
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybridized local and charge-transfer materials using pyrene as π-bridge towards negligible efficiency roll-off in non-doped OLED†
Yuqin Dua,
Yali Penga,
Wenjian Donga,
Huixia Xu *a,
Xinyu Lia,
Kang Chena,
Feng Wanga,
Yanqin Miao*ab,
Peng Tao*c,
Hua Wanga and
Junsheng Yud
*a,
Xinyu Lia,
Kang Chena,
Feng Wanga,
Yanqin Miao*ab,
Peng Tao*c,
Hua Wanga and
Junsheng Yud
aKey Laboratory of Interface Science and Engineering in Advanced Materials, Ministry of Education, Taiyuan University of Technology, Taiyuan 030024, China. E-mail: xuhuixia@tyut.edu.cn; miaoyanqin@tyut.edu.cn
bCollege of Physics and Optoelectronics, Taiyuan University of Technology, Taiyuan, 030024, China
cDepartment of Applied Biology and Chemical Technology, Research Institute for Smart Energy, The Hong Kong Polytechnic University, Hong Kong 999077, P. R. China. E-mail: pengtao@polyu.edu.hk
dState Key Laboratory of Electronic Thin Films and Integrated Devices, School of Optoelectronic Science and Engineering, University of Electronic Science and Technology of China (UESTC), Chengdu, 610054, China
First published on 20th May 2025
Abstract
The development of organic light-emitting diodes (OLED) with low efficiency roll-off at high brightness still remains a significant challenge. The hot-exciton mechanism provides a potential solution owing to its unique high-lying reverse intersystem crossing (hRISC) with a sufficiently large rate constant. Herein, two pyrene-based hot-exciton molecules, namely TPAPyCP and 2mTPAPyCP, were successfully designed using triphenylamine (TPA) as donor and benzonitrile as acceptor. The planar pyrene provided multiple fast hRISC channels, which effectively improved the photoluminescence quantum yield (PLQY) values to 73.2% and 65.8% in neat films. A non-doped device based on TPAPyCP demonstrated a maximum external quantum efficiency (EQE) of 9.41% and still maintained 8.95% at a high luminance of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2, highlighting a negligible efficiency roll-off of 4.97%.
000 cd m−2, highlighting a negligible efficiency roll-off of 4.97%.
Introduction
Organic light-emitting diodes (OLEDs) have attracted significant interests in the fields of display and lighting due to their potential advantages, such as flexibility, high contrast, and self-illumination.1–3 The development of efficient OLEDs relies strongly on the characteristics of the emitting materials, whose emission mechanisms mainly include traditional fluorescence,4 phosphorescence,5–8 thermally activated delayed fluorescence (TADF),9–11 and hot-excitons.12,13 Traditional fluorescent materials capture only 25% of singlet excitons, resulting in a upper limitation external quantum efficiency (EQE) of 5%. Although phosphorescent and TADF materials have shown satisfactory efficiency, such as over 30% EQE, they are confined by serious efficiency roll-off at high brightness because of the long-lived triplet excitons, which are significantly constraining the progress of commercial OLEDs. Therefore, it is crucial to develop high-performance and stable emitters.Hot-exciton materials, first reported by Ma Y. and coworkers, possess a hybridized local and charge-transfer (HLCT) excited state. These materials can simultaneously harness the non-radiative triplet excitons, thereby realizing 100% internal quantum efficiency (IQE).14 Furthermore, as shown in Scheme 1a, their triplet excitons are utilized via the high-lying triplet reverse intersystem crossing (hRISC) process to the singlet state (Tn → Sm, n ≥ 2, m ≥ 1), which can be completed in a few to tens of nanoseconds.15–19 The rate constant of hRISC (khRISC, 107–108 s−1) is much larger than that of RISC from T1 to S1 for TADF (104–105 s−1) emitters.20,21 Moreover, the fast radiative transition from S1 to S0 helps overcome triplet excitons accumulation. Another notable feature of hot-exciton emitters is their unique HLCT excited-state nature. The charge-transfer (CT) excited state effectively reduces the energy level difference between singlet and high-lying triplet states, thus enhancing triplet exciton utilization. The local-excited (LE) state with overlapped frontier molecular orbitals (FMOs) facilitates a fast radiative decay rate and high photoluminescence quantum yield (PLQY).22,23
 | ||
| Scheme 1 (a) Diagram of the reverse intersystem crossing channel (IC: internal conversion, Tn: high-lying triplet state, n ≥ 2; Sm: high-lying singlet state, m ≥ 1; F: fluorescence; DF: delayed fluorescence from Tn → Sm → S0; ISC: intersystem crossing); (b) the molecular structures and properties of hot-exciton molecules using anthracene and pyrene as π-bridge. | ||
