
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the covalency of versatile Pu(iii)-N bonds in a unique plutonium(iii) complex†
Zhuanling
Bai
 ,
Madeline C.
Martelles
,
Joseph M.
Sperling
* and
Thomas E.
Albrecht
*
,
Madeline C.
Martelles
,
Joseph M.
Sperling
* and
Thomas E.
Albrecht
*
Department of Chemistry, Nuclear Science & Engineering Center, Colorado School of Mines, Golden, Colorado 80401, USA. E-mail: jsperling@mines.edu; thomas.albrecht@mines.edu
First published on 6th May 2025
Abstract
A trivalent plutonium–pyrazinyl–tetrazolate complex Na2[Pu(Hdtp)(dtp)2(H2O)4]·9H2O (Pu_dtp, H2dtp = 2,3-di-1H-tetrazol-5-ylpyrazine) was synthesized through metathesis reaction of plutonium bromide and Na2(dtp)·2H2O in water. This structure is particularly notable among complexes formed by trivalent f-elements and the dtp2− ligand in aqueous media. In contrast to other trivalent f-elements, including all Ln3+ (with the exception of Pm3+) and Cm3+, preferentially coordinated with eight water molecules rather than the nitrogen donors of the dtp2− ligand, Pu3+ exhibits a distinct affinity for nitrogen coordination. This observation provides strong evidence that the 5f electrons in Pu3+ are more delocalized than other studied trivalent f-elements. In Pu_dtp, three distinct Pu(III)–N bonds are present: one Pu(III)–N5 from pyrazinyl, one Pu(III)–N4 from the least electronegative nitrogen in the tetrazolate, and three Pu(III)–N1/N2/N3 from the most electronegative nitrogens in the tetrazolate. Experimental Pu(III)–N bond lengths, Wiberg bond indices (WBI), natural localized molecular orbitals (NLMO), quantum theory of atoms in molecules (QTAIM), and energy decomposition analysis (EDA), reveal a covalency trend: Pu(III)–N from the most electronegative nitrogen in tetrazolate > Pu(III)–N from the least electronegative nitrogen in tetrazolate > Pu(III)–N from pyrazinyl. This trend arises from the increased negative charge on the most electronegative nitrogen atoms in the tetrazolate ring, enhancing electrostatic Pu–N1/N2/N3 interactions. These stronger electrostatic interactions lead to shorter bond lengths, thereby enhancing orbital overlap and greater covalency, compared to the less electronegative nitrogen in tetrazolate (Pu–N4) and the neutral pyrazinyl nitrogen (Pu–N5).
Introduction
Understanding metal–ligand interactions remains a critical frontier in f-element chemistry, providing key insights into covalency,1 electronic structure,2 and potential Ln/An and An/An separations.3 Unlike the predominantly ionic bonding observed in most lanthanides, actinides, particularly early actinides with high oxidation states, exhibit significant covalent character due to more accessible radial extension of their 5f and 6d orbitals.4 The extent of metal–ligand orbital mixing is proportional to their spatial overlap and inversely proportional to their energy difference.5 Since covalency is generally associated with orbital mixing, actinides display a dual mechanism for covalent bonding, driven both by energy degeneracy and orbital overlap. For example, computational studies on An3+:(DPA2−)n (An = Am, Cm, Bk, and Cf; n = 1–3) revealed that late actinides, with limited radial extension of their 5f orbitals, engage in covalent bonding primarily due to the energetic degeneracy between their 5f orbitals and ligand molecular orbitals, rather than spatial overlap.6 Furthermore, early actinides, where the 5f and 6d orbitals have similar energies, exhibit greater overlap between 6d orbitals and ligand orbitals than between 5f orbitals and ligand orbitals, as the 6d orbitals are more diffuse.5 A study on An–Cl covalency in AnCl62− (An4+ = Th, U, Np, Pu) demonstrated substantial mixing between Cl 3p orbitals and An4+ 5f and 6d orbitals, with the 6d orbitals forming stronger covalent bonds than the 5f orbitals.7A well-established trend in actinide-ligand bonding is the preference for softer ligands (e.g., N-, S-, or P-donors) to form more covalent bonds compared to harder ligands (e.g., F- and O-donors). This behaviour demonstrates that more itinerant 5f electrons in actinide ions exhibit greater affinity for softer donor atoms. For instance, Kaltsoyannis and coworkers found that in M[N(EPH2)2]3 (M = U and Pu) complexes, covalency in M–E bonds increases in the order S < Se < Te.8 Leveraging this preference for ligands with high polarizability has also been explored as a strategy for improving the separation of minor actinides from lanthanides in used nuclear fuel processing. In particular, heterocyclic N-donor ligands have been successfully employed to selectively extract actinides(III) over lanthanides(III), offering a promising route for enhancing nuclear waste management.9
While the impact of different donor ligands on covalency has been extensively studied, the influence of the local electronic and steric environment of a single ligand type on actinide-ligand covalency remains less explored. The 5f orbitals of plutonium(III) are sufficiently extended to participate in bonding. However, the covalency of Pu(III)–N bonds is poorly understood, because early actinide and lanthanide separations are relatively straightforward.10 In addition, experimental research on covalency of Pu3+ remains scarce, due to the combined challenges of requiring specialized facilities for safe handling of radiological materials, limited isotope availability,11,12 and the air-sensitive nature of Pu3+.13 To explore how the local electronic and steric environment of nitrogen-donor ligands influences covalent bonding with Pu3+, tetrazoles represent a promising class of ligands. These ligands offer unique advantages in coordination chemistry due to their versatile bonding modes and the ability to exist in both neutral and anionic forms, enabling fine-tuning of electronic properties and promoting structural diversity.14–16 Herein, 2,3-di-1H-tetrazolate-5-ylpyrazine (dtp2−) was chosen, with multiple nitrogen donor atoms, two from the pyrazine ring and eight from two tetrazolate rings. This provides a variety of nitrogen atoms with differing electron density for metal coordination.
