
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransforming 2D azolium salts to 3D caged tertiary amines via stereoselective dearomative cascade annulation†
Koushik
Patra
a,
Samiran
Deb
a,
Venkata Surya
Kumar Choutipalli
 c,
Sana
Mulani
a,
Sumitava
Mallik
a,
Venkatesan
Subramanian
bc and
Mahiuddin
Baidya
*a
c,
Sana
Mulani
a,
Sumitava
Mallik
a,
Venkatesan
Subramanian
bc and
Mahiuddin
Baidya
*a
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600 036, Tamil Nadu, India. E-mail: mbaidya@iitm.ac.in
bDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, Tamil Nadu, India
cCentre for High Computing, CSIR-Central Leather Research Institute, Chennai 600020, Tamil Nadu, India
First published on 26th March 2025
Abstract
Three-dimensional fused-ring frameworks, especially those incorporating heteroatoms, are fundamental to expanding chemical space and unlocking unique properties critical for drug discovery and functional materials, yet their synthesis remains a formidable challenge. Herein, we report for the first time the union of two distinct azolium salts as an efficient synthetic platform to access tertiary amine-caged frameworks under mild conditions. The strategy combines the masked nucleophilic and electrophilic properties of isoquinolinium and pyridinium salts, and avails double dearomatization guided inverse electron demand (4 + 2) or (3 + 2) annulation in a highly regio- and diastereoselective manner to construct the nitrogen caged motifs. Our methodology creates two new rings and four new bonds in a single operation and transforms flat-aromatic compounds into structurally unprecedented three-dimensional architectures with contiguous stereocenters in very high yields. DFT studies have shed light on the reaction mechanism, indicating that the annulation step is rate-limiting, with (4 + 2) annulation proceeding stepwise and (3 + 2) annulation following a concerted pathway.
Introduction
Transforming two-dimensional (2D) molecular frameworks into three-dimensional (3D) architectures by incorporating out-of-plane substituents can elevate the fraction of sp3 atoms (F sp3) in the resulting structures.1 This enhancement is a crucial design element in drug development, contributing to improved clinical outcomes such as better solubility, enhanced receptor–ligand interactions, target selectivity, and efficacy.1,2 In this context, tertiary amine-containing caged-like molecular structures offer exciting possibilities in pharmaceutical science and synthetic chemistry (Scheme 1a).3 They serve as fundamental backbones for various bioactive natural products, and many derivatives find widespread application as reagents, ligands, and catalysts in organic synthesis.3,4 Therefore, developing cogent strategies towards these intriguing, structurally complex caged-amine frameworks from readily available starting materials through simple experimental procedures is highly desirable.5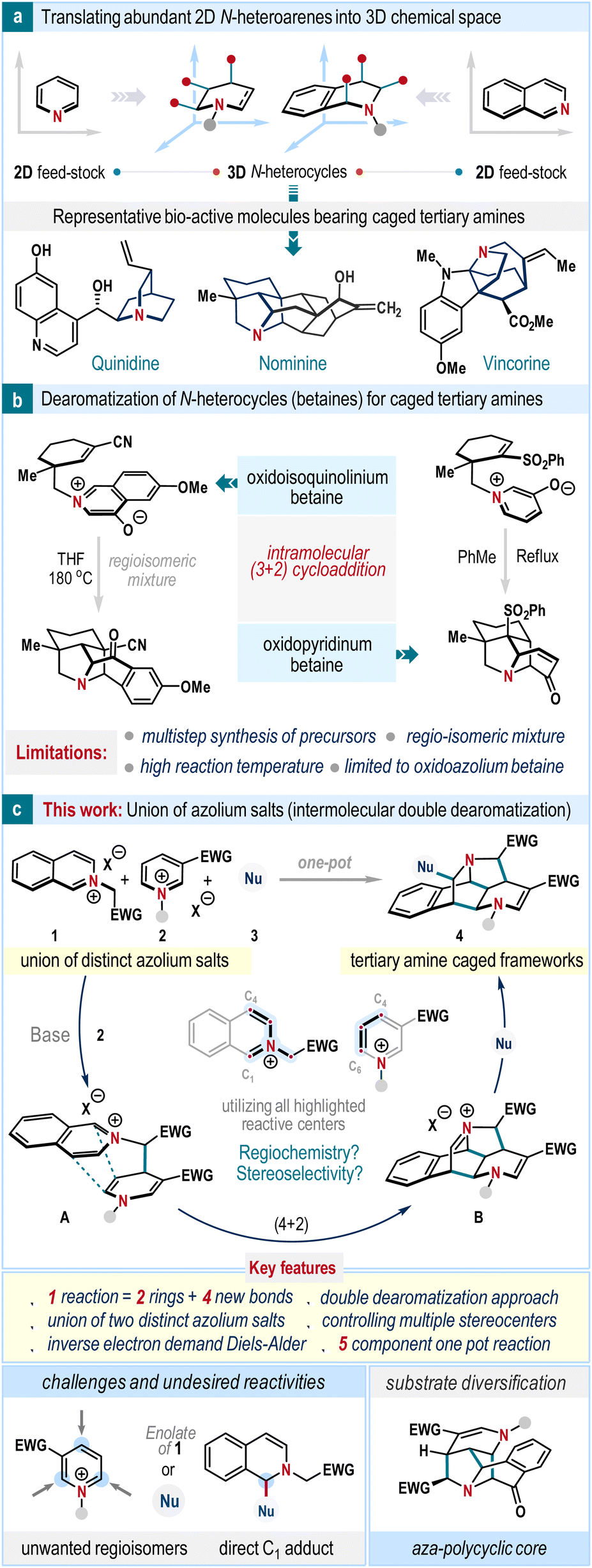 | ||
