
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen production via photocatalytic ammonia decomposition
Qijun
Pei
a,
Yongyu
Wang
ab,
Khai Chen
Tan
ab,
Jianping
Guo
 ab,
Teng
He
*abc and
Ping
Chen
abc
ab,
Teng
He
*abc and
Ping
Chen
abc
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy, of Sciences, Beijing 100049, China
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: heteng@dicp.ac.cn
First published on 24th April 2025
Abstract
Ammonia, as a carbon-free fuel and promising hydrogen carrier, has attracted significant attention in the context of a net-zero-emission scenario. Photocatalytic ammonia decomposition is a promising approach for hydrogen production, and much attention has been given to this area in recent years. This mini-review summarizes the latest research progress in photocatalytic ammonia decomposition for hydrogen production. We mainly focus on the photocatalytic decomposition of aqueous ammonia solution and gaseous ammonia. For aqueous ammonia solution, various semiconductor-based catalysts are introduced, and the role of water is discussed. The formation of the ˙NH2 radical as a key species in the decomposition was proposed by different groups. In the case of gaseous ammonia, different types of catalysts, including semiconductor-based and localized surface plasmon resonance (LSPR)-based ones, are described. The mechanisms of ammonia decomposition, such as N–N recombination and N2Hy dehydrogenation, are discussed. Methods for accurate temperature measurement in the photocatalytic process are summarized. We conclude that photocatalytic ammonia decomposition has unique advantages, including high activity, mild conditions, a green process, and fast response. Moreover, an excellent catalyst, efficient utilization of light, and suitable reactor design are critically important for the practical application of photocatalytic ammonia decomposition.
 Qijun Pei | Qijun Pei is an Assistant Professor at the Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences. He received a bachelor's degree from the College of Physics, Sichuan University. Subsequently, he pursued a doctoral degree at DICP and received his PhD in Physical Chemistry in 2020. After that, he joined the institute for a two-year postdoctoral fellowship. Since 2022, he has been working as an assistant professor at DICP. His research interests focus on the development of catalysts for liquid organic hydrogen carrier and ammonia decomposition. |
 Teng He | Teng He is currently a Professor at the Dalian Institute of Chemical Physics, Chinese Academy of Sciences (DICP, CAS). He pursued his PhD degree from 2006 to 2012 under the supervision of Prof. Ping Chen and Prof. Tao Zhang. Then, he was appointed as an Associate Professor immediately after receiving his PhD and joined DICP. He was a Visiting Scientist at the Pacific Northwest National Laboratory (PNNL) from 2017 to 2018. His research interests cover the development and characterization of novel hydrogen storage materials and catalysts for de/rehydrogenation, including B–N-based materials, liquid organic hydrogen carriers, metal–organic compounds, and ammonia decomposition catalysts. |
 Ping Chen | Ping Chen is a Professor and Division Head of Hydrogen Energy and Advanced Materials at the Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences. She received her PhD degree from Xiamen University in the field of physical chemistry in 1997 and was a faculty member in the Department of Physics, National University of Singapore, until 2008. Her primary research interests include materials development for hydrogen storage and catalysis. She pioneered research in the amide-hydride system for hydrogen storage and the alkali/alkaline earth hydride-transition metal composite system for ammonia synthesis. |
1. Introduction
Ammonia ranks among the most crucial chemicals for human beings.1–5 Ammonia is the second most produced chemical, with an annual output of approximately 180 million tons in 2015, and the estimated production capacity is around 290 million tons by 2030.6 The predominant use of produced ammonia is in fertilizer manufacturing, while there are still other applications for ammonia. Interestingly, ammonia as a fuel was proposed as early as 1822 and demonstrated in public transportation in 1942.7 However, its development stagnated until recent decades. The enthusiasm for ammonia as a fuel primarily stems from its carbon-free characteristic. In the context of a net-zero-emission scenario, it is estimated that over 10% of electricity generation and 20% of transportation fuel will derive from ammonia by 2050.8,9 Ammonia can be combusted directly or used to generate electricity via fuel cells, but several challenges remain. Moreover, ammonia is one of the promising hydrogen carriers owing to its high hydrogen content (17.6 wt%, 108 g-H2 per L-liquid ammonia). It can be easily converted into a liquid state, either at 10 bar pressure at room temperature or when cooled down to −33.5 °C at atmospheric pressure, making it convenient for storage and transportation. More importantly, the utilization of ammonia can benefit from its mature technology of production and transportation, thanks to the pipeline established in the existing industry. At the location where the hydrogen is used, hydrogen can be obtained via the decomposition of ammonia. Based on the existing reports, four methods have been extensively and intensively employed and investigated for ammonia cracking, including thermocatalysis, electrocatalysis, plasma-catalysis, and photocatalysis (Fig. 1). | ||
| Fig. 1 Thermocatalytic, electrocatalytic, plasma-catalytic, and photocatalytic ammonia decomposition for hydrogen production. Reproduced from ref. 10 with permission from Elsevier, Copyright 2021; ref. 11 with permission from the Royal Society of Chemistry, Copyright 2010; ref. 12 with permission from Elsevier, Copyright 2024; ref. 13 with permission from AAAS, Copyright 2022. | ||
Ammonia is a stable molecule, and its thermal decomposition requires high temperatures, which can be significantly reduced with the aid of an efficient catalyst. Nevertheless, to achieve a full conversion, the reaction temperature is typically above 500 °C, leading to low energy efficiency. Therefore, developing more efficient catalysts to decrease the reaction temperatures is a long-sought target in this area. Recently, some novel heating methods, such as Joule heating and magnetic induction heating,10,14 have been developed to address the issue of delayed response times associated with traditional electrical heating methods. Apart from thermocatalysis, alternative methods for external-field-assisted ammonia decomposition were also proposed, as listed in Table 1, facilitating ammonia decomposition under mild conditions.11–13,15–17 Theoretically, electrolysis of ammonia for hydrogen production requires an overall voltage of merely 0.077 eV,11 indicating that its energy expenditure is substantially lower than that of water. However, the real operating voltage is much higher than the theoretical one.11,16,18 Both cold and warm plasmas could dissociate stable NH3 molecules in the gas phase, therefore promoting the ammonia decomposition with the assistance of a catalyst.12,19–25 However, challenges still exist and more fundamental research is needed.
 | (1) |
 | (2) |
 | (3) |
 | (4) |
| Thermocatalysis | Electrocatalysis | Plasma-catalysis | Photocatalysis | |
|---|---|---|---|---|
| Advantages | Industrial maturity | Mild conditions | Rapid startup | Mild conditions |
| Catalyst diversity | Simplified production separation | High activity | Rapid startup | |
| High activity | Green process | |||
| Disadvantages | High energy demand | High overpotential | Complex equipment | Low efficiency |
| Poor stability | Scalability challenge | Low efficiency | Limited research |
The investigations of photochemical decomposition of ammonia have been carried out as early as the 1920s.26,27 In astrophysics and cosmology, it was found that ammonia can be photolyzed by sunlight irradiation, leading to the generation of nitrogen.28 Besides, NH3 and C2Hx mixtures under UV photoirradiation could produce nitriles, which are key reactants for the formation of amino acids.29,30 Photocatalytic ammonia decomposition is another promising approach for hydrogen production from ammonia. Since Fujishima and Honda reported water splitting under light in 1972,31 photocatalysis has been applied to numerous reactions.32–35 Photocatalysis can not only supply energy to overcome the barriers of reactions but also drive some thermodynamically unfavorable chemical reactions, such as water splitting (eqn (1)). As shown in eqn (2), the energy consumption for hydrogen generation from gaseous ammonia is 30.6 kJ/mol-H2, constituting only 10.7% of the 285.8 kJ/mol-H2 required for water splitting (eqn (1)). This indicates that hydrogen production from ammonia is an energy-efficient reaction. However, the reaction cannot occur spontaneously because of its positive Gibbs free energy at room temperature. Many researchers have attempted to conduct catalytic ammonia decomposition under light irradiation.13,17,36–44 If an efficient photocatalytic ammonia decomposition catalyst is successfully developed, the ammonia decomposition efficiency and the hydrogen production rate would be significantly boosted. Accordingly, such an advancement will definitely decrease hydrogen production costs, reduce reliance on fossil fuels, and accelerate the arrival of the ammonia–hydrogen economy. Although numerous reviews about catalytic decomposition have been reported very recently,1–4,45–48 reviews that focus on photocatalytic decomposition are scarce.36
In this mini-review, we comprehensively summarize the latest research advancements in photocatalytic ammonia decomposition for hydrogen production. We meticulously discuss the impact of catalyst preparation and structural properties on the performance of photocatalytic ammonia decomposition, along with in-depth investigations of the underlying mechanisms. Furthermore, issues and challenges encountered during the research and for the future are discussed, and potential catalysts for photocatalytic ammonia decomposition are proposed. We anticipate that this review can serve as a valuable reference for researchers working on the development of photocatalysts, reactor design, and mechanism exploration.