Na2[Pu(Hdtp)(dtp)2(H2O)4]·9H2O (Pu_dtp) was synthesized from the reaction of Na2(dtp)·2H2O with Pu3+ salt in aqueous solution. Notably, Pu_dtp does not have a structural congener among other reported trivalent f-element dtp complexes.17,18 Unlike other trivalent f-elements, including lanthanides (excluding Pm3+) and Cm3+ that preferentially coordinate with water rather than nitrogen atoms of this ligand, Pu3+ exhibits a distinct preference for nitrogen coordination. This observation indicates that 5f electrons in Pu3+ are more delocalized than the other studied trivalent f-elements.
As shown in Fig. S1.1,† this ligand exhibits three distinct coordination modes, forming three distinct Pu(III)–N bonds: three Pu(III)–N1/N2/N3 from the most negative charged nitrogens in tetrazolate, one Pu(III)–N4 from the least negative charged nitrogen in tetrazolate, and one Pu(III)–N5 from pyrazinyl, embedded in a resonance-stabilized aromatic ring. By integrating experimental bond length data with computational metrics—Wiberg bond indices (WBIs), natural localized molecular orbitals (NLMOs) analysis, the quantum theory of atoms in molecules (QTAIM), and energy decomposition analysis (EDA), covalent contributions to these five Pu(III)–N bonds were compared.
Experimental
General caution! Plutonium isotopes, including 239Pu (approximately 96% 239Pu, t1/2 = 24110 years), and 240Pu (approximately 4%, t1/2 = 6561 years) pose significant health hazards due to their α-particle emissions and the radiotoxicity of their decay products, α-, β-, and γ-emitting daughters. To address these risks, all plutonium-related reactions were carried out in a radiation laboratory featuring a high-efficiency particulate air (HEPA)-filtered fume hood. Additionally, to further reduce the potential for inhalation and exposure, plutonium materials were handled only under solvent or immersion oil. During all characterization processes, radiation levels were continuously and meticulously monitored using multiple instruments to ensure full compliance with established safety protocols.H2dtp (2,3-di-1H-tetrazol-5-ylpyrazine) is a high-energy material. While this ligand is thermally stable,19,20 explosions can occur under specific conditions. Therefore, it is essential to implement appropriate safety measures during experiments involving this ligand.
Materials
2,3-Pyrazinedicarbonitrile (98%, Sigma-Aldrich), NaN3 (99%, Sigma-Aldrich), DI H2O, anhydrous ZnCl2 (98%, Sigma-Aldrich), HCl (37%, Sigma-Aldrich), HBr(aq) (48%, Sigma-Aldrich), diethyl ether (99%, Fisher Scientific), NH3(aq) (32%, Sigma-Aldrich) are all used as received. Synthesis of Na2(dtp)·2H2O was carried out following a literature procedure developed by Bu et al. with minor modifications.21 For detailed experimental conditions, please refer to our previous report.18 The plutonium used in this experiment was utilized as a PuClx solution (10 mg mL−1 of plutonium content in 1 M HCl).22,23Synthesis of Na2[Pu(Hdtp)(dtp)2(H2O)4]·9H2O (Pu_dtp)
A 0.2 mL 239Pu stock solution (containing 2 mg 239Pu, 0.0083 mmol) evaporated to dryness in a 20 mL scintillation vial. The resulting residue was dissolved in 1 mL of water and transferred to a centrifuge tube. Plutonium hydroxide was precipitated by adding excess NH4OH to the solution. The precipitate was centrifuged and washed three times with 1 mL of water to eliminate excess NH4Cl. Excess HBr was then added to the precipitate, forming an aqueous solution of PuBr3. This solution was gently evaporated to dryness under a heat lamp with a slow stream of nitrogen, yielding a hydrated PuBr3 residue.The hydrated 239PuBr3·nH2O solid was washed with diethyl ether (3 × 2 mL) to remove any embedded Br2, formed due to the oxidation of HBr. The PuBr3·nH2O solid was then dissolved in 0.1 mL of DI water in a shell vial. A solution containing 8 mg (∼0.03 mmol) Na2(dtp)·2H2O, dissolved in 1.5 mL DI water, was added to the 239PuBr3 solution. The mixture was allowed to stand at room temperature, leading to the formation of crystals suitable for single-crystal X-ray diffraction within a day. A small portion of the crystals was isolated for characterization using single-crystal X-ray diffraction and solid-state UV-vis-NIR spectroscopy.