| Scheme 1 Dearomatization strategies towards three-dimensional cage amine scaffolds. | ||
The aromatic heterocycles, specifically N-heterocycles, are inexpensive building blocks and widely available in great structural diversity.6 We envisioned transforming these flat two-dimensional molecules into three-dimensional architectures as a synthetic maneuver to access tertiary amine caged-like frameworks. This strategic blueprint involves a dearomatization reaction, which is fundamental in the construction of diverse molecular scaffolds in organic synthesis;7 however, literature precedent is rare in the production of tertiary amine caged-like frameworks. The pioneering examples from Gin and co-workers showcasing the dearomatization of oxidoisoquinolinium and oxidopyridinium betaines stand alone in this realm as a direct and concise protocol (Scheme 1b).8a,b However, this strategy cannot be utilized with the less reactive parent isoquinolinium salt and is also restricted for intramolecular processes.8c–h
Nature often adopts the union of two or more simplified molecular progenitors in the form of cascade reactions to dispense complex architectures, which provide great impetus in devising biomimetic synthetic strategies.9 We hypothesized that an assembly of isoquinolinium (1) and pyridinium salts (2) could be a convenient route to make tertiary amine caged-like frameworks (Scheme 1b). In the presence of a base, the enolate thus generated from isoquinolinium salt 1 can react with pyridinium salt 2 to give intermediate A which possesses all characteristics to expedite an intramolecular inverse electron demand Diels–Alder reaction. Subsequently, the iminium ion intermediate B will be formed, which upon quenching with a suitable nucleophile would furnish the desired tertiary amine caged scaffold 4. This design is unique as it endorses the dearomatization event of both isoquinoline and pyridine heterocycles in a one-pot operation, which is so far unexplored. Also, it maximizes the use of potential reactive sites of isoquinoline (C1, C3, and C4 centers) and pyridine (C4, C5, and C6 positions) heterocycles in a cascade fashion (Scheme 1b). However, the successful execution of this plan must overcome several challenges. The pyridinium salts 2 are prototypes of ambident electrophiles and accept nucleophilic attack at C2, C4, and C6 centers (Scheme 1b, below). Hence, maintaining high regioselectivity during this cascade event is imperative to facilitate the formation of the caged framework. Further, a judicious choice of nucleophile (Nu) is critical so that its direct reaction with pyridinium salt as well as isoquinolinium salt can be outcompeted. Also, mild reaction conditions is necessary to suppress the potential self-dimerization process of isoquinolinium salt 1 and achieve high diastereoselectivity as the successful cascade would embrace four new bond formations with contiguous stereocenters.