In recent years, the majority of studies on hot-exciton materials have focused on donor (D)–acceptor (A) or D–π–A configurations.24–26 To improve charge-carrier mobility, the anthracene group with an alignment energy levels is suitable for hRISC.27 However, the large twisted structure between anthracene and adjacent groups usually causes a low PLQY in the aggregate state and a single hRISC channel.28,29 As shown in Scheme 1b, we previously reported two hot-exciton materials with an anthracene π-bridge, which showed a PLQY of 46.6% in neat films and a maximum EQE of 5.9% in the non-doped device.30
Considering the above dilemma, we proposed a molecular design strategy using pyrene as the π-bridge,31 which can suppress the twisted configurations, as shown in Scheme 1b. The remarkable advantage of pyrene for designing hot-exciton molecules lies in the minimal energy differences between several high-lying triplet states and Sm, as well as the relatively large energy gap between T1 and T2, which are favourable to intersystem crossing via multiple hRISC pathways and restrict the T2 → T1 internal conversion (IC) process.32 Additionally, the incorporation of an electron-donating triphenylamine (TPA) group enables precise modulation of π–π stacking and LE excited states.33,34 Whereas, the CT state and luminescence color can be adjusted by the introduction of an electron-withdrawing benzonitrile group. Therefore, TPAPyCP and 2mTPAPyCP (in Scheme 1b) exhibited moderately twisted structures and a PLQY of over 70%. Impressively, the maximum EQE values of 9.41% and 9.28% were achieved in non-doped OLEDs, and a device based on TPAPyCP still maintained 8.92% at a luminance of 10000 cd m−2, exhibiting a very low efficiency roll-off.
Results and discussion
The chemical molecular structures and the detailed synthesis processes of TPAPyCP and 2mTPAPyCP are depicted in Scheme S1.† Two molecules were synthesized by Suzuki–Miyaura coupling reactions. Their chemical structures were well confirmed by 1H NMR, 13C NMR, high-resolution mass spectroscopy, and single-crystal analysis. The detailed data and NMR spectra are listed in ESI (Fig. S1–S5).† As shown in Fig. S6a,† TPAPyCP and 2mTPAPyCP exhibited high thermal stability, with decomposition temperatures (Td, corresponding to 5% weight loss) of 460 °C and 451 °C, respectively. Additionally, high glass transition temperatures (Tg) of 109 °C and 114 °C (Fig. S6b†) were observed by studying the thermal gravimetric (TGA) and differential scanning calorimetry (DSC) analyses. In addition, their cyclic voltammetry (CV) testing curves were measured (Fig. S7†), indicating that the onset oxidation (EOXc) of TPAPyCP and 2mTPAPyCP were 0.97 and 0.83 V, respectively, corresponding to the highest occupied molecular orbital (HOMO) energy levels of −5.32 and −5.22 eV, according to the equations in ESI.†Single crystals of 2mTPAPyCP (CCDC: 2409186) were obtained from a mixed solution of dichloromethane (DCM)/methanol with a volume ratio of 1:2. Single-crystal data are summarized in Table S1† and the crystal structure is illustrated in Fig. 1. The X-ray single-crystal diffraction analysis reveals that 2mTPAPyCP adopts a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The torsion angles between pyrene and adjacent benzene rings were 44.6 and 53.6°, respectively, which were much smaller than those of hot-exciton molecules based on anthracene.30 A head-to-tail π–π stacking packing arrangement was observed between adjacent molecules with a distance of 3.395 Å and a slip angle of 46.3°, exhibiting a J-aggregation pattern. C–H⋯π interactions (2.316–2.844 Å) were found in the single-crystal packing. These arrangements ensured a rigid molecular structure to enhance PLQY and prevent concentration quenching in electroluminescence performance.
space group. The torsion angles between pyrene and adjacent benzene rings were 44.6 and 53.6°, respectively, which were much smaller than those of hot-exciton molecules based on anthracene.30 A head-to-tail π–π stacking packing arrangement was observed between adjacent molecules with a distance of 3.395 Å and a slip angle of 46.3°, exhibiting a J-aggregation pattern. C–H⋯π interactions (2.316–2.844 Å) were found in the single-crystal packing. These arrangements ensured a rigid molecular structure to enhance PLQY and prevent concentration quenching in electroluminescence performance.