Instrumentation
Solid-state UV-vis-NIR spectroscopy
Selected crystals were placed on a glass slide under Paratone-N oil and positioned under the light source of a microspectrophotometer. The software optimized the data collection time, and spectra were recorded from 320 to 1700 nm using a 75 W xenon lamp as the light source.Theory
All calculations were performed using the Amsterdam Density Functional (ADF) engine of the AMS 2024.106 software, utilizing the experimentally single crystal XRD determined crystal structures as a foundation. Wiberg bond indices (WBIs) and natural localized molecular orbitals (NLMOs) were carried out using NBO6.27 The electron density distribution of Pu_dtp was examined on the quantum theory of atoms in molecules (QTAIM).28 The numerical quality was set to “very good,” with the analysis level set as “Full”, source function and a grid spacing of 0.2 bohr. To compare the difference between those versatile Pu–N bonds, the energy of Pu_dtp was examined using energy decomposition analysis (EDA). We have calculated the EDA in terms of three reactions for which different fragments were defined to better capture the interaction of the different coordinating ligands (the details are shown in Section S8†):| Pu–N1/N5: [Pu(Hdtp)(dtp)(H2O)4] + dtp2− → [Pu(Hdtp)(dtp)2(H2O)4]2− |
| Pu–N2/N3: [Pu(Hdtp)(dtp)(H2O)4] + dtp2− → [Pu(Hdtp)(dtp)2(H2O)4]2− |
| Pu–N4: [Pu(dtp)2(H2O)4]− + Hdtp− → [Pu(Hdtp)(dtp)2(H2O)4]2− |
All calculations were generated employing the PBE0 functional,29 with no frozen-core approximation applied, paired with the TZ2P basis set.30 Scalar relativistic effects were incorporated through the zero-order regular approximation (ZORA).31 To ensure high precision, the computational settings were configured to be a “very good” accuracy level. More details are shown in ESI.†
Results and discussion
Structural characterization
The compound, Na2[Pu(Hdtp)(dtp)2(H2O)4]·9H2O (Pu_dtp) formed from the reaction of Na2(dtp)·2H2O and PuBr3 in aqueous solution. Single-crystal X-ray diffraction analysis reveals Pu_dtp crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2) consisting of one [Pu(Hdtp)(dtp)2(H2O)4]2− anion and two Na+ cations to neutralize the charge as well as nine noncoordinated water molecules to form a hydrogen framework in the asymmetric unit, shown in Sections S2 and S3.† In the [Pu(Hdtp)(dtp)2(H2O)4]2− anion (Fig. 1), the Pu3+ cation exhibits a coordination environment comprising four water molecules and 5 nitrogen atoms. The five nitrogen donors arise from two distinct bidentate moieties of the dtp2− dianion, a tetrazolate–tetrazolate (N^N, referring to a bidentate ligand with two nitrogen donor atoms) and a tetrazolate–pyrazinyl (N^N) as well as a monodentate Hdtp− anion. This arrangement results in a spherical capped square antiprismatic geometry with a deviation value of 0.928, from the analysis via SHAPE software.32,33 Additional structural information including coordination environment, hydrogen bonds, extended structures are shown in Sections S3 and S4.†
(#2) consisting of one [Pu(Hdtp)(dtp)2(H2O)4]2− anion and two Na+ cations to neutralize the charge as well as nine noncoordinated water molecules to form a hydrogen framework in the asymmetric unit, shown in Sections S2 and S3.† In the [Pu(Hdtp)(dtp)2(H2O)4]2− anion (Fig. 1), the Pu3+ cation exhibits a coordination environment comprising four water molecules and 5 nitrogen atoms. The five nitrogen donors arise from two distinct bidentate moieties of the dtp2− dianion, a tetrazolate–tetrazolate (N^N, referring to a bidentate ligand with two nitrogen donor atoms) and a tetrazolate–pyrazinyl (N^N) as well as a monodentate Hdtp− anion. This arrangement results in a spherical capped square antiprismatic geometry with a deviation value of 0.928, from the analysis via SHAPE software.32,33 Additional structural information including coordination environment, hydrogen bonds, extended structures are shown in Sections S3 and S4.†
 | ||
| Fig. 1 Thermal ellipsoid plots of Na2[Pu(Hdtp)(dtp)2(H2O)4]·9H2O, with probability 50% at 100 K. Non-coordinating H2O molecules and two Na+ cations are omitted for clarity. (Pu3+, blue; O, red; N, light blue; C, grey, H light gray). | ||
Bonding
Pu–H2O bonds ranging from 2.468(5) Å to 2.499(5) Å, is comparable with those observed in previously reported water-containing Pu3+ complexes.13,34–39 These three Pu–N1/N2/N3tetrazolate bonds involving the most electronegative nitrogen in the tetrazolate ring have bond lengths of 2.572(6) Å, 2.586(6) Å, and 2.597(6) Å, respectively. Those bonds are much shorter than Pu–N4tetrazolate (2.673(6) Å) from the least electronegative nitrogen in tetrazolate and Pu–N5 (2.757(6) Å) from pyrazinyl. They fall within the range of reported Pu–N bonds in recently reported Pu–tetrazolate complexes of [Pu(pmtz)3(H2O)3]·8H2O and [(Pu(pmtz)2(H2O)3)2(μ-pmtz)]2(pmtz)2·14H2O (pmtz− = 5-(pyrimidyl)tetrazolate).40 Pu–N4 is 0.16 Å shorter than that in [Pu(Hdtb)(H2O)8](dtb)·11H2O (dtb2− = 1,3-di(tetrazolate-5-yl)benzene) with IXPu(III)–N, where IX refers to the coordination number, (2.8338(15) Å) from least electronegative nitrogen in tetrazolate, likely due to less steric hindrance.41To further investigate the bonding of [Pu(Hdtp)(dtp)2(H2O)4]2−, Wiberg bond indices (WBI) and natural localized molecular orbitals (NLMOs) approximation were conducted, since it offers a more realistic picture of the chemical bonding, as reported in several studies.39,42–44 The Pu contribution to the NLMOs was decomposed into atomic orbital character, with detailed results provided in Section S8,†Fig. 2 and 3. The WBI analysis indicates that the Pu–N bonds, with average value of 0.2820 are stronger than Pu–H2O, with average values of 0.2350. In general, the total Pu contribution for the one σ and one weak π bonds to NLMOs of all four Pu–H2O interactions is less than 5.82% and average is 5.53%, that is observably less than plutonium contribution to NLMOs of five Pu–N interactions. This observation further supports the general trend that ligands with increased polarizability exhibit greater orbital overlap and stronger covalent interactions with actinide ions, consistent with previous studies.45,46