Herein, we report the development of this approach and demonstrate an unprecedented intermolecular annulative double dearomatization cascade of isoquinolinium and pyridinium salts to obtain tertiary cage amine scaffolds in high yields with excellent regio- and diastereoselectivity. Notably, the protocol can be performed as a one-pot five-component coupling without compromising efficacy and selectivity, highlighting synthetic versatility.10 Through the substrate design, we have also showcased the synthesis of the central aza-polycyclic core of hetisine-type natural alkaloids. We also provide a detailed elucidation of the reaction mechanism, supported by DFT studies, offering valuable insights into this transformative process.
Result and discussion
We began our investigation following the model reaction between isoquinolinium salt 1a and pyridinium salt 2a (Table 1). We consider ethyl alcohol as the reaction medium owing to its inability to undergo nucleophilic addition to azolium salts 1a and 2a; however, it could play the role of the third component and react with the proposed iminium intermediate to produce cage frameworks. Grounded on this understanding, a mixture of 1a and 2a in EtOH solvent was exposed to Na2CO3 base at room temperature (entry 1).|
|
|||
|---|---|---|---|
| Entry | Deviation from the standard conditions | Yield of 4ab (%) |
4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a′ :4a′ |
| a Reaction conditions: 1a (0.24 mmol), 2a (0.2 mmol), base (0.4 mmol), and EtOH (1.5 mL), 12 h, under N2 atmosphere. b Isolated yields were provided. c Ethanol (0.24 mmol) was used. DCE: 1,2-dichloroethane, GC: guanidine carbonate. | |||
| 1 | At rt for 30 h | 36 | — |
| 2 | None | 83 |
>10:1 |
| 3 | At 80 °C | 70 | >10:1 |
| 4c | DCE/THF/CH3CN instead of EtOH | 25/32/54 | 1:1 |
| 5 | K2CO3/Li2CO3/Ag2CO3 instead of Na2CO3 | 72/75/68 | >10:1 |
| 6 | DIPEA/GC/DBU instead of Na2CO3 | 77/64/41 | >10:1 |
| 7 | Without Na2CO3 | — | — |
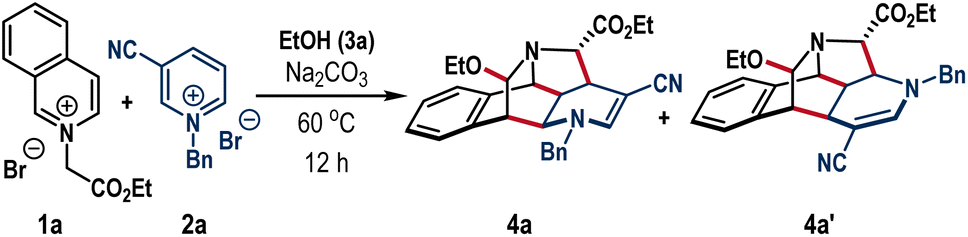
To our satisfaction, the formation of the caged scaffold took place, albeit the reaction was sluggish to render the desired annulation product 4a in 36% yield after 30 h. When the temperature was increased to 60 °C, the reaction was completed within 12 h, and product 4a was isolated as a single diastereomer with an improved yield of 83% (entry 2). At this juncture, a minor amount of other regioisomer 4a′ (arising from the initial C6-attack on 2a was also formed in 8% yield. Further increase of the reaction temperature rendered detrimental yield which can be attributed to the initiation of various undesired reactions as observed in the TLC (entry 3). The choice of solvent was crucial to preserve efficacy and selectivity. Examination of DCE, THF, and CH3CN solvents along with a reagent amount of EtOH provided an almost 1:1 mixture of 4a and 4a′ with poor yields (entry 4). The reaction was productive with different inorganic bases such as Li2CO3, K2CO3, and Cs2CO3 (entry 5). Organic bases also can promote this reaction. Hunig's base (iPr2NEt) and guanidine carbonate (GC) gave 4a in 77% and 64% yields, respectively, while the reaction yield was reduced to 41% for DBU (entry 6). Nonetheless, the regioisomeric ratio remained >10:1 for all these cases. Notably, the reaction completely shut down in the absence of a base (entry 7). Compound 4a was also crystalized and the single crystal X-ray analysis unambiguously confirmed the caged structure with defined regio- and stereoselectivity (Scheme 2, above).