 | ||
| Fig. 1 (a) Single-crystal structure and twisted angles; (b) the π–π interaction and slip angle; (c) packing manners of 2mTPAPyCP. | ||
Density functional theory (DFT) calculations and time-dependent density functional theory (TD–DFT) calculations were performed at B3LYP/6-31G(d) level to investigate the nature of the ground and excited states. As shown in Fig. 2a, both TPAPyCP and 2mTPAPyCP displayed moderately twisted molecular conformations with dihedral angles of ∼50° in the ground state, slightly larger than the corresponding angles in single crystals. Their HOMOs were primarily located at TPA and pyrene units, while the lowest unoccupied molecular orbitals (LUMOs) were predominantly distributed on pyrene and benzonitrile groups. The substantial overlaps between HOMO and LUMO occurred on the planar pyrene moieties.
 | ||
| Fig. 2 Theoretical calculations: (a) optimized molecular structures, FMO distributions, and HOMO and LUMO values; (b) NTO distributions, the CT, LE percentages; and (c) SOC matrix elements, the first five triplet and ten singlet levels of TPAPyCP and 2mTPAPyCP. | ||
The natural transition orbitals (NTOs) calculations were employed to investigate the transition characteristics of excited states. As shown in Fig. 2b, the NTOs of TPAPyCP in S1 and S2 states were characterized by the HLCT state with partly overlapped and partly separated NTO distributions with oscillator strengths (f) of 0.4351 and 0.6827. While the S1 of 2mTPAPyCP presented an HLCT state with a larger f of 1.4844 and the S2 state showed an LE state feature with overlapped NTOs. Their high-lying triplet states (Tm, m ≤ 5) all exhibited LE-pronounced HLCT states. The first ten triplet and singlet energy levels and their corresponding spin–orbital coupling matrix elements (SOC) were calculated and were shown in Fig. 2c. Both TPAPyCP and 2mTPAPyCP exhibited relatively small energy differences between S2 and Tm (2 < m ≤ 4) states and a large SOC values. Consequently, multiple hRISC channels, including of T4 → S2, T3 → S2, and T2 → S2, were available for TPAPyCP, which can substantially enhance the rate constant of hRISC. The S1 – T1 energy gaps for TPAPyCP and 2mTPAPyCP were estimated to be 0.94 and 0.56 eV, respectively, indicating that RISC processes from T1 to S1 states were suppressed. Their ΔET1–T2 values were 1.20 and 1.14 eV, suggesting that the IC processes from T2 to T1 were inefficient. Hence, the large SOC and the corresponding energy level distributions of TPAPyCP and 2mTPAPyCP implied a hot-exciton mechanism. These results indicated that introducing pyrene as the π-bridge can not only realize HLCT characteristics but also adjust LE components, f and SOC matrix elements, which are favorable for achieving high PLQY and facilitating the hRISC process.