2. Photocatalytic decomposition of aqueous ammonia solution
Photocatalytic ammonia decomposition can be divided into liquid-phase and gas-phase processes. Significant progress has been made in the past two years, with records of the ammonia decomposition rate being continuously updated. Table 2 enumerates the current performance of photocatalytic ammonia decomposition for hydrogen production. Generally, the hydrogen production rate from gas-phase ammonia decomposition significantly exceeds that of the liquid phase, thanks to the synergy between photogenerated charges and photo-induced heat during the photocatalytic decomposition of ammonia in the gaseous phase, which greatly accelerates the reaction rate. In the following sections, a detailed account of the advancements in ammonia decomposition in the aqueous phase will be elaborated, emphasizing the development of catalysts and the mechanistic understanding of this reaction.| Catalyst | C NH3 (mol/L) | C Cat (g/L) | Light source | Reaction temp. (°C) | H2 rate (mol/(g h)) | Ref. | |
|---|---|---|---|---|---|---|---|
| a C Cat: the concentration of the catalyst in the aqueous ammonia solution. b rGO/TiO2 NWs: TiO2 nanowire-intercalated reduced graphene oxide. c NH4Cl aq. (pH 11 adjusted with NaOH). d Mw-Pt–TiO2: Pt–TiO2 prepared by using a microwave-assisted deposition method. e WHSV: weight hourly space velocity. f η: light-to-H2 energy efficiency. g Ru NPs/GaN NWs/Si: GaN nanowire-supported Ru nanoparticles on silicon. h Cu–Fe-AR: Cu–Fe antenna-reactor complex. i Ni-MCN: macroporous carbon-nitride-supported Ni single-atom catalyst. | |||||||
| Aqueous phase catalysis | Pt0.9Au0.1/TiO2 | 0.261 | 1 | Solar simulator (AM1.5G) | 25 | 1.6 × 10−4 | 38 |
| rGO/TiO2 NWsb | 0.052 | 0.05 | Hg lamp, 8 W 254 nm UV | Room temperature | 2.08 × 10−4 | 49 | |
| Ru–ZnS | 0.02c | 1 | Solar simulator (AM1.5G) | Room temperature | 4.0 × 10−6 | 40 | |
| Ni/TiO2 | 0.59 | 4 | Xe lamp through a color filter | Room temperature | 4.39 × 10−5 | 50 | |
| ZnO | 0.052 | 1 | Hg lamp, 8 W 254 nm UV | Room temperature | 1.03 × 10−5 | 41 | |
| 1.2 wt% Ce/TiO2 | 0.052 | 1 | Hg lamp, 8 W 254 nm UV | Room temperature | 0.106 | 51 | |
| Pt/Fe–TiO2 | 0.59 | 8 | Xe lamp through a color filter | Room temperature | 5.6 × 10−6 | 52 | |
| 0.1 wt% Pt/TiO2(A) | 15 | 100 | Xe lamp | Room temperature | 5.07 × 10−4 | 53 | |
| Pt/TiO2 | 0.1 | 10 | Hg lamp, λ > 300 nm | Room temperature | 4.5 × 10−4 | 42 | |
| Mw-Pt–TiO2d | 0.02 | 10 | Hg lamp, λ > 300 nm | Room temperature | 1.92 × 10−4 | 43 | |
| RuO2–NiO–SrTiO3 | 0.66 | 100 | Hg lamp | 40 | 1.0 × 10−5 | 44 | |
| Catalyst | WHSVe (mL/(g h)) | η (%) | Light source | Catalyst temp. (°C) | H2 rate (mol/(g h)) | Ref. | |
|---|---|---|---|---|---|---|---|
| Gaseous phase catalysis | Ru NPs/GaN NWs/Sig | — | 5 | Xe lamp, full-spectrum, 5 W cm−2 | 409.7 | 11.06 | 17 |
| Cu–Fe-ARh | — | 15.6 | White-light laser, 7.63 W cm−2 | 352 | 1.678 | 13 | |
| Cu–Ru-AR | — | 18 | White-light laser, 9.6 W cm−2 | 475.4 | 4.32 | 39 | |
| Ni-MCNi | — | — | Xe lamp, λ ≥ 420 nm | 52 | 3.56 × 10−5 | 54 | |
| Co@C | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
— | Xe lamp, full-spectrum, 3.3 W cm−2 | 250 | 0.503 | 55 | |
| Fe@C | 20000 |
— | Xe lamp, full-spectrum, 4.65 W cm−2 | 250 | 0.326 | 56 | |
| 0.1 wt% Pt/TiO2(A) | 100 | — | Xe lamp | 50 | 6.5 × 10−4 | 53 |
2.1 Catalysts for the photocatalytic decomposition of aqueous ammonia solution
Most of the catalysts for photocatalytic decomposition of aqueous ammonia solutions are TiO2-based materials, which are widely applied in various reactions thanks to their chemical stability, nontoxicity, low cost, and high reactivity.34,57 However, pristine TiO2 suffers from low light harvesting and a high recombination rate of photogenerated charge carriers. It was reported that cocatalysts or dopants could enhance their photocatalytic performance via extending light absorption (Fig. 2a), optimizing the adsorption of reactant/products, and/or promoting the efficiency of charge-carrier separation.33,51,58 Transition metals as cocatalysts demonstrate significant improvements for TiO2 photocatalysts, with Pt being the most effective cocatalyst (Fig. 2b).42,53 The ratio of H2 to N2 was 3.2 during the 4 h photocatalytic reaction, which agreed with the stoichiometry of ammonia decomposition (Fig. 2c). Platinum nanoparticles with a large work function would contribute to the efficient separation of photogenerated electrons. Furthermore, as shown in Fig. 2d, alloying Pt with Au further enhances the ability of charge separation.38 In the case of Pt1/TiO2, the high Schottky barrier (φB) suppresses electron transfer from the TiO2 conduction band (CB) to Pt. In contrast, for Au1/TiO2, the very low φB promotes electron transfer while simultaneously encouraging reverse electron transfer from Au to the TiO2 CB. By alloying Pt with Au, Pt0.9Au0.1/TiO2 exhibits an optimized φB for efficient separation of photogenerated electrons. Among the non-noble metals, Ni was the most efficient cocatalyst, exhibiting the highest yield of H2 in an aqueous ammonia solution under UV irradiation at room temperature.50 The bandgap of TiO2 is around 3.0–3.2 eV, which is much higher than the theoretical potential for photocatalytic ammonia decomposition. Doping TiO2 with additives could effectively decrease its bandgap and improve its light absorption capabilities. For instance, the light absorption could be optimized from ultraviolet to the visible light region via doping TiO2 with Ce, N, and/or Fe, resulting in increased H2 production efficiency.51,52,59 Jung et al. reported that the N and Fe co-doped TiO2 photocatalyst exhibited a bandgap as low as 2.4 eV, which was a visible light-sensitive catalyst.59 Wu et al. found that a mixture of N doped graphene and TiO2 nanowires in aqueous ammonia solution produced H2 at a rate of 208 μmol/(g h) under 254 nm UV irradiation, which is about 14-fold and 30-fold higher than that of the rates of pristine P25-TiO2 and TiO2 nanowires, respectively, under the same conditions.49 | ||
| Fig. 2 (a) Absorption edges of TiO2 doped with different Ce loadings. Reproduced from ref. 51 with permission from Elsevier, Copyright 2015. (b) Correlation between photocatalytic H2 evolution from an aqueous ammonia suspension of M/TiO2 (P25) particles and hydrogen overvoltage of metal electrodes. (c) Photocatalytic decomposition of ammonia in an aqueous suspension of Pt/TiO2 (P25) particles. (b and c) Reproduced from ref. 42 with permission from Elsevier, Copyright 2012. (d) Schematic representation of the electronic structures of Pt1/TiO2, Pt0.9Au0.1/TiO2, and Au1/TiO2 catalysts. Reproduced from ref. 38 with permission from the American Chemical Society, Copyright 2020. | ||
Other semiconductor catalysts were also developed for the photocatalytic decomposition of aqueous ammonia solutions. Reli et al. observed a high yield of hydrogen with a ZnO catalyst prepared by thermal annealing of zinc acetate in air. The activity of ZnO was even higher than that of commercial TiO2 Evonik P25, as the prepared ZnO possessed the lowest concentration of oxygen vacancies, thus suppressing the recombination of photo-induced electrons and holes.41 Iwase et al. prepared an Ru–ZnS cocatalyst that showed efficacy in the photocatalytic decomposition of aqueous ammonia solution under simulated sunlight irradiation.40 Dzíbelová et al. reported a green synthesis of 2D hematene (α-Fe2O3) from earth-abundant iron oxide ore using an ultrasound-assisted exfoliation method in an aqueous solution.60 The catalyst was prepared by doping hematene with ruthenium, which successfully decomposed ammonia into hydrogen and nitrogen. Dye can serve as a photosensitizer in the photocatalytic decomposition of aqueous ammonia solution. For example, Tajima et al. carried out photocatalytic ammonia decomposition using dye-encapsulated single-walled carbon nanotubes, which exhibited an apparent quantum yield of 0.22% at 510 nm.61
2.2 Proposed mechanisms
It is widely acknowledged that the mechanism of heterogeneous photocatalysis involves four consecutive steps: (1) light harvesting; (2) charge excitation/separation; (3) charge migration, transport, and recombination; and (4) charge utilization (surface electrocatalytic reduction and oxidation). Thus, the overall photocatalysis efficiency greatly depends on the combined effects of these steps (Fig. 3a).62 The photocatalytic decomposition of ammonia in aqueous solution is an uphill reaction (eqn (2)) that converts photon energy to chemical energy driven by one-step photoexcitation. Fig. 3b illustrates that the reduction and oxidation potentials of ammonia should lie between the conduction band and the valence band of the photocatalyst. However, different nitrogen-containing products (i.e., N2, NO2−, NO3−, and NOx) can be produced in the presence of O2 because of their similar redox potentials. Since ammonia is a pollutant, there are numerous studies on converting ammonia into non-toxic substances; for example, ammonia could be oxidized to N2 and H2O, which is thermodynamically favorable at room temperature (eqn (4)).63,64 Since the hydrogen production does not occur in such reactions, it will therefore not be discussed in this review. A mutually beneficial approach to managing waste ammonia is to transform it into harmless nitrogen and useful hydrogen as the sole end products. Therefore, the photocatalytic decomposition of aqueous ammonia solution is commonly conducted under deaerated conditions to avoid extensive oxidation of ammonia. | ||
| Fig. 3 (a) Four different stages involved in heterogeneous photocatalysis. (b) Illustration of the process of photocatalytic ammonia conversion. Reproduced from ref. 62 and ref. 36 with permission from the Royal Society of Chemistry, Copyright 2016 and 2020. | ||
Investigations have indicated that water is helpful for photocatalytic aqueous ammonia decomposition.44,53 Yuzawa et al. proposed a mechanism for photocatalytic ammonia decomposition on platinum-loaded titanium oxide, both in the presence and absence of water (Fig. 4). They applied in situ EPR to detect the amide radical (·NH2) and in situ FT-IR to observe the activation of the N–H bond. The dominant reaction path was proposed as follows: the photogenerated hole oxidizes NH3 to form a ·NH2 radical and a proton, which subsequently forms hydrazine that can be decomposed to N2 and H2. Then, the photogenerated electron reduces the protons to form hydrogen. In this mechanism, water is needed, where water inhibited the accumulation of NH4+ on the TiO2 surface and provided a continuous reaction, since the in situ formed NH4+ during the reaction is consistently considered an unfavorable byproduct (Fig. 4b).