 | ||
| Fig. 2 Selected α and β spin natural localized molecular orbitals (NLMOs) and metal contributions to the NLMOs along with their hybrid contributions showing the Pu–N bonding [Pu(Hdtp)(dtp)2(H2O)4]2−. | ||
 | ||
| Fig. 3 Average (α + β spin) plutonium and specific orbitals (6d, 5f and 7s) contributions to the Pu–N NLMOs vs. respective bond lengths. | ||
A comparative analysis of these five Pu–N bonds reveals differences in their bonding characteristics. The WBI bond order analysis indicates that the Pu–N1/N2/N3 bonds are stronger than Pu–N4 bond (ΔWBI > 0.04) and are stronger than Pu–N5 bond (ΔWBI > 0.07), shown in Table 1. As illustrated in Fig. 3, further evaluation of the covalency in the Pu–N bonds using NLMO analysis, is consistent with the WBI findings. The results indicate that the contribution of plutonium to the bonding orbitals is inversely correlated with the Pu–N bond lengths. The average α + β spin contribution of Pu to the NLMOs is approximately 8% for Pu–N1/N2/N3 bonds, higher than the 6.46% for Pu–N4 and decreasing to 5.90% for Pu–N5. A breakdown of this metal contribution into 6d, 5f, and 7s components, with a negligible 6p contribution, reveals that all these NLMOs exhibit stronger 6d involvement than 5f due to the greater diffusivity of 6d orbitals. The 7s contribution remains relatively consistent across the five Pu–N bonds, while 6d and 5f contributions show a slight decrease from Pu–N1/N2/N3 to Pu–N4/5. These findings demonstrate a distinct trend in covalency: Pu–N1/N2/N3 from the most negative charged nitrogen in tetrazolate > Pu–N4 from the least negative charged nitrogen in tetrazolate > Pu–N5 from neutral pyrazinyl. Here, a hypothesis is proposed that this observed decrease in covalency correlates with the electronegativity of the respective nitrogen atoms, highlighting the influence of electronic charge distribution on the strength of covalent interactions.
| Bond | WBI |
|---|---|
| M–N1 | 0.3236 |
| M–N2 | 0.3095 |
| M–N3 | 0.2979 |
| M–N4 | 0.2518 |
| M–N5 | 0.2273 |
To further elucidate trends in covalency and investigate the underlying factors contributing to these trends, QTAIM analysis was employed including several metrics.47 The electron density at the bond critical point (BCP), ρ(r), serves as an indicator of electron accumulation, with higher values signifying stronger bonding interactions. The potential energy density, V(r), is associated with attractive interactions, where more negative values reflect stabilization of electron density concentration. An excess of kinetic energy density, G(r), suggests pronounced closed-shell characteristics, while a more negative total energy density, H(r), is indicative of stronger covalent bonding. The bond strength parameter, H(r)/ρ(r), provides further insight into covalency, with more negative values corresponding to increased covalent character. The delocalization index, δ(r), quantifies electron delocalization between atomic basins, with higher values implying stronger bonding and enhanced electron sharing. The localization index, λ(M), represents the electron population assigned to an atom M based on QTAIM partitioning. It is used calculating the oxidation state, OS(M) through the equation OS(M) = Z(M) – λ(M), where Z(M) is the nuclear charge of atom M. A greater degree of covalent character can result in deviations from classical oxidation state values.48Table 2 summarizes the main QTAIM metrics for Pu–H2Oaverage and five Pu–N interactions, while detailed metrics for individual Pu–H2O bond are provided in Section S5.†
| Pu–Oavg | Pu–N1 | Pu–N2 | Pu–N3 | Pu–N4 | Pu–N5 | |
|---|---|---|---|---|---|---|
| Distance/Å | 2.484(5) | 2.572(6) | 2.586(6) | 2.597(6) | 2.673(6) | 2.757(6) |
| ρ(r) | 0.3276 | 0.3354 | 0.3273 | 0.3079 | 0.2584 | 0.2351 |
| V(r) | −964.60 | −905.97 | −864.64 | −804.37 | −639.09 | −529.51 |
| G(r) | 942.50 | 786.12 | 744.42 | 719.26 | 615.19 | 491.56 |
| H(r) | −22.10 | −119.85 | −120.21 | −85.11 | −23.91 | −37.94 |
| H(r)/ρ(r) | −67.18 | −357.28 | −367.25 | −276.38 | −92.61 | −161.36 |
| δ(r) | 0.2638 | 0.3053 | 0.2987 | 0.2852 | 0.2268 | 0.2217 |
| λ(Pu) | 90.4908 | |||||
| OS(Pu) | 3.5 |
QTAIM analysis demonstrates that Pu–N bonds exhibit a greater degree of covalency compared to Pu–H2O bonds. Being consistent with NLMO results, among five Pu–N bonds, Pu–N1/N2/N3, associated with the most electronegative nitrogen sites in tetrazolate, exhibit the stronger covalent character, as evidenced by their higher electron density ρ(r), more negative total energy density H(r), more negative bond strength parameter H(r)/ρ(r), and greater delocalization indices δ(r), compared with those of Pu–N4/N5. In addition, QTAIM analysis also reveals that Pu–N4 is slightly stronger than Pu–N5 due to slightly higher electron density (ρ(r) = 0.2584 vs. 0.2351 e Å−3) and slightly higher degree of electron delocalization (δ(r) = 0.2268 vs. 0.2217). However, Pu–N4 possess less negative total energy density H(r) (H(r) = −23.91 vs. −37.94 kJ mol−1 Å−3) and less negative bond strength parameter (H(r)/ρ(r) = −92.61 vs. −161.36 kJ mol−1 e−1). The observed deviation in the total energy density for Pu–N4 and N5 can be rationalized by the chelate effect, that enhances the affinity of a chelating ligand Pu–N5 for a metal ion compared to its monodentate counterparts Pu–N4.