The normalized ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra of TPAPyCP and 2mTPAPyCP in toluene solution (10−5 M) and in neat films were measured (Fig. 3a), and the corresponding data were summarized in Table S2.† Both molecules displayed two prominent absorption bands. The absorption peaks around 305–360 nm could be attributed to the n–π* transitions of the TPA group, while the absorption bands in the range 350–450 nm were ascribed to the π–π* transitions of cyanobenzene and pyrene groups. The absorption peaks in neat films showed a slight redshift relative to that in solution. Compared with TPAPyCP, 2mTPAPyCP showed slightly redshifted absorption spectra due to the enhanced electron-donating ability. The maximum emission peaks of 2mTPAPyCP and TPAPyCP in toluene solution were observed at 478 and 488 nm and shifted to 511 and 522 nm in neat films because of the compact molecular stacking in the aggregate states. The energy gaps of 2.76 eV were determined using the absorption onset value measured via absorption spectra. Therefore, the LUMO energy levels can be deduced to be −2.56 and −2.46 eV for TPAPyCP and 2mTPAPyCP, respectively, according to the equation in ESI.†
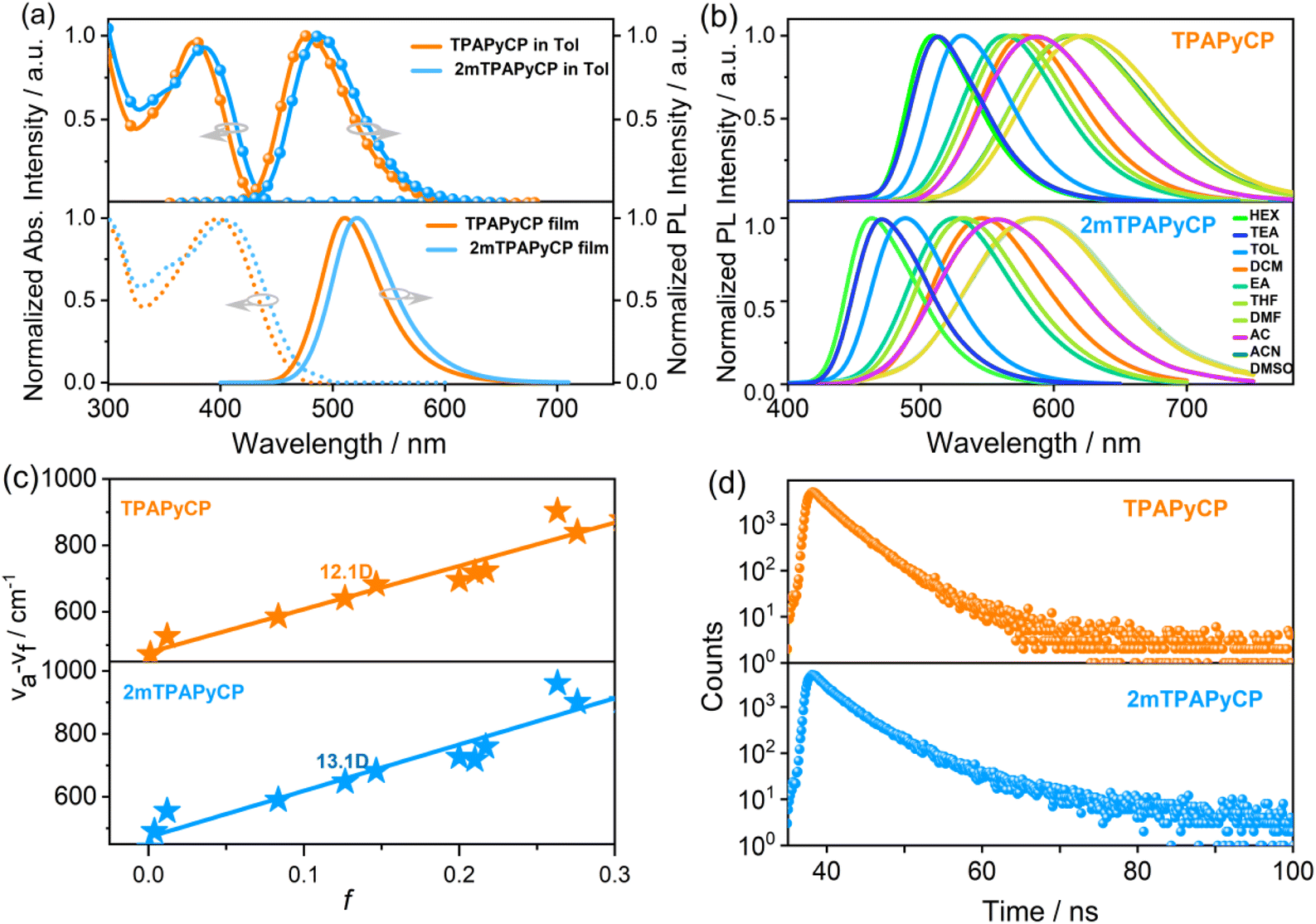 | ||
| Fig. 3 (a) PL and UV-visible absorption spectra in toluene and neat films; (b) PL spectra in different solvents (TEA: triethylamine, Tol: toluene, DCM: dichloromethane, EA: ethyl acetate, THF: tetrahydrofuran, DMF: N,N-dimethylformamide, AC: acetone, ACN: acetonitrile, DMSO: dimethyl sulfoxide); (c) linear fit of solvent polarities versus Stokes shift; (d) transient decay spectra in neat films of TPAPyCP and 2mTPAPyCP. | ||
To characterize the ground and excited state properties of the two molecules, the UV-vis and PL spectra of TPAPyCP and 2mTPAPyCP in solvents with different polarities were tested, as shown in Fig. 3b and S8.† Their absorption spectra of both molecules exhibited tiny changes with increasing polarity, indicating that polarity had essentially no effect on the ground state. The excited state showed high sensitivity to changes in polarity. From low-polarity n-hexane (HEX) to high-polarity dimethyl sulfoxide (DMSO), the emission spectra of TPAPyCP exhibit a large redshift of 113 nm from 455 to 568 nm, while the maximum emission peak of 2mTPAPyCP redshifted from 462 to 604 nm. Moreover, their emission spectra tended to be broadened with increasing solvent polarity, indicating a strong CT state component. The excited-state dipole moment (μe) of the S1 state can be estimated from the plot of Stokes shift (νa − νf) against solvent polarity according to the Lippert–Mataga equation.35 As shown in Fig. 3c, their dipole moments (μe) were determined to be 12.1 and 13.1 D, respectively, which were larger than those of LE emitters (<10 D), indicating CT-dominated excited-state characters.36 The transient PL decay curves of 2mTPAPyCP and TPAPyCP in low-polarity HEX, medium-polarity tetrahydrofuran (THF), and high-polarity acetonitrile (ACN) solutions were fitted to a single exponential process with nanosecond lifetime (Fig. S9†), indicating that their emission originates from a single excited state. The transient PL decay curves in neat films were fitted to a double-exponential decay lifetime (Fig. 3d) with lifetimes of 1.77/2.49 and 1.81/2.25 ns, both of which were short lifetimes, ruling out the TADF mechanism.