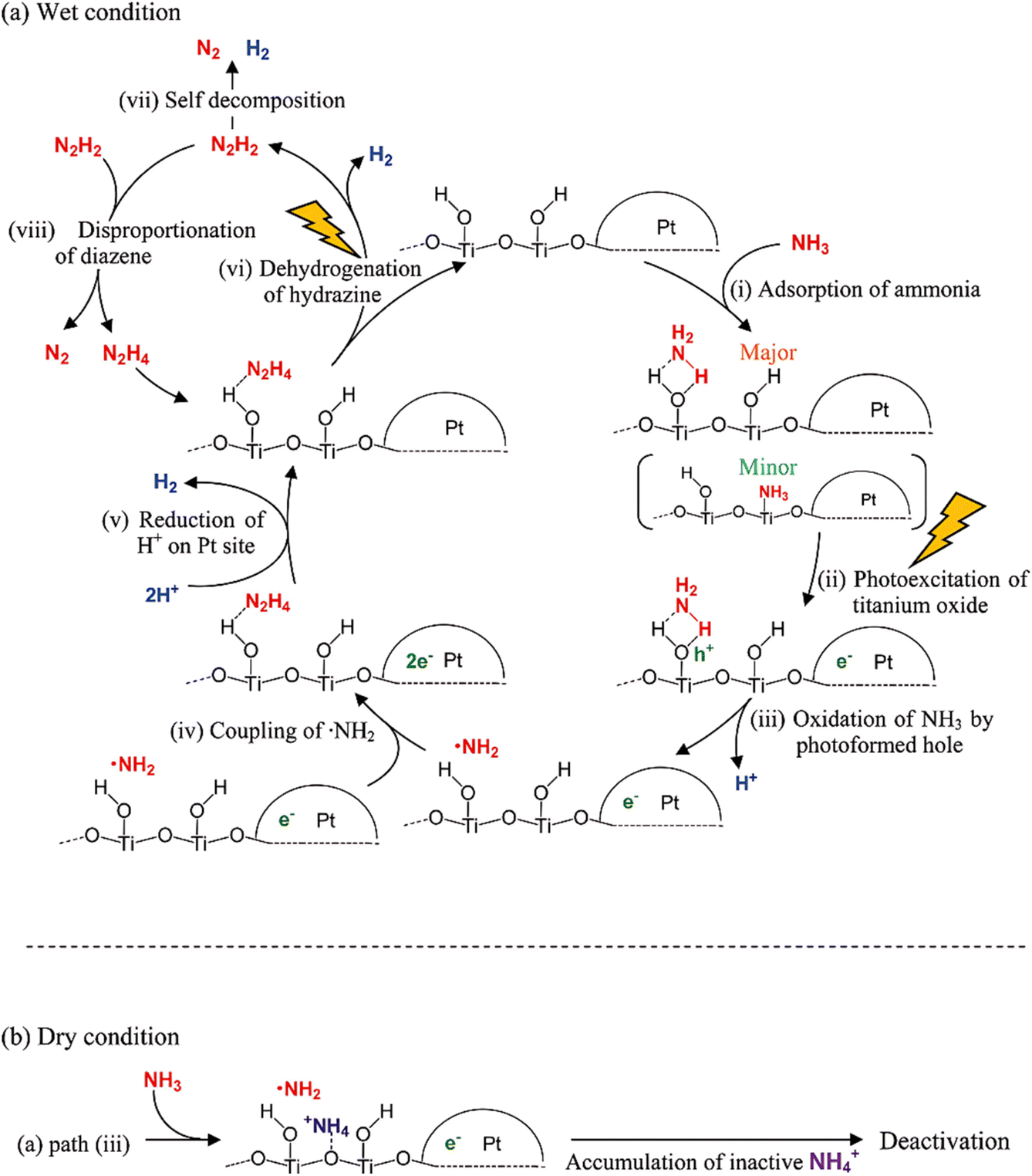 | ||
| Fig. 4 Proposed dominant reaction path of photocatalytic ammonia decomposition on platinum loaded titanium oxide in the presence (a) and absence of water (b). Reproduced from ref. 53 with permission from the American Chemical Society, Copyright 2012. | ||
Utsunomiya et al. presented a detailed energy profile of photocatalytic ammonia decomposition on TiO2.50 They indicated that an ·NH2 radical was formed as a dominant intermediate, as confirmed by an electron spin resonance (ESR) signal at g = 2.004. Besides, reaction pathways via ·NH2 radical formation were also thoroughly investigated using density functional theory, resulting in the proposal of three reaction pathways (Fig. 5): Route 1 involves the sequential extraction of H atoms from the NHx species until N radicals are formed. The activation energy for this route is estimated to be as high as 236 kcal/mol. Route 2 reveals that NH3 photo-decomposition may occur via the formation of H2N–NH2, which is formed through the coupling of adjacent ·NH2 radicals. The activation energy for Route 2 is approximately 74.4 kcal/mol. In Route 2′, the reaction pathway may alternatively occur via the formation of H2N–NH3, generated from the reaction of one ·NH2 radical and one gaseous NH3 molecule, which differs from the process in Route 2. The activation energy for Route 2′ is estimated to be 65.8 kcal/mol, which is more energetically favorable compared with Route 2. These two activation energies were not significantly different from each other. Therefore, it is possible that NH3 decomposition may have occurred via both Routes 2 and 2′, resulting in the formation of H2N–NH2.
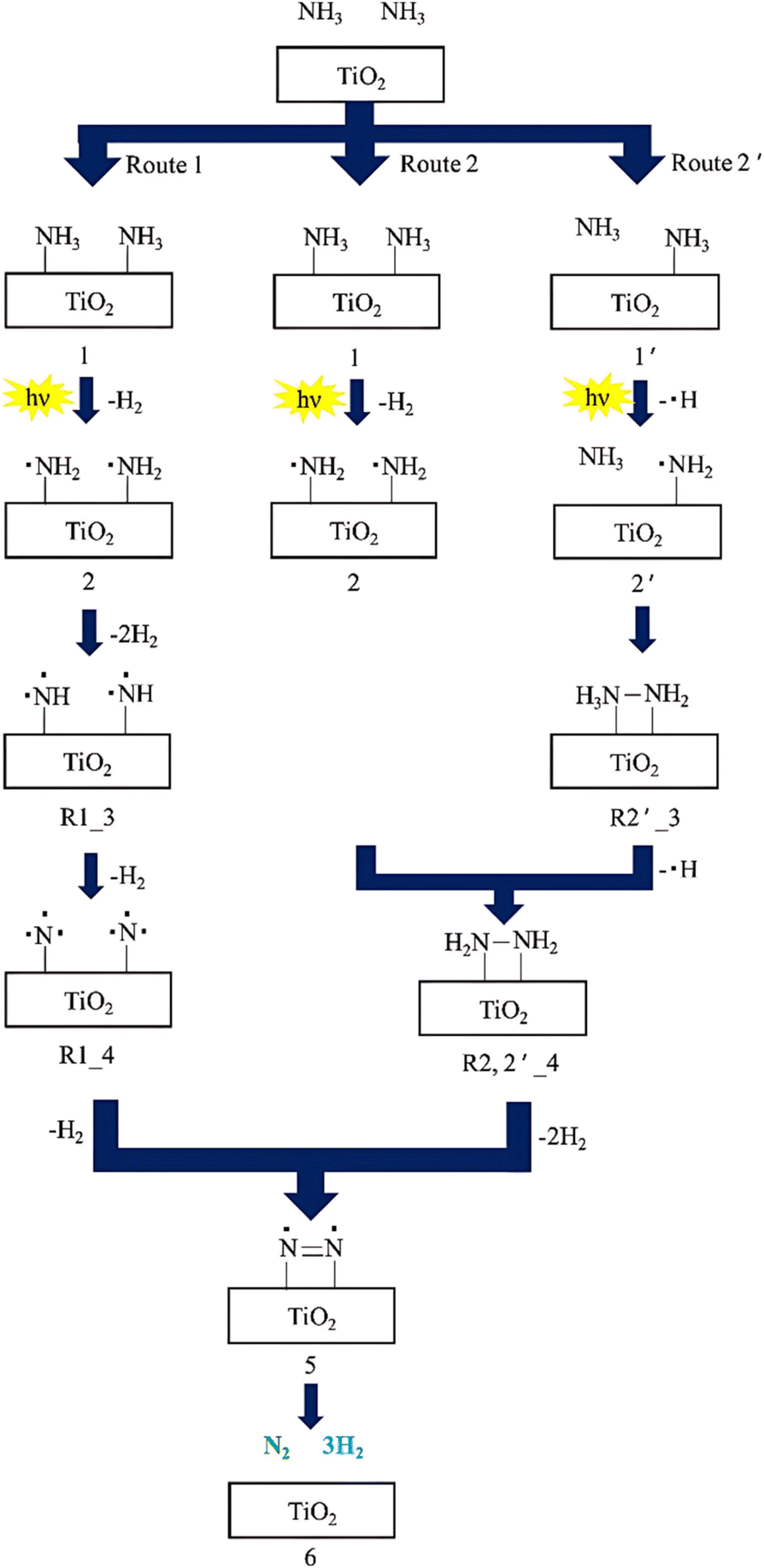 | ||
| Fig. 5 Suggested reaction mechanism for NH3 decomposition to form N2 and H2 over a TiO2 photocatalyst. Reproduced from ref. 50 with permission from Elsevier, Copyright 2017. | ||
3. Photocatalytic decomposition of gaseous ammonia
The photocatalytic decomposition of gaseous ammonia is a gas–solid heterogeneous catalytic reaction. Under light illumination, the catalyst absorbs light energy, converting it to heat on its surface. At high light intensities, photo-induced heat may play an important role in enhancing the activity of ammonia decomposition. Therefore, in many cases, photocatalytic ammonia decomposition is actually a photothermal catalytic process, characterized as a synergetic process between the photochemical effect and the photo-induced thermal effect.17,55,56 Usually, the activity of photocatalytic ammonia decomposition in the gaseous phase is higher than that of the liquid phase, with the difference potentially reaching several orders of magnitude (Table 2).During the photothermal catalytic decomposition of gaseous ammonia, the main roles of light can be broadly classified into two categories: photogenerated charges and photo-induced heat. Photogenerated charges include holes and electrons because of the excitation of semiconductors,34,57,65,66 as well as hot carriers induced by localized surface plasmon resonance (LSPR) when the catalyst surface absorbs light beams.39,67–69 The sources of photo-induced heat mainly consist of non-radiative relaxation of excited electrons in the semiconductor, energy transfer from hot carriers generated by LSPR to the catalyst lattice, and inelastic photon-phonon scattering between light and the catalyst lattice.70–73
It should be particularly noted that the photothermal catalysis discussed in this article refers to a process that integrates both the photochemical effect and the photo-induced thermal effect, excluding catalytic processes that rely solely on photo-induced heat as an energy source for thermocatalysis.74–76
3.1 Catalysts for the photocatalytic decomposition of gaseous ammonia
In the realm of photocatalysis, it is a prevalent methodology to employ semiconductor materials to absorb light, thereby generating carriers that drive chemical reactions. Li et al. fabricated an Ru NPs/GaN NWs/Si composite catalyst.17 Primarily, GaN was utilized to absorb light and generate electrons and holes. Under the influence of photogenerated carriers and the localized heat generated by the Si support, ammonia was activated and decomposed into nitrogen and hydrogen. As shown in Fig. 6a, the hydrogen production rate by the illuminated catalyst experienced an approximate 1000-fold enhancement compared to that produced by thermocatalysis at the same temperature, attaining 11.06 mol/(g h). Concurrently, the activation energy of the reaction decreased from 1.08 eV to 0.22 eV (Fig. 6b). Lin et al. implemented macroporous carbon nitride (MCN) as a photocatalyst for ammonia decomposition.54 To enhance the performance