The observation of both potential energy density, V(r) and kinetic energy density G(r) of Pu–N1/N2/N3 are higher than that of Pu–N4/N5 and reveals that the negative charge from the tetrazolate ring enhances the electrostatic interaction of the ligand and Pu3+, that makes more Pu3+ preference to the ligand (shorter Pu–N bond length), that in turn lead to more orbital overlap and more covalency. Even though N4 is less negatively charged than N1/N2/N3 due to a resonance and charge distribution of tetrazolate, electronegativity of N4 from the delocalized change tetrazolate is more preferred for Pu3+ than neutral N5. Therefore, there are more potential energy density (V(r) = −639.09 vs. −529.51 kJ mol−1 Å−3) and kinetic energy density (G(r) = 615.19 vs. 491.56 kJ mol−1 Å−3) for N4 than that of neutral N5 with Pu3+. This corresponds to shorter bond length of Pu–N4 (2.673 Å vs. 2.757 Å for Pu–N5) further supports that Pu–N4, exhibits a greater degree of covalency than Pu–N5. This further affirms the covalency trend from NLMO data and experimental bond lengths.
In addition, the integrated oxidation state for Pu3+ is notably elevated to 3.5, that may be attributed to Pu3+/4+ redox potentials (−1.04 V, 1 M HClO4, vs. SHE),49 leading to electron density donation from Pu(III) to the local environment. This increase in the integrated oxidation state is likely driven more by redox behaviour than by Pu–L bonding characteristics.
To further validate the hypothesis, a more comprehensive comparison of the electrostatic and orbital energy contributions between the Pu–N1/N2/N3tetrazolate and Pu–N5pyridinyl interactions was conducted using energy decomposition analysis (EDA), shown in Table 3.50 This method divides the interaction energy (ΔEint) of chemical bonds between two or more molecular fragments in a fixed geometry into four main components, as expressed in the following equation:51
| ΔEint = ΔEelast + ΔEPauli + ΔEorb + ΔEdisp |
| kJ mol−1 | Pu–N1/N5 | Pu–N2/N3 | Pu–N4 |
|---|---|---|---|
| Pauli repulsion | 365.13 | 2260.17 | 191.53 |
| Electrostatic | −506.76 | −1067.58 | −179.42 |
| Orbital | −137.94 | −2406.8 | 22.99 |
| Solvation (el) | −1018.67 | −1017.36 | −1018.66 |
| Total | −1283.81 | −2217.15 | −969.14 |
Electrostatic interaction (ΔEelestat) represents the classical Coulomb attraction between the charge distributions of the interacting fragments. This term is dominant in ionic bonds and describes stabilization due to electrostatic forces. Pauli repulsion (ΔEPauli) accounts for the repulsive interactions caused by the overlap of occupied orbitals, due to the Pauli exclusion principle, that comprises the destabilizing interactions between electrons of the same spin on either fragment. Orbital interaction (ΔEorb) describes the mixing of molecular orbitals, including covalent bonding effects, charge transfer, and polarization. This term plays a crucial role in covalent bonding and stabilizes the system beyond electrostatics. Dispersion energy (ΔEdisp) represents van der Waals interactions due to instantaneous dipole–induced dipole attractions, particularly significant for non-covalent interactions.
Comparing the EDA results for Pu–N1/N5 and Pu–N2/N3 reveals that the longer Pu–Npyrazinyl bond length than Pu–Ntetrazolate results in a significant decrease in ΔEPauli. However, the negative charge of tetrazolate provides substantially greater electrostatic stabilization, particularly in the Pu–N2/N3 bonds. Additionally, the Pu–Ntetrazolate interactions exhibit stronger orbital contributions to the total bonding interactions than Pu–Npyrazinyl. Therefore, in Pu_dtp, compared to the Pu–Npyrazinyl interaction, the Pu–Ntetrazolate interaction is predominantly stabilized by stronger electrostatic interactions along with significant orbital contributions.
All these computational data on these three distinct Pu(III)–N bonds reveal a covalency strength trend: Pu(III)–Ntetrazolate from the most electronegative nitrogen in tetrazolate > Pu(III)–Ntetrazolate from the least electronegative nitrogen in tetrazolate > Pu(III)–Npyrazinyl from pyrazinyl. This trend arises from the increased negative charge on the tetrazolate ring, enhancing their electrostatic interactions and shortening the bond lengths with Pu3+. This, in turn, promotes better orbital overlap and results in greater covalency compared to the neutral pyrazinyl nitrogen.