The absolute PLQYs of TPAPyCP and 2mTPAPyCP in neat films were estimated to be 73.2 and 65.8%, respectively, which were much higher than those of many An-based hot-exciton materials.28,30 The high PLQYs were attributed to the introduction of an intermediate pyrene, which promotes a relatively more planar conformation. Combined with the high PLQY and short lifetimes (τ) (Table S2†), large radiative rate constant (kr) values of 2.90 × 108 and 2.05 × 108 s−1 and hRISC rate constants (khRISC) values of 9.53 × 108 and 7.19 × 108 s−1 were calculated for TPAPyCP and 2mTPAPyCP, respectively. Notably, the calculated kr and khRISC were found to be of the same order of magnitude of 108 s−1, indicating that the conversions between triplet and singlet excitons of S1 → S0 and Tn → S1 during the emission process for these two molecules were inclined to realize a relative equilibrium.
To further evaluate the potentials of TPAPyCP and 2mTPAPyCP, the non-doped OLEDs were fabricated with optimized structures of ITO/MoO3 (3 nm)/NPB (40 nm)/TCTA (10 nm)/TPAPyCP or 2mTPAPyCP (25 nm)/TPBi (45 nm)/LiF (1 nm)/Al (100 nm) (Fig. 4a). Indium tin oxide (ITO) and aluminum metal (Al) were used as the anode and cathode. MoO3 and lithium fluoride (LiF) were chosen as the hole-injection and electron-injection layers. N,N′-Bis(1-naphthalenyl)-N,N′-bisphenyl-(1,1′-biphenyl)-4,4′-diamine (NPB) and 3,5-tris(1-phenyl-1H-benzimidazol-2-yl) benzene (TPBi) were employed as the hole-transport and electron-transport materials, respectively. [4-(Carbazol-9-yl)phenyl]amine (TCTA) was adopted for electron-blocking layers. The synthesized TPAPyCP and 2mTPAPyCP were deposited as the emitting layers. The corresponding molecular structures and the energy level diagrams of each functional layer in non-doped devices are depicted in Fig. 4a and S10.†
 | ||
| Fig. 4 (a) Device structure and energy diagram; (b) EL spectra under a voltage of 5 V; (c) EQE versus luminance curves; insert: corresponding CIE coordinates for the devices, (d) the reported maximum EQE based on hot-exciton materials with the EL emission peaks in the range of 470–520 nm. | ||
The non-doped devices of TPAPyCP and 2mTPAPyCP demonstrated blue and green light with emission peaks at 496 and 511 nm, corresponding to the Commission Internationale de l'Éclairage (CIE) coordinates of (0.21, 0.46) and (0.27, 0.56) (in Fig. 4b and c), respectively, which were close to the PL emission peaks in neat films. Notably, the EL spectra exhibited remarkable stability with tiny changes when the operating voltage increased from 4 to 9 V, indicating effective carrier injection and transport (Fig. S11†). As shown in Fig. 4c, a maximum luminance (Lmax) of 39470 cd m−2, a maximum current efficiency (CEmax) of 17.21 cd A−1, a maximum power efficiency (PEmax) of 14.86 m W−1, and a maximum external quantum efficiency (EQEmax) of 9.41% were achieved for non-doped TPAPyCP devices. The Lmax, CEmax, PEmax and EQEmax of the devices based on 2mTPAPyCP were 29340 cd m−2, 17.82 cd A−1, 14.641 m W−1, and 9.28%, respectively. The detailed EL performances are listed in Table 1. As shown in Fig. 4d, their maximum EQE were higher than those of many reported non-doped hot-exciton devices with the maximum EL peaks in the range of 475–520 nm,30,37–44 which were mainly attributed to the improved PLQY and rapid hRISC process. It was especially noteworthy that both devices exhibited an extremely small efficiency roll-off. Even when the luminance reached 10000 cd m−2, the EQEs were still maintained at 8.95 and 8.77% with efficiency roll-off values of 4.97 and 5.54%, respectively, which were also much smaller than those of many reported non-doped devices. The relatively high PLQYs, and fast and balanced kr and khRISC rate constants contributed to significant suppression of exciton quenching, resulting in an exceptionally low efficiency roll-off.