of the catalyst, a series of transition metals on MCN were loaded. Among these, Ni-MCN demonstrated the most optimal photocatalytic activity for ammonia decomposition (Fig. 6c). Subsequent characterization indicated that Ni-MCN functions as a single-atom catalyst, featuring Ni–N4 sites within its structure. These sites effectively enhanced the optical characteristics of the catalyst, expedited the separation and transfer of photogenerated carriers, and ultimately hastened the rate of ammonia decomposition. Sousa et al. synthesized an Fe/C catalyst from an iron metal–organic framework (MOF),56 exhibiting remarkable photothermal catalytic activity for ammonia decomposition (Fig. 6d). This Fe/C catalyst contained Fe3O4, a semiconductor that is capable of generating charge carriers upon exposure to light irradiation. These charge carriers effectively provide the necessary energy for the intermediate species involved in ammonia decomposition, thereby facilitating the ammonia decomposition.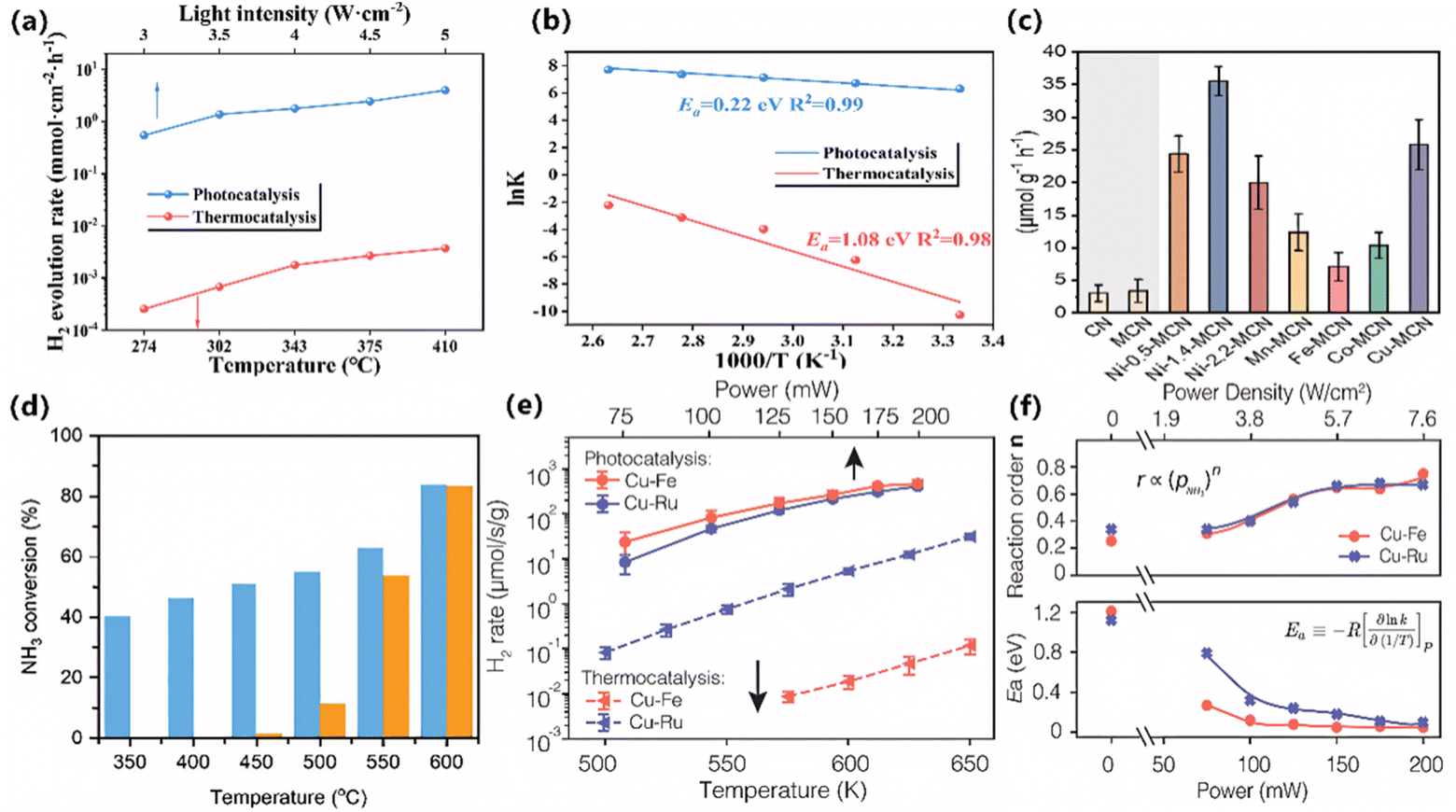 | ||
| Fig. 6 (a) H2 evolution rates over Ru NPs/GaN NWs/Si under photocatalysis and thermocatalysis. (b) Arrhenius plots for the H2 evolution rate under dark and light conditions over Ru NPs/GaN NWs/Si. (a and b) Reproduced from ref. 17 with permission from Springer Nature, Copyright 2024. (c) H2 production rates of CN, MCN, and Ni-MCNs with different Ni loadings and TMs-MCNs. Reproduced from ref. 54 with permission from the American Chemical Society, Copyright 2023. (d) Performance of the Fe@C catalyst under dark (orange) and light (blue) conditions. Reproduced from ref. 56 with permission from Wiley, Copyright 2025. (e) Comparison of photocatalysis and thermocatalysis for both antenna reactors (ARs). (f) Macroscopic kinetics studies: (top) the reaction orders of the reactant, (bottom) the apparent activation barrier, Ea. (e and f) Reproduced from ref. 13 with permission from AAAS, Copyright 2022. | ||
Notwithstanding, research endeavors focused on harnessing semiconductors to absorb light, generate electrons and holes, and facilitate gaseous ammonia decomposition remain scarce. Nevertheless, future catalyst design can draw inspiration from the research on photocatalytic water splitting. Given that the redox potential of ammonia decomposition is substantially lower than that of water decomposition, the bandgap of the semiconductor catalyst does not need be overly wide. Theoretically, a value exceeding 0.077 V is sufficient, which is significantly lower than the 1.23 V requisite for water splitting to produce hydrogen.11 Consequently, narrow bandgap semiconductors with enhanced light absorption capabilities can therefore be selected. Moreover, semiconductors with even narrower bandgaps, achieved through doping or the construction of heterojunctions, can be utilized in photocatalytic ammonia decomposition reactions.36
Currently, in the context of photocatalytic ammonia decomposition, alternative photocatalysts predominantly leverage the LSPR effect to expedite the rate of ammonia decomposition. The research work by Halas et al. has made a profound contribution to the study of photocatalytic ammonia decomposition.13,39 They engineered a Cu–Ru antenna reactor (Cu–Ru-AR), thereby enabling the catalytic decomposition of ammonia upon illumination. The active sites in Cu–Ru-AR comprise Cu, which exhibits the LSPR effect, and Ru, which is renowned for its thermal catalytic activity. Under light illumination, hot carriers are efficiently transferred from Cu to Ru, thus activating the ammonia molecules adsorbed onto Ru. It was found that the photocatalytic activity of Cu–Ru-AR was about one to two orders of magnitude higher than the thermal catalytic process at the same temperature. Furthermore, the Cu–Fe-AR catalyst, devoid of noble metals, exhibited photocatalytic activity for ammonia decomposition, which was comparable to that of the Cu–Ru-AR catalyst (Fig. 6e). Notably, in comparison to the thermal catalytic activity of the Cu–Fe-AR, the photocatalytic activity experienced an upsurge of around four orders of magnitude. The kinetic study reveals that, in contrast to thermocatalysis, photocatalytic ammonia decomposition has lower reaction orders of ammonia and higher apparent activation barriers (Fig. 6f). Cu–Fe-AR, an abundant and cost-effective plasmonic photocatalyst, holds great promise for sustainable hydrogen production via catalytic ammonia decomposition under sunlight.77 In addition, Sousa et al. used the LSPR effect of Co to achieve photocatalytic ammonia decomposition for hydrogen production.55 Their findings unequivocally demonstrated that the photocatalytic rate of this catalyst substantially surpasses that of thermocatalysis.