Solid-state UV-vis-NIR spectrum
The characteristic Laporte-forbidden 5f → 5f transition peaks observed in Pu_dtp are consistent with those reported for PuCl3 thin films by Carnall and co-workers, and the following discussion is based on the spectral assignments for that compound.52 As illustrated in Fig. 4, the spectral profile exhibits three distinct electronic transition groups (L, M, and K), as diagnostic markers for the Pu3+ compounds: group L and M (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 822–16828 cm−1), namely these 5f → 5f transition peaks as 6H5/2 → 4L13/2, 4K11/2, 4I9/2, 4P5/2, and group K (16828–15765 cm−1), as 6H5/2 → 4M15/2. These 5f → 5f transition peaks align with prior observations in Pu3+ compounds, such as Pu2[C6(CO2)6](H2O)9·H2O, Pu2[C6(CO2)6](H2O)8·2H2O,37 and [(H3O)(18-crown-6)][Pu(H2O)4(18-crown-6)](ClO4)4·2H2O.39 Additionally, other high-intensity 5f → 5f transitions characteristic of Pu3+ are evident, including group H (15424–14637 cm−1, 6H5/2 → 6F11/2), group F (13323–11714 cm−1, 6H5/2 →6H15/2, 6F9/2), group E (11655–10520 cm−1, 6H5/2 → 6H13/2), group D (10127–9541 cm−1, 6H5/2 → 6F7/2), group C (9422–8397 cm−1, 6H5/2 → 6H11/2), and group B (7400–6000 cm−1, 6H5/2 → 6F3/2, 6H5/2, 6H9/2). Furthermore, as shown in Fig. S5.1,† compared with the solid-state absorption spectrum of [Pu(H2O)9]3+,38 all these 5f → 5f transition peaks exhibit red shifts due to more covalency of Pu–N bonds than Pu–H2O bonds. In addition, the broad band from 32000 cm−1 to 20000 cm−1, corresponding to the metal to ligand charge transfer due to the accessible Pu3+/4+, (−1.04 V, 1 M HClO4, vs. SHE),49 has also been observed in other Pu3+ complexes, such as Pu2[C6(CO2)6](H2O)9·H2O, Pu2[C6(CO2)6](H2O)8·2H2O,37 [Pu(pmtz)3(H2O)3]·8H2O and [(Pu(pmtz)2(H2O)3)2(μ-pmtz)]2(pmtz)2·14H2O.40
822–16828 cm−1), namely these 5f → 5f transition peaks as 6H5/2 → 4L13/2, 4K11/2, 4I9/2, 4P5/2, and group K (16828–15765 cm−1), as 6H5/2 → 4M15/2. These 5f → 5f transition peaks align with prior observations in Pu3+ compounds, such as Pu2[C6(CO2)6](H2O)9·H2O, Pu2[C6(CO2)6](H2O)8·2H2O,37 and [(H3O)(18-crown-6)][Pu(H2O)4(18-crown-6)](ClO4)4·2H2O.39 Additionally, other high-intensity 5f → 5f transitions characteristic of Pu3+ are evident, including group H (15424–14637 cm−1, 6H5/2 → 6F11/2), group F (13323–11714 cm−1, 6H5/2 →6H15/2, 6F9/2), group E (11655–10520 cm−1, 6H5/2 → 6H13/2), group D (10127–9541 cm−1, 6H5/2 → 6F7/2), group C (9422–8397 cm−1, 6H5/2 → 6H11/2), and group B (7400–6000 cm−1, 6H5/2 → 6F3/2, 6H5/2, 6H9/2). Furthermore, as shown in Fig. S5.1,† compared with the solid-state absorption spectrum of [Pu(H2O)9]3+,38 all these 5f → 5f transition peaks exhibit red shifts due to more covalency of Pu–N bonds than Pu–H2O bonds. In addition, the broad band from 32000 cm−1 to 20000 cm−1, corresponding to the metal to ligand charge transfer due to the accessible Pu3+/4+, (−1.04 V, 1 M HClO4, vs. SHE),49 has also been observed in other Pu3+ complexes, such as Pu2[C6(CO2)6](H2O)9·H2O, Pu2[C6(CO2)6](H2O)8·2H2O,37 [Pu(pmtz)3(H2O)3]·8H2O and [(Pu(pmtz)2(H2O)3)2(μ-pmtz)]2(pmtz)2·14H2O.40
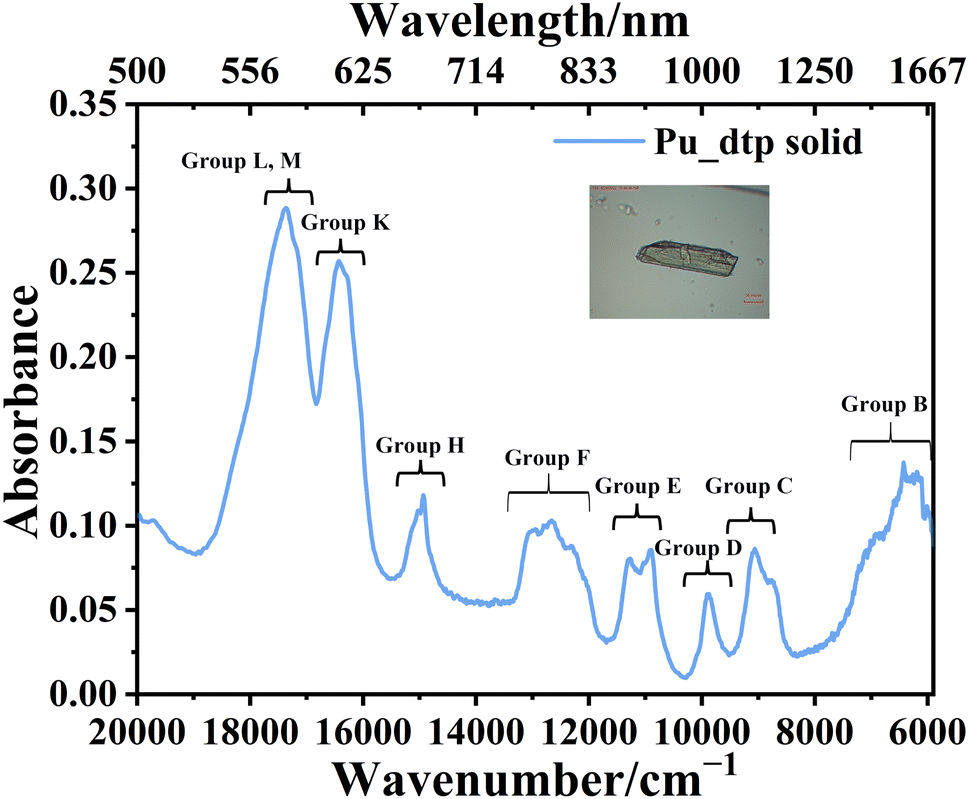 | ||
| Fig. 4 Experimental solid-state UV-vis-NIR absorption spectrum of Pu_dtp. | ||
Conclusions