 | ||
| Scheme 2 Exploration of the reaction scopes. aReaction conditions: 1 (0.24 mmol), 2 (0.2 mmol), Na2CO3 (0.4 mmol), and alcohol solvent 3 (1.5 mL), under N2 atmosphere. Isolated yields were provided. bReaction time was 18 h. | ||
With the optimized reaction conditions (Table 1, entry 2), we then explored the substrate scope (Scheme 2). Initially, different isoquinolinium salts (1) were examined. Isoquinolinium salts obtained from various α-bromo esters of primary (4b–4d), secondary (4e, 4f), and tertiary (4g) alcohols smoothly delivered caged molecules in very high yields. The product with allyl ester 4h was also obtained in 74% yield. Similarly, isoquinolinium salts prepared from phenacyl bromide and its derivatives having electron-donating and electron-deficient functionalities at meta- and para-positions in the aryl ring were suitable substrates to offer 4i–4m in high yields. Also, substrates with bulky 1-naphthyl ketone and heterocyclic thienyl ketone motifs gave 4n and 4o in 62% and 64% yields, respectively. The bromine functional group in the isoquinoline ring (4p) can also be accommodated, leaving a synthetic handle for further transformation. The protocol was also effective with phenolic ester-embedded isoquinolinium salts; however, for these cases, we observed concomitant transesterification with ethanol solvent leading to 4a in high yields (Scheme 2). The generality of other alcoholic solvents as a third component in this synthesis was also examined. Linear alcohols such as propanol, butanol, and branch alcohol isopropanol delivered 4q–4s in 70–77% yields. With bulky alcohols, for example, tertiary butanol and tert-amyl alcohol, desired products 4t and 4u were isolated in good yields.
Next, the reaction scope was surveyed with different pyridinium salts 2 (Scheme 2). A wide variety of N-benzyl analogs of 3-cyanopyridinium salts bearing electronically different substitutions at ortho-, meta-, and para-positions of the aryl ring furnished tertiary amine caged compounds 4v–4ae in uniformly high yields (70–77%). Reactions of N-methyl (4af), ethyl (4ag), butyl (4ah), and allyl (4ai) pyridinium salts also led to high annulation yields. The 3-nitropyridinium salt also readily participated in this reaction and the corresponding caged amine product 4aj was isolated in 74% yield. It is worth noting that an activated pyridinium salt was essential for the synthesis of caged amine framework, while parent pyridinium salt without any activating group was not productive (4ak). Satisfyingly, all products were prepared as a single diastereomer with >10:1 regioselectivity.
To garner diversity in the synthesis, we employed 3-methyl substituted isoquinolinium salts 5. We hypothesized that the presence of methyl group could be beneficial to neutralize the iminium ion intermediate C generating from the (4 + 2) annulation process and thereby a tertiary amine cage framework bearing an exocyclic double bond could be within reach (Scheme 3). Accordingly, isoquinolinium salt 5a was reacted with pyridinium salt 2a under standard reaction conditions where desired product 6 was formed in 57% yield. The combination of other isoquinolinium salts 5b and 5c with pyridinium salt 2a was also fruitful, albeit the process was associated with a transesterification step leading to 6 in synthetically useful yields (Scheme 3).