As mentioned above, the majority of photocatalytic gaseous ammonia decomposition processes fall under the purview of photothermal catalysis. The temperature engendered by the photo-induced heat can reach relatively high levels. Consequently, catalysts with superior thermocatalytic activity for ammonia decomposition can be viewed as active components in photocatalysis. This encompasses metals such as Ru, Ni, Co, Fe, and Mo, as well as bimetallic or multimetallic configurations formed by combinations of these metals with others, and the incorporation of promoters like K, Cs, and Ba.4,5,7,78–81 Moreover, alkali metal amides and imides, which exert a substantial promotional effect on ammonia decomposition, may potentially be contemplated as active components for photocatalytic ammonia decomposition.82–84
The decomposition of ammonia represents the reverse reaction of ammonia synthesis. Catalysts demonstrating high activity in photocatalytic ammonia synthesis may also exhibit discernible performance in photocatalytic ammonia decomposition.82 Presently, research on photocatalytic ammonia decomposition catalysts remains relatively sparse compared to the extensive investigations on photocatalytic ammonia synthesis catalysts. Thus, the design paradigm of photocatalytic ammonia synthesis catalysts holds the potential to inspire contemporary research endeavors in photocatalytic ammonia decomposition, potentially expediting the development of highly efficient photocatalysts.85,86
Most transition metals are capable of forming nitrides, which may undergo nitridation during ammonia decomposition reactions. For instance, nitrides are likely to be prevalent species in ammonia decomposition. Therefore, some nitrides function as semiconductors, exemplified by gallium nitride;17,87 others exhibit a robust LSPR effect, such as titanium nitride,88 and may serve as an efficient ammonia decomposition catalyst. These nitrides possess the potential of catalyzing ammonia decomposition when exposed to light. Therefore, the exploration of nitrides as active species for photocatalytic ammonia decomposition represents a worthy area of investigation.
3.2 Proposed mechanisms
The ammonia decomposition mechanism can usually be classified into two categories: the N–N recombination mechanism and the N2Hy dehydrogenation mechanism.2,89 In the ammonia decomposition process involving photogenerated charges, it primarily adheres to the N2Hy dehydrogenation mechanism, as mentioned previously, in the aqueous state. This mechanism indicates that during the ammonia decomposition process, NHx species will combine with each other to form N2Hy species. Subsequently, the N2Hy species generate N2 through a dehydrogenation process.89–91 The N–N recombination mechanism is quite common in the gaseous ammonia decomposition reactions. The mechanism was proposed earlier and has been widely accepted.1,3,5,48,92 Specifically, the steps of ammonia decomposition are as follows: | (5) |
 | (6) |
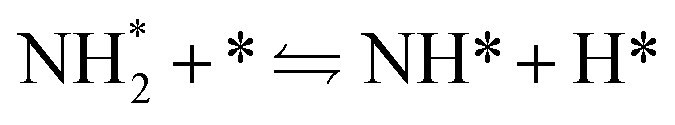 | (7) |
NH* + * ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) N* + H* N* + H* | (8) |
| 2N* 2* + N2 | (9) |
| 2H* 2* + H2 | (10) |
The rate-determining step in ammonia decomposition often involves either the cleavage of the N–H bond in the initial stage or the associative desorption of N* in the final stage. Nevertheless, due to the influence exerted by the characteristics of the catalyst and reaction conditions, the rate-determining step of ammonia decomposition may deviate from these common scenarios to other steps within the ammonia decomposition pathway.
The ammonia decomposition reaction, facilitated by semiconductor-based photocatalysts, mainly derives its photochemical effect from the electron–hole pairs produced during the photoexcitation of semiconductors. The electron–hole pairs serve as the driving force for the ammonia decomposition reaction. Nevertheless, semiconductor catalytic materials inherently possess several drawbacks. For instance, they often exhibit a wide band gap, leading to inadequate light absorption efficiency. Additionally, severe recombination of electrons and holes occurs after charge separation, and the charge transfer process is relatively sluggish. As a consequence, the photogenerated electron–hole pairs cannot be efficiently transported to the active sites to participate in the chemical reaction. Loading metals onto semiconductor materials represents an effective approach to overcoming these challenges. Wang et al. conducted theoretical calculations and revealed that ammonia decomposition on the surface of the semiconductor GaN adheres to the N–N recombination mechanism.17 The cleavage of the first N–H bond in the adsorbed  is likely the rate-determining step in this decomposition process, with an energy difference of 1.78 eV (Fig. 7b). When Ru is introduced onto the GaN surface, the adsorption of NH3 on the catalyst surface is remarkably enhanced, facilitating the cleavage of the first N–H bond. Subsequently, the potential-limiting step of the reaction shifts to the desorption of the adsorbed H* generated after the cleavage of the second N–H bond, with the adsorption energy barrier for this step being merely 0.58 eV. Moreover, the incorporation of Ru could increase the density of electronic states around the Fermi level of GaN (Fig. 7a), thereby promoting electron transfer and enabling a greater number of photogenerated electrons to engage in the NH3 decomposition reaction. Lin et al. carried out an investigation on the impact of different transition metals on the photocatalytic ammonia decomposition activity of carbon nitride.54 Accordingly, the research findings indicate that transition metals can alter the electronic structure, electrical conductivity, charge distribution, band gap of carbon nitride, and the adsorption energy of ammonia decomposition intermediate species. Among these transition metals, single-atom Ni demonstrates the most remarkable enhancement effect. The introduction of single-atom Ni will reconstruct the local electronic structure and yield a relatively narrow band gap, hence facilitating improved light absorption. It also exhibits a high electrical conductivity and an optimal charge distribution, expediting the transfer of photogenerated charges. In addition, it exhibited an appropriate adsorption energy for ammonia decomposition intermediate species. These properties collectively minimize the energy required for the potential-determined step of ammonia decomposition, thereby effectively accelerating the photocatalytic ammonia decomposition reaction.
is likely the rate-determining step in this decomposition process, with an energy difference of 1.78 eV (Fig. 7b). When Ru is introduced onto the GaN surface, the adsorption of NH3 on the catalyst surface is remarkably enhanced, facilitating the cleavage of the first N–H bond. Subsequently, the potential-limiting step of the reaction shifts to the desorption of the adsorbed H* generated after the cleavage of the second N–H bond, with the adsorption energy barrier for this step being merely 0.58 eV. Moreover, the incorporation of Ru could increase the density of electronic states around the Fermi level of GaN (Fig. 7a), thereby promoting electron transfer and enabling a greater number of photogenerated electrons to engage in the NH3 decomposition reaction. Lin et al. carried out an investigation on the impact of different transition metals on the photocatalytic ammonia decomposition activity of carbon nitride.54 Accordingly, the research findings indicate that transition metals can alter the electronic structure, electrical conductivity, charge distribution, band gap of carbon nitride, and the adsorption energy of ammonia decomposition intermediate species. Among these transition metals, single-atom Ni demonstrates the most remarkable enhancement effect. The introduction of single-atom Ni will reconstruct the local electronic structure and yield a relatively narrow band gap, hence facilitating improved light absorption. It also exhibits a high electrical conductivity and an optimal charge distribution, expediting the transfer of photogenerated charges. In addition, it exhibited an appropriate adsorption energy for ammonia decomposition intermediate species. These properties collectively minimize the energy required for the potential-determined step of ammonia decomposition, thereby effectively accelerating the photocatalytic ammonia decomposition reaction.
 | ||
| Fig. 7 (a) Local density of states (LDOS) for pristine GaN and Ru/GaN, respectively. The black dashed line indicates the position of the Fermi level. (b) The calculated free energy ΔG diagrams for NH3 decomposition on GaN and Ru/GaN. The values in the figures indicate the energy difference for the potential-limiting step of the reaction. (a and b) Reproduced from ref. 17 with permission from Springer Nature, Copyright 2024. (c) Schematic comparison of the energy landscape for photocatalysis (excited states) and thermocatalysis (ground state) on Cu–Fe- and Cu–Ru-ARs. (c) Reproduced from ref. 13 with permission from AAAS, Copyright 2022. | ||
Recently, Halas et al. reported a plasmonic antenna-reactor for photocatalysis.13,39,93,94 Compared to thermocatalysis, the plasmonic antenna-reactor catalysts (Cu–Ru-AR, Cu–Fe-AR) exhibit much higher activity in the ammonia decomposition reaction, by one to four orders of magnitude.13,39 The hot carriers produced by the antenna metal Cu can be transferred to the active transition metals, Ru or Fe. In ground-state thermocatalysis, the associative desorption of N2 on Cu–Ru-AR or Cu–Fe-AR constitutes the rate-determining step, with barriers of 2.67 and 2.84 eV, respectively. However, in photocatalysis, the barrier for the associative desorption of N2 decreases to 0.50 and 0.65 eV, respectively (Fig. 7c). In this scenario, the activation barriers for the dissociative adsorption of NH3 are 0.53 and 0.95 eV, respectively. Finally, the reaction rate is greatly enhanced upon illumination. Wen et al. used embedded correlated wavefunction (ECW) theory to predict the rate-determining step for plasmon-driven ammonia decomposition on Pd (111).95 It is found that the rate-determining step is altered by illumination, i.e., thermocatalysis (dissociating the first N–H bond,  , in the ground state) contrasts with photocatalysis (dissociating the second N–H bond,
, in the ground state) contrasts with photocatalysis (dissociating the second N–H bond,  , in the excited state).