| Device | λ (nm) | Vona (V) | Lmax (cd m−2) | CEmax (cd A−1) | PEmax (lm W−1) | EQEmax/10000b (%) | CIE (x, y) | EUE (%) |
|---|---|---|---|---|---|---|---|---|
| a Voltage at a luminance of 1 cd m2.b Maximum external quantum efficiency (EQE)/EQE at a luminance of 10000 cd m−2. |
||||||||
| TPAPyCP | 496 | 3.3 | 39470 |
17.21 | 14.86 | 9.41/8.95 | (0.21, 0.46) | 64.3 |
| 2mTPAPyCP | 511 | 2.7 | 29340 |
17.82 | 14.64 | 9.28/8.77 | (0.27, 0.56) | 70.5 |
The exciton utilization efficiency (EUE) of non-doped devices based on TPAPyCP and 2mTPAPyCP were estimated to be 64.3% and 70.5% according to the equation EQEmax = γ × ΦPL × EUE × ηout,45 where γ, ΦPL and ηout are the recombination efficiency of the injected charge carriers (ideally 100%), PLQY in neat films, and the efficiency of optical output coupling (20%). Clearly, the EUE were beyond the upper limit of 25% for conventional fluorescent materials, indicating that a significant number of triplet excitons were involved in the light-emitting process. Therefore, low-temperature fluorescence and phosphorescence of TPAPyCP and 2mTPAPyCP were recorded. As shown in Fig. 5a and b, the maximum emission peaks without any delay were located at 458 and 468 nm in 2-MeTHF solution at 77 K, corresponding to the S1 energy levels of 2.70 and 2.60 eV, respectively. No long-lived phosphorescence emission was directly observed due to the inefficient intersystem crossing (ISC) process from S1 to T1 states. To obtain their T1 energy levels of TPAPyCP and 2mTPAPyCP, the phosphorescent spectra including of PtOEP&TPAPyCP, PtOEP&2mTPAPyCP and PtOEP were characterized using PtOEP (T1 = 1.91 eV) as sensitizer. The new emission bands with 10 ms delayed were observed at 716 and 704 nm with a delay time of 10 ms, which were attributed to the transition from the T1 of PtOEP to the T1 of emitters. So, the T1 energy levels of TPAPyCP and 2mTPAPyCP were estimated to be 1.73 and 1.76 eV, respectively. The sensitizing mechanisms are shown in Fig. 5c. These results demonstrated a lower T1 energy level of ≈1.70 eV and large singlet–triplet energy gaps (ΔEST) of >0.80 eV. Thus, the triplet excitons utilized in the emission process should originate from the high-lying triplet states.
 | ||
| Fig. 5 (a) and (b) Fluorescence spectra of the two emitters, phosphorescence spectra with 10 ms delay of emitters & PtOEP, and PtOEP at 77 K in 2-MeTHF solution, (c) schematic diagram of the determination of triplet state (Dexter ET: Dexter energy transfer, ISC: intersystem crossing, Phos: phosphorescent). | ||
Conclusions
In conclusion, two hot-exciton molecules of TPAPyCP and 2mTPAPyCP using pyrene as a π-bridge were designed and synthesized. The presence of the planar pyrene group reduced the twisted structures of emitters, enhanced thermal stability and provided multiple hRISC channels. Thus, TPAPyCP and 2mTPAPyCP achieved the high PLQY values of up to 73.2 and 65.8% in neat films along with a rapid radiative rate. The T1/S1 energy levels of TPAPyCP and 2mTPAPyCP were estimated to be 1.73/2.70 and 1.76/2.60 eV, respectively. Non-doped OLED devices based on TPAPyCP and 2mTPAPyCP demonstrated EQEmax of 9.41 and 9.28% with very low efficiency roll-off. This study provides an effective molecular design strategy for constructing efficient and stable hot-exciton emitters.Data availability
The data supporting this article have been included as part of the ESl.† Crystallographic data for 2mTPAPyCP has been deposited at the CCDC under numbers 2409186 and can be obtained from .https://www.ccdc.cam.ac.uk/data.Author contributions
H. Wang and J. Yu supervised the project. Y. Du made the investigations and wrote the original draft. P. Tao and X. Xu reviewed and edited the manuscript. W. Dong and X. Li conducted the synthesis, basic property characterization. K. Chen made the theoretical calculations. Y. Peng performed the fabrication and characterization of the devices. All the authors participated in the discussion of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Joint Funds of the National NSFC (U21A20492); National Natural Science Foundation of China (62074109); Key R&D program of Shanxi Province (International Cooperation, 201903D421087, 201903D121100); Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (20230007).References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS PubMed.