 | ||
| Scheme 3 Synthesis of cage tertiary amines with exocyclic double bond. | ||
As an alternative route to cage tertiary amine framework, we plan to alter the reaction modality from (4 + 2) annulation to (3 + 2) annulation. It was envisioned that incorporation of a suitable electron-donating functionality at the C4 position of isoquinolinium salt will increase the nucleophilic character at the C3 position and such a scenario is expected to favor (3 + 2) annulation towards desired cage framework (Scheme 4a). In view of that, isoquinolinium salt 7 having a methoxy group at C4 position was prepared and reacted with 2a under standard reaction conditions. However, we observed formation of cage molecule 8 in 80% yield via (4 + 2) annulation discussed in the preceding section, indicating further substrate modification was required (Scheme 4b). Accordingly, the reaction was revisited with isoquinolinium salt 9 bearing a free hydroxyl functionality at C4 position. At this juncture, dearomatization guided (3 + 2) annulation proceeded cleanly producing densely functionalized cage molecule 10a in 70% yield as a single diastereomer. A minor amount of other regioisomer 11a (arising from the initial C6-attack on 2a) was also formed in 14% yield. Reactions of other pyridinium salts (2) also led to the production of 10b–10f in high yields with moderate regio-selectivity (Scheme 4c). With ester based isoquinolinium salts 11a–11b, reactions were also successful; however, as indicated above, the concomitant transesterification took place to give 10a in high yields (Scheme 4d).
 | ||
| Scheme 4 Structural tuning and dearomatization guided (3 + 2) annulation for cage frameworks. | ||
To highlight synthetic expediency, a one-pot five-component protocol was established. We recognized that synthesis of isoquinolinium salt proceeds at room temperature while the preparation of pyridinium salts requires an elevated temperature. Such reactivity distinction suggested that these azolium salts can be prepared sequentially and engaged in the synthesis of tertiary amine cage scaffolds by controlling the reaction temperature as depicted in Scheme 5a. First, isoquinoline and ethyl bromoacetate were stirred in ethanol for 2 h at room temperature. Then, 3-cyanopyridine, benzyl bromide, and Na2CO3 were added and the mixture was heated at 60 °C, where the desired reaction also proceeded effectively to give 4a in 71% yield without affecting the regio- and diastereoselectivity.
 | ||
| Scheme 5 One pot approach and gram–scale reaction. | ||
Following the same sequence, various cage scaffolds were also made in good to high yields without compromising selectivities (Scheme 5a). Scale-up was also suitable. A gram-scale reaction rendered 4a in 75% yield as a single diastereomer with >10:1 rr (Scheme 5b).
To gain deeper mechanistic insights, we conducted density functional theory (DFT) calculations (Scheme 6a). The formation of intermediate A from the reaction of enolate, generated in situ from isoquinolinium salt 1a, with pyridinium salt 2a can occur through two distinct pathways: one in which the isoquinoline motif is distal to the nitrile group of the pyridine (blue line), and the other in which the isoquinoline motif is in proximity to the nitrile group of the pyridine unit (red line). Our calculations revealed that the former pathway, with a lower activation barrier (TS1R = 4.75 kcal mol−1), is more favorable compared to latter (TS1S = 5.51 kcal mol−1) and it can be atributed to the distabilizing steric interaction of isoquinolium ring with cyano group of the pyridinium salt. These pathways lead to highly exergonic formation of intermediates AR and AS, each releasing approximately 15 kcal mol−1 of energy. Next, the intermediate AR undergoes a (4 + 2) inverse electron demand Diels–Alder reaction to yield iminium intermediate B. It is interesting to note that this (4 + 2) cycloaddition reaction proceeds in a stepwise manner rather than through a concerted mechanism, and the activation barriers for the two carbon–carbon bond formations are 18.18 (TS2a) and 5.84 (TS2b) kcal mol−1, respectively.