, in the excited state).
3.3 Temperature measurement
As mentioned above, both the photo-induced thermal effect and the photochemical effect are indispensable for promoting photocatalytic ammonia decomposition. It is scientifically important to distinguish between the photo-induced thermal effect and the photochemical effect.37,39 Usually, the catalytic process with the photochemical effect differs from thermocatalysis in the aspects of catalyst activity, activation energy, reaction order, and so on. In fact, unlike the relatively uniform temperature field of a traditional thermocatalytic system, the temperature distribution of a typical gas–solid photothermal system varies according to various parameters, including photon flux density, catalyst absorbance, and photothermal conversion efficiency.96 Particularly, ammonia decomposition involves hydrogen, a gas with extremely high thermal conductivity (H2: 163 mW/(m K), N2: 23 mW/(m K), NH3: 24 mW/(m K)). The temperature of the catalyst may decrease as the amount of hydrogen produced from ammonia decomposition increases. These factors lead to significant temperature gradients within the catalyst layer (Fig. 8a). Therefore, a biased assessment of photocatalytic ammonia decomposition performance is likely to result in misjudgment of catalyst activity and an unreasonable comparison between photocatalysis and thermocatalysis, thus exaggerating the advantages of photocatalytic ammonia decomposition.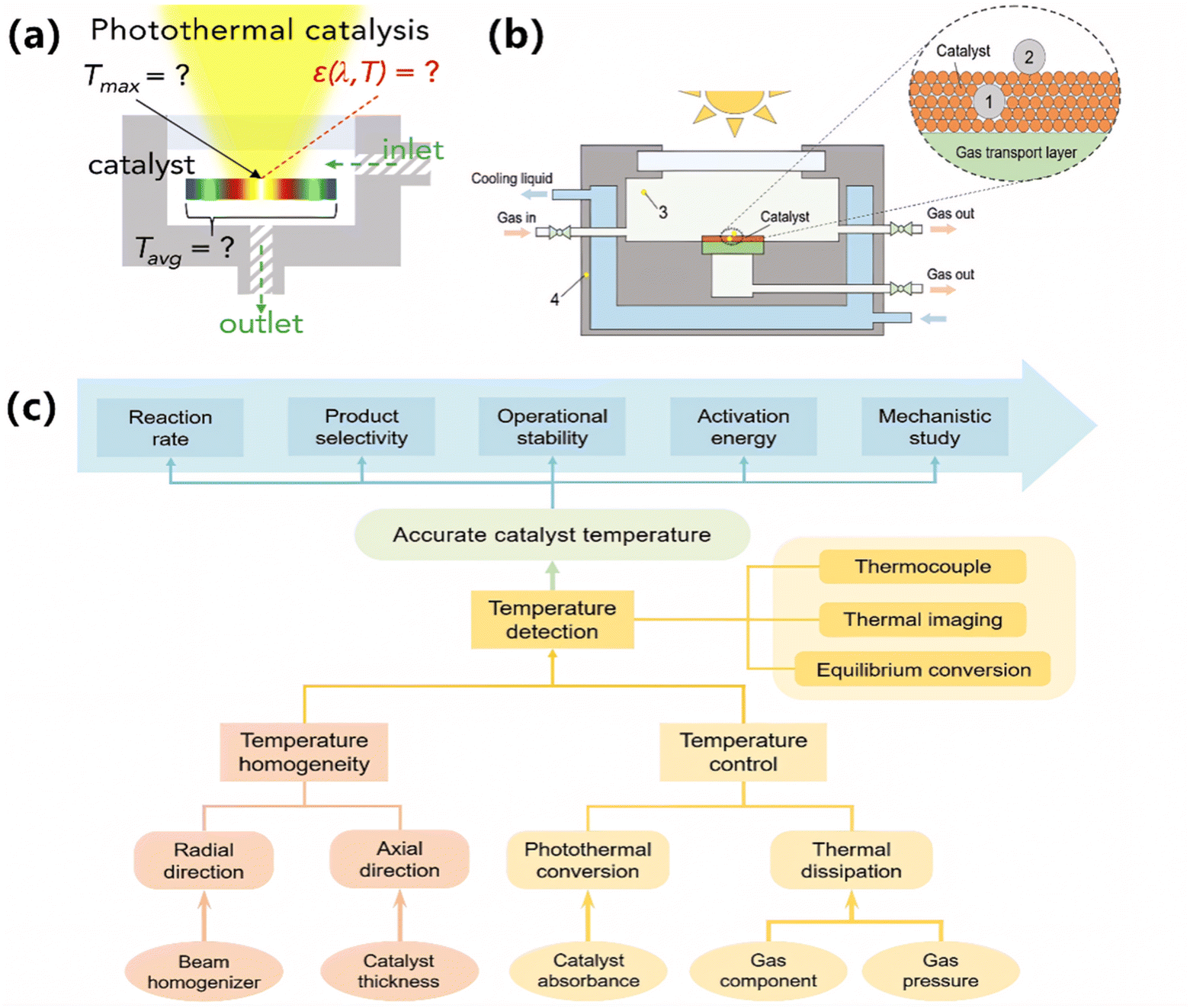 | ||
| Fig. 8 (a) Challenges for the accurate temperature measurements in gas–solid photothermal catalysis. Reproduced from ref. 97 with permission from Elsevier, Copyright 2022. (b) Schematic diagram of a photothermal catalysis device marked with several temperature measurement positions. (c) Recommendations for reliable temperature measurements in gas–solid photothermal catalysis. (b and c) Reproduced from ref. 96 with permission from Wiley, Copyright 2023. | ||
Bian et al. provided guidance for researchers to avoid the misconceptions of temperature evaluation in photothermal catalysis (Fig. 8c).96 As a premise, the actual temperature of a catalyst should be taken for a reliable temperature measurement in photothermal catalysis. Suggestions to improve the accuracy of thermocouples are as follows. The thermocouple, having a diameter significantly smaller than the thickness of the catalyst layer, is embedded into the catalyst layer (position 1 shown in Fig. 8b) to ensure good thermal contact. Using a beam homogenizer and reducing the thickness of the catalyst layer can create a more uniform temperature distribution. Moreover, a thermal imaging camera can easily monitor and check the actual temperature distribution of the catalyst layer. Besides, a properly designed temperature compensation device is essential for regulating temperature, thereby mitigating fluctuations induced by atmospheric changes, variations in catalyst absorbance, or the endothermic nature of ammonia decomposition.
Moreover, the catalyst temperature for photocatalytic ammonia decomposition can be calibrated using various alternative methods: (1) determining the temperature by exploiting the correlation between the equilibrium conversion of ammonia decomposition and temperature.98 (2) Conducting control experiments with catalysts lacking photochemical effects or by covering photocatalysts with light-absorbing materials that exhibit no ammonia decomposition activity.37,56,99 (3) Depicting the reaction temperature profile through numerical simulation to obtain the catalyst surface temperature during photocatalysis.13 Accurate temperature assessment is essential for reliably distinguishing the thermal and photochemical contributions in photocatalytic ammonia decomposition.