In summary, [Pu(Hdtp)(dtp)2(H2O)4]2− has been synthesized and characterized by X-ray single crystal diffraction and solid-state UV-vis-NIR absorption spectroscopy. Analysis of experimental bond lengths, WBIs, NLMOs and QTAIM reveals that the negative charge on the most electronegative nitrogen atom within the tetrazolate ring enhances electrostatic interactions (Pu–N1, N2, and N3). These stronger electrostatic interactions result in shorter bond lengths that in turn enhance orbital overlap and increase covalency, compared to the interactions involving the less electronegative nitrogen atom of the tetrazolate ring (Pu–N4) and the neutral pyrazinyl nitrogen (Pu–N5). This observed trend highlights a direct correlation between the electronegativity of the coordinating nitrogen atoms and the degree of Pu–N covalency, demonstrating that the strength of Pu(III)–N bonding is influenced by the electronic properties of the nitrogen donors. These findings underscore the complexity of Pu–N bonding in Pu3+ complexes with nitrogen-containing ligands of varying electronic structures, providing valuable insights for the rational design of ligands to further investigate covalency and separation processes in actinide chemistry.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for Pu_dtp has been deposited at the CCDC under 2421228.Author contributions
Zhuanling Bai and Joseph M. Sperling carried out the synthesis and characterization of Pu_dtp. Zhuanling Bai and Madeline C. Martelles performed the computational studies. Zhuanling Bai, Joseph M. Sperling, and Thomas E. Albrecht jointly conceived and organized the project. Zhuanling Bai wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Elements Chemistry Program, under the award number DE-SC0023693.Notes and references
- L. I. Katzin, Nature, 1950, 166, 605 CrossRef.
- S. K. Cary, M. Vasiliu, R. E. Baumbach, J. T. Stritzinger, T. D. Green, K. Diefenbach, J. N. Cross, K. L. Knappenberger, G. Liu, M. A. Silver, A. E. DePrince, M. J. Polinski, S. M. Van Cleve, J. H. House, N. Kikugawa, A. Gallagher, A. A. Arico, D. A. Dixon and T. E. Albrecht-Schmitt, Nat. Commun., 2015, 6, 6827 CrossRef CAS.
- K. L. Nash, C. Madic, J. N. Mathur and J. Lacquement, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer Netherlands, Dordrecht, 2006, pp. 2622–2798 Search PubMed.
- G. Schreckenbach, P. J. Hay and R. L. Martin, J. Comput. Chem., 1999, 20, 70–90 CrossRef CAS.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- M. P. Kelley, J. Su, M. Urban, M. Luckey, E. R. Batista, P. Yang and J. C. Shafer, J. Am. Chem. Soc., 2017, 139, 9901–9908 CrossRef CAS PubMed.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS.
- K. I. M. Ingram, M. J. Tassell, A. J. Gaunt and N. Kaltsoyannis, Inorg. Chem., 2008, 47, 7824–7833 CrossRef CAS PubMed.
- Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS.
- A. Kovács, C. Apostolidis and O. Walter, Inorganics, 2019, 7, 26 CrossRef.
- J. Su, T. Cheisson, A. McSkimming, C. A. P. Goodwin, I. M. DiMucci, T. Albrecht-Schönzart, B. L. Scott, E. R. Batista, A. J. Gaunt, S. A. Kozimor, P. Yang and E. J. Schelter, Chem. Sci., 2021, 12, 13343–13359 RSC.
- N. K. Gupta, J. Mol. Liq., 2018, 269, 72–91 CrossRef CAS.
- S. Wang, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Inorg. Chem., 2011, 50, 2079–2081 CrossRef CAS PubMed.
- F. R. Benson, Chem. Rev., 1947, 41, 1–61 CrossRef CAS.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS.
- P. Pachfule, R. Das, P. Poddar and R. Banerjee, Cryst. Growth Des., 2010, 10, 2475–2478 CrossRef CAS.
- Z. Bai, M. Redington, S. Haldar, N. B. Beck, J. M. Sperling, B. Scheibe, J. P. Brannon, E. Zurek, L. Gagliardi and T. E. Albrecht, J. Am. Chem. Soc., 2025, 147, 6137–6148 CrossRef CAS.
- Z. Bai, B. Scheibe, J. M. Sperling and T. E. Albrecht-Schönzart, Inorg. Chem., 2022, 61, 19193–19202 CrossRef CAS PubMed.
- D.-P. Chi, L. Yang, J.-M. Han, G.-Y. Zhang, J.-C. Liu, Z.-Z. Yan, J. Qin and W.-C. Tong, Main Group Chem., 2020, 19, 105–116 CrossRef CAS.
- C. Qiao, L. Lü, W. Xu, Z. Xia, C. Zhou, S. Chen and S. Gao, Acta Phys.-Chim. Sin., 2020, 36, 190508 Search PubMed.
- J.-R. Li, Y. Tao, Q. Yu, X.-H. Bu and S. Kitagawa, Chem.–Eur. J., 2008, 14, 2271–2776 Search PubMed.