 | ||
| Scheme 6 Mechanistic investigation through DFT study. aAll the quantum chemical calculations were performed using Gaussian 16 program package, employing the density functional theory (DFT) based M062X functional in conjunction with 6-31G(d,p) basis set. Geometries were fully relaxed in solvent (methanol) medium using polarized continuum model (PCM) and characterized by frequency analysis to confirm they are true minima or saddle points on potential energy surface. Relative free energies are given in kcal mol−1. | ||
Despite numerous attempts, a concerted transition state for the (4 + 2) cycloaddition was not observed. As expected, the subsequent nucleophilic attack of ethanol on the intermediate B happens from the less hindered side, which is kinetically more favorable (TS3F, 4.66 kcal mol−1vs.TS3C, 22.46 kcal mol−1), leading to the selective formation of the caged amine product 4a. Overall, the (4 + 2) cycloaddition step is identified as the rate-limiting step for this reaction (Scheme 6a). When investigating the annulation reaction involving the enolate arising from the isoquinolinium salt 9, which has an electron-releasing hydroxy group, and the pyridinium salt 2a (Scheme 6b), we found that the initial nucleophilic attack in both pathways are energetically comparable (TS1REDG = 2.25 kcal mol−1vs.TS1sEDG = 2.45 kcal mol−1). It is only marginally more favorable to produce the intermediate AREDG, where the isoquinoline motif and the nitrile group of pyridine are positioned distally from each other. However, unlike the preceding (4 + 2) annulation reaction, a (3 + 2) annulation predominates here, and it proceeds in a concerted fashion via the TScEDG with an activation barrier of 24.34 kcal mol−1 to give (3 + 2) cycloaddition product 10a, which is exothermic by −34.96 kcal mol−1. The (3 + 2) cycloaddition step is also the rate-limiting step for this annulation, as apparent from Scheme 6b.
Conclusions
In summary, we have demonstrated a novel cascade strategy featuring an unexplored intermolecular annulated coupling of two distinct azolium salts derived from isoquinoline and pyridine heterocycles. This innovative approach effectively transforms flat 2D N-heterocycles into complex 3D molecular frameworks with regio- and diastereoselectivity, facilitating the synthesis of challenging caged tertiary amines. By capitalizing on the inherent reactivity of these azolium salts and maximizing their reactive sites, we expedite a double dearomatization-guided (4 + 2) or (3 + 2) annulation reaction, yielding intricate nitrogen-caged motifs, including aza-polycyclic cores akin to hetisine-type alkaloids, in high yields. DFT studies have provided insights into the underlying mechanism of the reaction, revealing that the annulation step is rate-limiting, where (4 + 2) annulation proceeds in a stepwise fashion while corresponding (3 + 2) annulation follows a concerted reaction pathway. To further streamline the synthetic process, we developed a five-component one-pot sequential addition strategy. Importantly, these maneuvers maintain the selectivity of the reaction while emphasizing the efficient utilization of resources (formation of two new rings and four new bonds) and the pot economy. Applications of this concept to access other three-dimensional high-value scaffolds are currently ongoing in our laboratory.Data availability
General information, experimental procedures, characterization data for all new compounds, and NMR spectra are in the ESI.† Data for the crystal structure reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number CCDC: 2190610.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. M. B. and K. P. have conceptualized the idea. K. P., S. D., S. M. and S. M. carried out the experiments, mechanistic investigations, and analyzed experimental data. V. S. K. C and V. S. conducted the computational studies. All the authors discussed the results and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. P. acknowledges the PMRF fellowship from MHRD, Government of India. We gratefully acknowledge IIT Madras ERP-project (RF23241505CYRFER008650) and IOE-project SB20210792CYMHRD008189) for the financial support.Notes and references
- For selected reviews, see: (a) W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 CAS; (b) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917–2940 CrossRef CAS PubMed; (c) C. J. Huck and D. Sarlah, Chem, 2020, 6, 1589–1603 CrossRef CAS; (d) C. Zheng and S.-L. You, ACS Cent. Sci., 2021, 7, 432–444 CrossRef CAS; (e) Y.-Z. Cheng, Z. Feng, X. Zhang and S.-L. You, Chem. Soc. Rev., 2022, 51, 2145–2170 RSC; (f) M. Escolano, D. Gaviña, G. Alzuet-Piña, S. Díaz-Oltra, M. Sánchez Roselló and C. d. Pozo, Chem. Rev., 2024, 124, 1122–1246 CrossRef CAS PubMed . For selected examples, see: ; (g) M. W. Gribble Jr, S. Guo and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 5057–5060 CrossRef PubMed; (h) J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown, K. N. Houk and F. Glorius, Science, 2021, 371, 1338–1345 CrossRef CAS; (i) Z. Siddiqi, T. W. Bingham, T. Shimakawa, K. D. Hesp, A. Shavnya and D. Sarlah, J. Am. Chem. Soc., 2024, 146, 2358–2363 CrossRef CAS PubMed.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS; (b) F. Lovering, MedChemComm, 2013, 4, 515–519 RSC; (c) K. E. Prosser, R. W. Stokes and S. M. Cohen, ACS Med. Chem. Lett., 2020, 11, 1292–1298 Search PubMed.