4. Conclusions and outlook
In the current context of decarbonization, ammonia, as a carbon-free fuel, exhibits great potential in the energy supply chain. Ammonia decomposition for hydrogen production is indispensable when ammonia is employed as a hydrogen carrier or as fuel in internal combustion engines. Among external-field-assisted ammonia decompositions, photo-driven ammonia decomposition shows unique advantages over thermocatalysis, particularly with high activity and an environmentally friendly process. Moreover, photoenergy could be regionally applied to the catalyst area, leading to a rapid response for hydrogen production, which is a disadvantage for thermocatalysis. Fig. 9 summarizes the functions and advantages of light in photocatalytic ammonia decomposition. Herein, we propose that the following areas should be the focus of future research. | ||
| Fig. 9 Photocatalytic ammonia decomposition by coupling the photochemical effect and photo-induced thermal effect. | ||
4.1 Catalyst design and mechanism exploration
In the pursuit of efficient photocatalysts, superior catalysts in thermocatalysis merit consideration. Their active sites may retain functionality under light illumination, leveraging photo-induced heat. Significantly, the active component of a catalyst in thermocatalysis is often a transition metal, which may exhibit a pronounced LSPR effect, while the catalyst support is commonly a semiconductor such as TiO2. Therefore, an excellent catalyst in thermocatalysis may also function as a robust photocatalyst for ammonia decomposition by virtue of the synergy between photo-induced heat and charge carriers generated by LSPR or an excited semiconductor. Moreover, metal nitrides, amides, and imides require particular attention. These materials are anticipated to form in an ammonia atmosphere and may possess semiconductor properties or exhibit the LSPR effect. These metal nitrides possess metal-like characteristics and are expected to exhibit a strong LSPR effect,100 resulting in high activity through the synergistic combination of the photo-induced thermal effect and photochemical effect. Hence, metal nitrides, as photocatalysts for ammonia decomposition, are worthy of increased research attention. Concurrently, elucidating the mechanism of the photocatalytic process is essential, which can be achieved by integrating advanced characterization techniques with sophisticated theoretical calculations. With a clear mechanism, the development of efficient photocatalysts can be expedited. Furthermore, the application of advanced artificial intelligence (AI) in the design of efficient photocatalysts presents great promise and could potentially revolutionize this domain, where more but accurate data are needed.4.2 Convincing evidence of the photochemical effect
To improve the utilization of sunlight, ammonia decomposition through the synergistic catalysis of the photochemical effect and photo-induced thermal effect under full-spectrum illumination may be one of the excellent approaches.70–72,101 Theoretically, the photo-induced thermal effect takes advantage of the high temperature generated on the surface of the photocatalyst. In this scenario, the performance of the catalyst is solely related to the temperature rather than directly associated with light, and its catalytic behavior is identical to that of the thermocatalytic process. In contrast, the photochemical effect drives the reaction by harnessing photogenerated electrons and holes in semiconductors or hot electrons produced by the LSPR effect. Under the influence of the photochemical effect, the thermodynamic equilibrium of the reaction will be changed. Ammonia decomposition, for example, has a higher equilibrium conversion rate than that of thermocatalytic ammonia decomposition. And the photochemical effect is generally validated by comparing it with thermocatalysis in terms of activity, selectivity, reaction order, activation energy, and so on. Consequently, accurately determining the temperature on the catalyst surface under light illumination is particularly important. Therefore, it is crucial to collect accurate and correct temperatures. In addition, ammonia may be photolyzed when exposed to specific light sources like deep ultraviolet radiation. Therefore, it is scientifically important to distinguish the processes of photolysis and photocatalysis. Only the acquisition of rules and conclusions derived from accurate experimental results can help elucidate a clearer mechanism of photocatalytic ammonia decomposition and facilitate the development of more efficient catalysts.4.3 Key points for practical applications
In the use of photocatalytic ammonia decomposition, reactor design and optimization, especially for light source selection and photoenergy utilization, are as crucial as catalyst development. Numerous aspects need to be considered in reactor design, such as reaction chamber configuration, catalyst layout, and the integration of light-transmitting components to ensure optimal catalyst illumination and mass transfer. For example, the selection of a light source necessitates aligning spectral characteristics with the absorption range of the photocatalyst while also taking into account light intensity, stability, and energy consumption. More importantly, the light-to-hydrogen energy efficiency should be considered, which is defined as the efficiency of photons impinging on the catalyst surface being converted into chemical energy by comparing the converted heat values of ammonia (18.65 kJ/g) and hydrogen (119.96 kJ/g), i.e., the energy of hydrogen production divided by the sum of the light energy and ammonia energy.13 So far, the highest reported energy efficiency of photocatalysis is 18%.39 There is still ample room for improvement. The overall system must feature a high hydrogen production rate, elevated energy efficiency, stability, and minimal cost. The applicable scenarios of photocatalytic ammonia decomposition hold substantial importance. For instance, leveraging its features such as rapid response and high hydrogen production rate, it can be applied in hydrogen-production scenarios where frequent and prompt start-up and shut-down operations are demanded. By advancing the implementation of photocatalytic ammonia decomposition, the arrival of the ammonia-hydrogen economy can be accelerated.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
P. Chen and T. He conceived the idea of the review. Q. Pei wrote the manuscript. T. He revised the manuscript. Y. Wang conducted data search and verification. K. C. Tan carried out the language polishing of the manuscript. J. Guo put forward suggestions on the idea of the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key R&D Program of China (2023YFE0198900) and the Major Science and Technology Project of Liaoning Province (Grant No. 2024JH1/11700016). T. He and Q. Pei acknowledge the support provided by the National Natural Science Foundation of China (52171226 and U24A20499).References
- B. S. Solanki, H. Lim, S. J. Yoon, H. C. Ham, H. S. Park, H. E. Lee and S. H. Lee, Renewable Sustainable Energy Rev., 2025, 207, 114974 CrossRef CAS.
- X. Hu, B. Guan, J. Chen, Z. Zhuang, C. Zheng, J. Zhou, T. Su, C. Zhu, S. Zhao, J. Guo, H. Dang, Y. Zhang, Y. Yuan, C. Yi, C. Xu, B. Xu, W. Zeng, Y. He, Z. Wei and Z. Huang, Fuel, 2025, 381, 133134 CrossRef CAS.
- Z. Zhao, M. Zhang, Y. Wu, W. Song, J. Yan, X. Qi, J. Yang, J. Wen and H. Zhang, Ind. Eng. Chem. Res., 2024, 63, 8003 CrossRef CAS.
- Z. Lu, B. Jiang, Z. Chen, J. Shi, D. Jing, Y. Lu and M. Liu, Innov. Energy, 2024, 1, 100056 CrossRef.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 60, 18560 CrossRef CAS.
- Chemical Industry, Production capacity of ammonia worldwide from 2018 to 2021, with a forecast for 2026 and 2030, 2022, https://www.statista.com/statistics/1065865/ammonia-production-capacity-globally/(accessed on 18 September 2022).
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63 CrossRef.
- International Energy Agency, IEA Report, 2021, https://iea.blob.core.windows.net/assets/deebef5d-0c34-4539-9d0c-10b13d840027/NetZeroby2050-ARoadmapfortheGlobalEnergySector_CORR.pdf (accessed on 15 September 2022) Search PubMed.
- T. Autrey and P. Chen, J. Energy Chem., 2023, 77, 119 CrossRef.
- A. Badakhsh, Y. Kwak, Y.-J. Lee, H. Jeong, Y. Kim, H. Sohn, S. W. Nam, C. W. Yoon, C. W. Park and Y. S. Jo, Chem. Eng. J., 2021, 426, 130802 CrossRef CAS.
- N. Hanada, S. Hino, T. Ichikawa, H. Suzuki, K. Takai and Y. Kojima, Chem. Commun., 2010, 46, 7775 RSC.
- I. Fedirchyk, I. Tsonev, R. Quiroz Marnef and A. Bogaerts, Chem. Eng. J., 2024, 499, 155946 CrossRef CAS.
- Y. Yuan, L. Zhou, H. Robatjazi, J. L. Bao, J. Zhou, A. Bayles, L. Yuan, M. Lou, M. Lou, S. Khatiwada, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2022, 378, 889 CrossRef CAS PubMed.
- R. Zhang, X. Liu, N. Song, J. He, Z. Cen, C. Li, M. Wang, H. Tang, W. Liu, X. Ren and D. Ma, J. Am. Chem. Soc., 2024, 146, 28635 CrossRef CAS PubMed.
- Y. Ofuchi, K. Mitarai, S. Doi, K. Saegusa, M. Hayashi, H. Sampei, T. Higo, J. G. Seo and Y. Sekine, Chem. Sci., 2024, 15, 15125 RSC.
- C. Mi, R. Ghazfar, M. R. Smith and T. W. Hamann, Joule, 2022, 6, 772 CrossRef CAS.
- J. Li, B. Sheng, Y. Chen, J. Yang, P. Wang, Y. Li, T. Yu, H. Pan, L. Qiu, Y. Li, J. Song, L. Zhu, X. Wang, Z. Huang and B. Zhou, Nat. Commun., 2024, 15, 7393 CrossRef CAS.
- B. K. Boggs and G. G. Botte, J. Power Sources, 2009, 192, 573 CrossRef CAS.
- G. Chen, J. Qu, P. Cheah, D. Cao, Y. Zhao and Y. Xiang, Ind. Eng. Chem. Res., 2022, 61, 11436 CrossRef CAS.
- L. Wang, Y. Yi, Y. Zhao, R. Zhang, J. Zhang and H. Guo, ACS Catal., 2015, 5, 4167 CrossRef CAS.
- N. Zhu, Y. Hong, F. Qian and J. Liang, Int. J. Hydrogen Energy, 2024, 59, 791 CrossRef CAS.
- S. Meng, S. Li, S. Sun, A. Bogaerts, Y. Liu and Y. Yi, Chem. Eng. Sci., 2024, 283, 119449 CrossRef CAS.
- S. Bang, R. Snoeckx and M. S. Cha, J. Phys. Chem. A, 2023, 127, 1271 CrossRef CAS PubMed.
- J. A. Andersen, K. van 't Veer, J. M. Christensen, M. Østberg, A. Bogaerts and A. D. Jensen, Chem. Eng. Sci., 2023, 271, 118550 CrossRef CAS.
- J. A. Andersen, J. M. Christensen, M. Østberg, A. Bogaerts and A. D. Jensen, Int. J. Hydrogen Energy, 2022, 47, 32081 CrossRef CAS.
- L. S. Kassel and W. A. Noyes Jr, J. Am. Chem. Soc., 1927, 49, 2495 CrossRef CAS.
- W. Kurn, C. R. Acad. Sci., Ser. Gen. Vie Sci., 1923, 177, 956 Search PubMed.
- J. P. Ferris and D. E. Nicodem, Nature, 1972, 238, 268 CrossRef CAS.
- A. Canta, K. I. Öberg and M. Rajappan, Astrophys. J., 2023, 953, 81 CrossRef CAS.
- J. P. Ferris and Y. Ishikawa, Nature, 1987, 326, 777 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- X. Li, C. Wang and J. Tang, Nat. Rev. Mater., 2022, 7, 617 CrossRef CAS.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919 CrossRef CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229 CrossRef CAS PubMed.
- S. Zhang, Z. He, X. Li, J. Zhang, Q. Zang and S. Wang, Nanoscale Adv., 2020, 2, 3610 RSC.
- A. Beck, J. Marlowe, M. J. Gordon and P. Christopher, J. Phys. Chem. C, 2024, 128, 8590 CrossRef CAS.
- Y. Shiraishi, S. Toi, S. Ichikawa and T. Hirai, ACS Appl. Nano Mater., 2020, 3, 1612 CrossRef CAS.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69 CrossRef CAS PubMed.
- A. Iwase, K. Ii and A. Kudo, Chem. Commun., 2018, 54, 6117 RSC.
- M. Reli, M. Edelmannová, M. Šihor, P. Praus, L. Svoboda, K. K. Mamulová, H. Otoupalíková, L. Čapek, A. Hospodková, L. Obalová and K. Kočí, Int. J. Hydrogen Energy, 2015, 40, 8530 CrossRef CAS.
- H. Kominami, H. Nishimune, Y. Ohta, Y. Arakawa and T. Inaba, Appl. Catal., B, 2012, 111–112, 297 CrossRef CAS.