- Y. Im, S. Y. Byun, J. H. Kim, D. R. Lee, C. S. Oh, K. S. Yook and J. Y. Lee, Adv. Funct. Mater., 2017, 27, 1603007 CrossRef.
- Y. Cao, I. D. Parker, G. Yu, C. Zhang and A. J. Heeger, Nature, 1999, 397, 414 CrossRef CAS PubMed.
- Y. Ma, H. Zhang, J. Shen and C. Che, Synth. Met., 1998, 94, 245 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- J. S. Wilson, A. S. Dhoot, A. J. A. B. Seeley, M. S. Khan, A. Kohler and R. H. Friend, Nature, 2001, 413, 828 CrossRef CAS PubMed.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- K. Xu, N. Li, Z. Ye, Y. Guo, Y. Wu, C. Gui, X. Yin, J. Miao, X. Cao and C. Yang, Chem. Sci., 2024, 15, 18076 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- H. Jiang, P. Tao and W.-Y. Wong, ACS Mater. Lett., 2023, 5, 822 CrossRef CAS.
- S. Du, M. Luo, D. Li, L. Lyu, W. Li, M. Zhao, Z. Wang, J. Zhang, D. Liu, Y. Li, S.-J. Su and Z. Ge, Adv. Mater., 2023, 35, 2303304 CrossRef CAS PubMed.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797 CrossRef CAS.
- C. Lin, P. Han, S. Xiao, F. Qu, J. Yao, X. Qiao, D. Yang, Y. Dai, Q. Sun, D. Hu, A. Qin, Y. Ma, B. Tang and D. Ma, Adv. Funct. Mater., 2021, 31, 2106912 CrossRef CAS.
- Y. Gao, S. Zhang, Y. Pan, L. Yao, H. Liu, Y. Guo, Q. Gu, B. Yang and Y. Ma, Phys. Chem. Chem. Phys., 2016, 18, 24176 RSC.
- F. Liu, H. Liu, Y. Chen, X. He, Z. Cheng, X. Ma, X. Qiao, D. Ma and P. Lu, CCS Chem., 2025 DOI:10.31635/ccschem.025.202405291.
- X. Guo, P. Yuan, J. Fan, X. Qiao, D. Yang, Y. Dai, Q. Sun, A. Qin, B. Z. Tang and D. Ma, Adv. Mater., 2021, 33, 2006953 CrossRef CAS PubMed.
- Y. Pan, W. Li, S. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 510 CrossRef CAS.
- K. Chen, Q. Luo, Y. Du, Y. Peng, X. Li, H. Xu, Y. Miao, H. Wang, R. Chen, J. Yu and D. Ma, Chem. Eng. J., 2025, 509, 161133 CrossRef CAS.
- R. Zhong, M. Wang, X. Wang, S. Wang and L. Wang, Chem. Sci., 2024, 15, 13290 RSC.
- Y. Xu, X. Liang, X. Zhou, P. Yuan, J. Zhou, C. Wang, B. Li, D. Hu, X. Qiao, X. Jiang, L. Liu, S. J. Su, D. Ma and Y. Ma, Adv. Mater., 2019, 31, 1807388 CrossRef PubMed.
- X. H. Lv, L. Xu, M. Cang, R. Z. Wang, M. Z. Sun, H. Y. Zhou, Y. Yu, Q. K. Sun, Y. Y. Pan, Y. W. Xu, D. H. Hu, S. F. Xue and W. J. Yang, CCS Chem., 2020, 3, 2557 CrossRef.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609 CrossRef CAS.
- S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755 CrossRef CAS.
- H. Zhang, B. Zhang, Y. Zhang, Z. Xu, H. Wu, P.-A. Yin, Z. Wang, Z. Zhao, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 2002323 CrossRef CAS.
- Y. Xu, X. Liang, Y. Liang, X. Guo, M. Hanif, J. Zhou, X. Zhou, C. Wang, J. Yao, R. Zhao, D. Hu, X. Qiao, D. Ma and Y. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 31139 CrossRef CAS PubMed.
- Q. Luo, Y. Gao, H. Xu, S. Zhao, W. Dong, Y. Miao, Y. Wang, H. Wang and J. Yu, J. Mater. Chem. C, 2023, 11, 15918 RSC.
- K. J. Kim, J. Kim, J. T. Lim, J. Heo, B. J. Park, H. Nam, H. Choi, S. S. Yoon, W. Kim, S. Kang and T. Kim, Mater. Horiz., 2024, 11, 1484 RSC.
- W. Dong, W. Bai, Ya. Wang, S. Zhao, H. Xu, Y. Miao, Q. Luo, H. Wang and J. Yu, Sci. China Mater., 2024, 67, 197 CrossRef CAS.
- Y. Yu, P. Xu, Y. Pan, X. Qiao, L. Ying, D. Hu, D. Ma and Y. Ma, Adv. Opt. Mater., 2023, 11, 2202217 CrossRef CAS.
- L. Xu, Y. Yu, M. Li, Y. Li, W. Tan, B. Wang, D. Yang, D. Hu, L. Ying and Y. Ma, Adv. Opt. Mater., 2024, 12, 2401275 CrossRef CAS.
- G. Chen, J. Wang, W.-C. Chen, Y. Gong, N. Zhuang, H. Liang, L. Xing, Y. Liu, S. Ji, H.-L. Zhang, Z. Zhao, Y. Huo and B. Z. Tang, Adv. Funct. Mater., 2023, 33, 2211893 CrossRef CAS.
- S. Zeng, C. Xiao, J. Zhou, Q. Dong, Q. Li, J. Lim, H. Ma, J. Y. Lee, W. Zhu and Y. Wang, Adv. Funct. Mater., 2022, 2113183 CrossRef CAS.
- Z. Zhong, X. Zhu, X. Wang, Y. Zheng, S. Geng, Z. Zhou, X. J. Feng, Z. Zhao and H. Lu, Adv. Funct. Mater., 2022, 32, 2112969 CrossRef CAS.
- L. Xu, M. Sun, Y. Zhou, J. Lou, M. Xie, Z. Li, Q. Sun, Y. Pan, S. Xue and W. Yang, Org. Chem. Front., 2023, 10, 490 RSC.
- H. Zhang, J. Zeng, W. Luo, H. Wu, C. Zeng, K. Zhang, W. Feng, Z. Wang, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 6359 RSC.
- C. Zhou, X. Zhang, G. Pan, X. Tian, S. Xiao, H. Liu, S. Zhang and B. Yang, Org. Electron., 2019, 75, 105414 CrossRef CAS.
- J. Liu, Z. Li, T. Hu, X. Wei, R. Wang, X. Hu, Y. Liu, Y. Yi, Y. Yamada-Takamura, Y. Wang and P. Wang, Adv. Opt. Mater., 2018, 7, 1801190 CrossRef.
- W. Z. Yuan, X. Bin, G. Chen, Z. He, J. Liu, H. Ma, Q. Peng, B. Wei, Y. Gong, Y. Lu, G. He and Y. Zhang, Adv. Opt. Mater., 2017, 5, 1700466 CrossRef.
- C. Ma, M. Sun, Y. Zhou, L. Chu, J. Song, L. Zhang, Q. Sun, W. Yang and S. Xue, Dyes Pigments, 2025, 235, 112647 CrossRef CAS.
- J. Jayabharathi, J. Anudeebhana, V. Thanikachalam and S. Sivaraj, RSC Adv., 2020, 10, 8866 RSC.
- X. Qiu, T. Li, C. Liu, X. Liu, J. Li, S. Xue and Y. Pan, Dyes Pigments, 2023, 208, 110814 CrossRef.
- C. Wang, X. Li, Y. Pan, S. Zhang, L. Yao, Q. Bai, W. Li, P. Lu, B. Yang, S. Su and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 3041 CrossRef CAS PubMed.
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2409186. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01756d |
| This journal is © The Royal Society of Chemistry 2025 |