- CrysAlisPro, Version 1.171.41.115a, Rigaku Oxford Diffraction, Oxford, UK, 2021 Search PubMed.
- APEX4, Version 2021.4-0, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. I. Rodríguez, R. F. W. Bader, P. W. Ayers, C. Michel, A. W. Götz and C. Bo, Chem. Phys. Lett., 2009, 472, 149–152 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS.
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Comput. Chem., 1994, 101, 9783–9792 CAS.
- D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem.–Eur. J., 2005, 11, 1479–1494 CrossRef CAS PubMed.
- A. Ruiz-Martínez, D. Casanova and S. Alvarez, Chem.–Eur. J., 2008, 1291–1303 CrossRef.
- S. K. Cary, S. S. Galley, M. L. Marsh, D. L. Hobart, R. E. Baumbach, J. N. Cross, J. T. Stritzinger, M. J. Polinski, L. Maron and T. E. Albrecht-Schmitt, Nat. Chem., 2017, 9, 856–861 CrossRef CAS.
- R. G. Surbella, L. C. Ducati, K. L. Pellegrini, B. K. McNamara, J. Autschbach, J. M. Schwantes and C. L. Cahill, Chem. Commun., 2017, 53, 10816–10819 RSC.
- W. Runde, L. F. Brodnax, G. Goff, A. C. Bean and B. L. Scott, Inorg. Chem., 2009, 48, 5967–5972 CrossRef CAS PubMed.
- J. M. Sperling, A. N. Gaiser, C. J. Windorff, B. E. Klamm, M. A. Whitefoot, A. T. Chemey, B. N. Long, J. G. Campbell and T. E. Albrecht-Schmitt, Inorg. Chem., 2020, 59, 3085–3090 CrossRef CAS.
- J. H. Matonic, B. L. Scott and M. P. Neu, Inorg. Chem., 2001, 40, 2638–2639 CrossRef CAS.
- K. Li, S. Hu, Q. Zou, Y. Zhang, H. Zhang, Y. Zhao, T. Zhou, Z. Chai and Y. Wang, Inorg. Chem., 2021, 60, 8984–8989 CrossRef CAS.
- Z. Bai, B. Scheibe, J. M. Sperling, N. B. Beck, J. P. Brannon, D. Gomez Martinez, B. M. Rotermund and T. E. Albrecht-Schönzart, Inorg. Chem., 2024, 63, 1266–1273 CrossRef CAS PubMed.
- Z. Bai, J. M. Sperling and T. E. Albrecht, Inorg. Chem., 2025, 64(16), 8164–8173 CrossRef CAS.
- Z. K. Huffman, J. M. Sperling, C. J. Windorff, B. N. Long, L. Cordova, H. Ramanantoanina, C. Celis-Barros and T. E. Albrecht-Schönzart, Chem. Commun., 2022, 58, 11791–11794 RSC.
- B. N. Long, M. J. Beltrán-Leiva, C. Celis-Barros, J. M. Sperling, T. N. Poe, R. E. Baumbach, C. J. Windorff and T. E. Albrecht-Schönzart, Nat. Commun., 2022, 13, 201 CrossRef CAS.
- J. M. Sperling, E. J. Warzecha, C. Celis-Barros, D.-C. Sergentu, X. Wang, B. E. Klamm, C. J. Windorff, A. N. Gaiser, F. D. White, D. A. Beery, A. T. Chemey, M. A. Whitefoot, B. N. Long, K. Hanson, P. Kögerler, M. Speldrich, E. Zurek, J. Autschbach and T. E. Albrecht-Schönzart, Nature, 2020, 583, 396–399 CrossRef CAS PubMed.
- Y.-M. Chen, C.-Z. Wang, L. Zhang, Q.-Y. Wu, J.-H. Lan, Z.-F. Chai and W.-Q. Shi, Dalton Trans., 2024, 53, 7406–7413 RSC.
- C. A. P. Goodwin, A. W. Schlimgen, T. E. Albrecht-Schönzart, E. R. Batista, A. J. Gaunt, M. T. Janicke, S. A. Kozimor, B. L. Scott, L. M. Stevens, F. D. White and P. Yang, Angew. Chem., Int. Ed., 2021, 60, 9459–9466 CrossRef CAS PubMed.
- Q.-R. Huang, J. R. Kingham and N. Kaltsoyannis, Dalton Trans., 2015, 44, 2554–2566 RSC.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, Chem. Phys., 2002, 117, 5529–5542 CAS.
- S. W. Rabideau and J. F. Lemons, J. Am. Chem. Soc., 1951, 73, 2895–2899 CrossRef CAS.
- D. Rodrigues Silva, L. De Azevedo Santos, M. P. Freitas, C. F. Guerra and T. A. Hamlin, Chem.–Asian J., 2020, 15, 4043–4054 CrossRef CAS.
- P. Jerabek, P. Schwerdtfeger and G. Frenking, J. Comput. Chem., 2019, 40, 247–264 CrossRef CAS PubMed.
- W. T. Carnall, P. R. Fields and R. G. Pappalardo, J. Chem. Phys., 1970, 53, 2922–2938 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Coordination modes of 2,3-di-1H-tetrazolate-5-ylpyrazine (dtp2−), images of reaction and crystals, additional crystal structures, crystallographic data, spectra of Na2[Pu(Hdtp)(dtp)2(H2O)4]·9H2O and [Pu(H2O)9]3+, additional theory details, including WBI, QTAIM, NLMO, and EDA. CCDC 2421228. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01808k |
| This journal is © The Royal Society of Chemistry 2025 |