- K. Fuku, T. Kamegawa, K. Mori and H. Yamashita, Chem.–Asian J., 2012, 7, 1366 CrossRef CAS PubMed.
- Q.-s. Li, K. Domen, S. Naito, T. Onishi and K. Tamaru, Chem. Lett., 1983, 12, 321 CrossRef.
- N. Li, C. Zhang, D. Li, W. Jiang and F. Zhou, Chem. Eng. J., 2024, 495, 153125 CrossRef CAS.
- A. Kumar, V. Vibhu, J.-M. Bassat, L. Nohl, L. G. J. de Haart, M. Bouvet and R.-A. Eichel, ChemElectroChem, 2024, 11, e202300845 CrossRef CAS.
- M. Asif, S. Sidra Bibi, S. Ahmed, M. Irshad, M. Shakir Hussain, H. Zeb, M. Kashif Khan and J. Kim, Chem. Eng. J., 2023, 473, 145381 CrossRef CAS.
- X. Huang, K. Lei, Y. Mi, W. Fang and X. Li, Molecules, 2023, 28, 5245 CrossRef CAS PubMed.
- Z. Wu, N. Ambrožová, E. Eftekhari, N. Aravindakshan, W. Wang, Q. Wang, S. Zhang, K. Kočí and Q. Li, Emergent Mater., 2019, 2, 303 CrossRef CAS.
- A. Utsunomiya, A. Okemoto, Y. Nishino, K. Kitagawa, H. Kobayashi, K. Taniya, Y. Ichihashi and S. Nishiyama, Appl. Catal., B, 2017, 206, 378 CrossRef CAS.
- M. Reli, N. Ambrožová, M. Šihor, L. Matějová, L. Čapek, L. Obalová, Z. Matěj, A. Kotarba and K. Kočí, Appl. Catal., B, 2015, 178, 108 CrossRef CAS.
- K. Obata, K. Kishishita, A. Okemoto, K. Taniya, Y. Ichihashi and S. Nishiyama, Appl. Catal., B, 2014, 160–161, 200 CrossRef CAS.
- H. Yuzawa, T. Mori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 4126 CrossRef CAS.
- J. Lin, Y. Wang, W. Tian, H. Zhang, H. Sun and S. Wang, ACS Catal., 2023, 13, 11711 CrossRef CAS.
- A. Sousa, A. Rendon Patino, L. Garzon Tovar, D. Mateo, J. Gascon and A. Bavykina, ChemSusChem, 2024, e202401896 Search PubMed.
- A. Sousa, D. Mateo, L. Garzon-Tovar, K. Brennan, A. Rendón-Patiño, N. Morlanés, X. Wang, J. C. Navarro, J. Ruiz-Martinez, M. García-Melchor and J. Gascon, Small, 2025, 2411468 CrossRef CAS PubMed.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, 1901997 CrossRef CAS PubMed.
- W. Fang, J. Yan, Z. Wei, J. Liu, W. Guo, Z. Jiang and W. Shangguan, Chin. J. Catal., 2024, 60, 1 CrossRef CAS.
- S.-C. Jung, K.-H. Chung, J. Choi, Y.-K. Park, S.-J. Kim, B.-J. Kim and H. Lee, Catal. Today, 2022, 397–399, 165 CrossRef CAS.
- J. Dzíbelová, S. M. H. Hejazi, V. Šedajová, D. Panáček, P. Jakubec, Z. Baďura, O. Malina, J. Kašlík, J. Filip, Š. Kment, M. Otyepka and R. Zbořil, Appl. Mater. Today, 2023, 34, 101881 CrossRef.
- T. Tajima, K. Yano, K. Mukai and Y. Takaguchi, Catalysts, 2024, 14, 715 CrossRef CAS.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603 RSC.
- Y. Shu, D. Wang, J. Wang and H. Huang, Chem. Eng. J., 2024, 498, 154925 CrossRef CAS.
- K. Vikrant, K.-H. Kim, F. Dong and D. A. Giannakoudakis, ACS Catal., 2020, 10, 8683 CrossRef CAS.
- L. Zhang, J. Zhang, H. Yu and J. Yu, Adv. Mater., 2022, 34, 2107668 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411 CrossRef CAS.
- S. Luo, X. Ren, H. Lin, H. Song and J. Ye, Chem. Sci., 2021, 12, 5701 RSC.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656 CrossRef.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25 CrossRef CAS.
- C. Xu, Q. Tang, W. Tu and L. Wang, Energy Environ. Sci., 2024, 17, 4461 RSC.
- J. Zhang, H. Chen, X. Duan, H. Sun and S. Wang, Mater. Today, 2023, 68, 234 CrossRef CAS.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem Catal., 2022, 2, 52 CrossRef CAS.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173 RSC.
- Y. Li, Q. Guan, G. Huang, D. Yuan, F. Xie, K. Li, Z. Zhang, X. San and J. Ye, Adv. Energy Mater., 2022, 12, 2202459 CrossRef CAS.
- T. Hu and Y. Wang, J. Energy Eng., 2020, 146, 04020018 CrossRef.
- B. Wang, H. Kong, H. Wang, Y. Wang and X. Hu, Int. J. Hydrogen Energy, 2019, 44, 26874 CrossRef CAS.
- J. Liu, J. Feng, Z. Zou and Z. Li, Sci. Bull., 2024, 69, 1 CrossRef CAS.
- X. Ju, L. Liu, P. Yu, J. Guo, X. Zhang, T. He, G. Wu and P. Chen, Appl. Catal., B, 2017, 211, 167 CrossRef CAS.
- S.-F. Yin, Q.-H. Zhang, B.-Q. Xu, W.-X. Zhu, C.-F. Ng and C.-T. Au, J. Catal., 2004, 224, 384 CrossRef CAS.
- S. Podila, H. Driss, S. F. Zaman, A. M. Ali, A. A. Al-Zahrani, M. A. Daous and L. A. Petrov, Int. J. Hydrogen Energy, 2020, 45, 873 CrossRef CAS.
- J. Zhang, J.-O. Müller, W. Zheng, D. Wang, D. Su and R. Schlögl, Nano Lett., 2008, 8, 2738 CrossRef CAS.
- J. Guo and P. Chen, Acc. Chem. Res., 2021, 54, 2434 CrossRef CAS.
- J. Guo, P. Wang, G. Wu, A. Wu, D. Hu, Z. Xiong, J. Wang, P. Yu, F. Chang, Z. Chen and P. Chen, Angew. Chem., 2015, 127, 2993 CrossRef.
- W. I. F. David, J. W. Makepeace, S. K. Callear, H. M. A. Hunter, J. D. Taylor, T. J. Wood and M. O. Jones, J. Am. Chem. Soc., 2014, 136, 13082 CrossRef CAS PubMed.
- Y. Guan, H. Wen, K. Cui, Q. Wang, W. Gao, Y. Cai, Z. Cheng, Q. Pei, Z. Li, H. Cao, T. He, J. Guo and P. Chen, Nat. Chem., 2024, 16, 373 CrossRef CAS PubMed.
- J. Di, J. Xia, M. F. Chisholm, J. Zhong, C. Chen, X. Cao, F. Dong, Z. Chi, H. Chen, Y.-X. Weng, J. Xiong, S.-Z. Yang, H. Li, Z. Liu and S. Dai, Adv. Mater., 2019, 31, 1807576 CrossRef PubMed.
- A. Zakutayev, J. Mater. Chem. A, 2016, 4, 6742 RSC.
- G. V. Naik, J. L. Schroeder, X. Ni, A. V. Kildishev, T. D. Sands and A. Boltasseva, Opt. Mater. Express, 2012, 2, 478 CrossRef CAS.
- X. Zou, H.-Y. Su, X. Sun, W. Pang, X. Hao, Y. Xu and K. Sun, Appl. Surf. Sci., 2024, 649, 159175 CrossRef CAS.
- J.-J. Zhong, S.-P. Huang, J.-F. Gu, Y. Li, K.-N. Ding, Y.-F. Zhang, W. Lin and W.-K. Chen, Appl. Surf. Sci., 2023, 609, 155280 CrossRef CAS.
- H. Gerischer and A. Mauerer, J. Electroanal. Chem., 1970, 25, 421 CrossRef CAS.
- M. I. Temkin, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weez, Academic Press, 1979, ch. 173, vol. 28, pp. 173–291 Search PubMed.
- L. Yuan, J. Zhou, M. Zhang, X. Wen, J. M. P. Martirez, H. Robatjazi, L. Zhou, E. A. Carter, P. Nordlander and N. J. Halas, ACS Nano, 2022, 16, 17365 CrossRef CAS PubMed.
- D. F. Swearer, H. Zhao, L. Zhou, C. Zhang, H. Robatjazi, J. M. P. Martirez, C. M. Krauter, S. Yazdi, M. J. McClain, E. Ringe, E. A. Carter, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8916 CrossRef CAS PubMed.
- X. Wen, J. M. P. Martirez and E. A. Carter, ACS Catal., 2024, 14, 9539 CrossRef CAS.
- X. Bian, Y. Zhao, C. Zhou and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202219340 CrossRef CAS.
- L. Mascaretti, A. Schirato, T. Montini, A. Alabastri, A. Naldoni and P. Fornasiero, Joule, 2022, 6, 1727 CrossRef.
- L. Wang, Y. Dong, T. Yan, Z. Hu, F. M. Ali, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS.
- X. Li, X. Zhang, H. O. Everitt and J. Liu, Nano Lett., 2019, 19, 1706 CrossRef CAS.
- P. Patsalas, N. Kalfagiannis, S. Kassavetis, G. Abadias, D. V. Bellas, C. Lekka and E. Lidorikis, Mater. Sci. Eng., 2018, 123, 1 CrossRef.
- M. Shi and X. Meng, Int. J. Hydrogen Energy, 2023, 48, 34659 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |