G. F. Fitzgerald, K. F. Chater and D. van Sinderen, Microbiol. Mol. Biol. Rev., 2007, 71, 495–548 CrossRef CAS PubMed.
- P. R. O. De Montellano, Cytochrome P450: structure, mechanism, and biochemistry, Springer Science & Business Media, 2007 Search PubMed.
- T. Jovanovic, R. Farid, R. A. Friesner and A. E. McDermott, J. Am. Chem. Soc., 2005, 127, 13548–13552 CrossRef CAS PubMed.
- A. Ghith and S. G. Bell, J. Steroid Biochem. Mol. Biol., 2023, 235, 106406 CrossRef CAS PubMed.
- T. Devamani, A. M. Rauwerdink, M. Lunzer, B. J. Jones, J. L. Mooney, M. A. O. Tan, Z.-J. Zhang, J.-H. Xu, A. M. Dean and R. J. Kazlauskas, J. Am. Chem. Soc., 2016, 138, 1046–1056 CrossRef CAS PubMed.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS.
- K. L. Harris, R. E. S. Thomson, Y. Gumulya, G. Foley, S. E. Carrera-Pacheco, P. Syed, T. Janosik, A.-S. Sandinge, S. Andersson, U. Jurva, M. Bodén and E. M. J. Gillam, Mol. Biol. Evol., 2022, 39, msac116 CrossRef CAS.
- P. Hartz, S. J. Strohmaier, B. M. EL-Gayar, A. Abdulmughni, M. C. Hutter, F. Hannemann, E. M. J. Gillam and R. Bernhardt, FEBS J., 2021, 288, 6510–6527 CrossRef CAS.
- F. Mahmoodani, C. O. Perera, B. Fedrizzi, G. Abernethy, H. Chen, T. Varaksa, S. Bukhdruker, I. Grabovec, E. Marin, A. Kavaleuski, A. Gusach, K. Kovalev, I. Maslov, A. Luginina, D. Zabelskii, R. Astashkin, M. Shevtsov, S. Smolskaya, A. Kavaleuskaya, P. Shabunya, A. Baranovsky, V. Dolgopalets, Y. Charnou, A. Savachka, R. Litvinovskaya, A. Hurski, E. Shevchenko, A. Rogachev, A. Mishin, V. Gordeliy, A. Gabrielian, D. E. Hurt, B. Nikonenko, K. Majorov, A. Apt, A. Rosenthal, A. Gilep, V. Borshchevskiy and N. Strushkevich, J. Mol. Biol., 2021, 433, 166763 CrossRef.
- F. Mahmoodani, C. O. Perera, B. Fedrizzi, G. Abernethy and H. Chen, Food Chem., 2017, 219, 373–381 CrossRef CAS PubMed.
- M.-Y. Yang, C.-Y. Huang, T. H. T. Chiu, K.-C. Chang, M.-N. Lin, L.-Y. Chen and A. Hu, J. Food Drug Anal., 2019, 27, 494–501 CrossRef CAS PubMed.
- S. J. Gaskell, A. G. Smith and C. J. W. Brooks, Biomed. Mass Spectrom., 1975, 2, 148–155 CrossRef CAS.
- A. V. Vasilevskaya, A. V. Yantsevich, G. V. Sergeev, A. P. Lemish, S. A. Usanov and A. A. Gilep, J. Steroid Biochem. Mol. Biol., 2017, 169, 202–209 CrossRef CAS PubMed.
- B. S. Jones, C. M. Ross, G. Foley, N. Pozhydaieva, J. W. Sharratt, N. Kress, L. S. Seibt, R. E. Thomson, Y. Gumulya and M. A. Hayes, Angew. Chem., Int. Ed., 2024, 63, e202314869 CrossRef CAS PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo and N. Mudie, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary information including bioinformatic analysis, sequence alignments, the entire phylogenetic tree used for ancestral sequence reconstruction, UV-vis spectroscopy, GC-MS chromatograms of enzyme reactions and MS data, X-ray crystallographic data is available. See DOI: https://doi.org/10.1039/d5sc01863c |
| ‡ Daniel Z. Doherty Current address; Department of Physics, IBILCE, Universidade Estadual Paulista (UNESP), São Jose do Rio Preto, São Paulo, Brazil. |
| This journal is © The Royal Society of Chemistry 2025 |