- For selected books, see: (a) G. L. Adams and A. B. Smith, The Chemistry of the Akuammiline Alkaloids, Elsevier Ltd, 2016 Search PubMed; (b) T. Yin, H. Zhang, W. Zhang and Z. Jiang, RSC Adv., 2021, 11, 36023–36033 CAS; (c) N. A. Doering, R. Sarpong and R. W. Hoffmann, Angew. Chem., Int. Ed., 2020, 59, 10722–10731 CAS . For selected examples, see: ; (d) Y. Yasui, T. Kinugawa and Y. Takemoto, Chem. Commun., 2009, 4275–4277 Search PubMed; (e) H. Arai, Y. Hirasawa, A. Rahman, I. Kusumawati, N. Cholies, S. Sato, C. Aoyama, J. Takeo and H. Morita, Bioorg. Med. Chem., 2010, 18, 2152–2158 CAS; (f) R. Eckermann, M. Breunig and T. Gaich, Chem.–Eur. J., 2017, 23, 3938–3949 CAS; (g) X. Zong, X. Yan, J. Wu, Z. Liu, H. Zhou, N. Li and L. Liu, J. Nat. Prod., 2019, 82, 980–989 CAS; (h) M. M. Pompeo, J. H. Cheah and M. Movassaghi, J. Am. Chem. Soc., 2019, 141, 14411–14420 CAS.
- For selected books, see: (a) A. Berkessel and H. Groger, Asymmetric Organocatalysis – From Biomimetic Concepts to Applications in Asymmetric Synthesis, 2005 Search PubMed; (b) C. E. Song, Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis, Wiley, 2009. For selected review, see: Search PubMed; (c) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CAS.
- For selected Reviews, see: (a) A. M. Hamlin, J. K. Kisunzu and R. Sarpong, Org. Biomol. Chem., 2014, 12, 1846–1860 CAS; (b) A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Nat. Prod. Rep., 2016, 33, 491–522 CAS . For selected examples, see: ; (c) M. Zhang, X. Huang, L. Shen and Y. Qin, J. Am. Chem. Soc., 2009, 131, 6013–6020 CrossRef CAS; (d) A. M. Hamlin, F. D. J. Cortez, D. Lapointe and R. Sarpong, Angew. Chem., Int. Ed., 2013, 125, 4954–4957 CrossRef; (e) B. D. Horning and D. W. C. Macmillan, J. Am. Chem. Soc., 2013, 135, 6442–6445 CrossRef CAS; (f) R. Eckermann, M. Breunig and T. Gaich, Chem. Commun., 2016, 52, 11363–11365 RSC; (g) S. Cathafoline, J. Moreno, E. Picazo, L. A. Morrill, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 1162–1165 CrossRef; (h) X. Xie, B. Wei, G. Li and L. Zu, Org. Lett., 2017, 19, 5430–5433 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed; (b) S. Sowmiah and J. M. S. S. Esperança, Org. Chem. Front., 2018, 5, 453–493 RSC; (c) Y. Xie and Q. Wang, Org. Biomol. Chem., 2021, 19, 3960–3982 RSC; (d) S. Das, Org. Biomol. Chem., 2022, 20, 1838–1868 RSC; (e) N. Kratena and T. J. Donohoe, Chem. Sci., 2022, 13, 14213–14225 RSC . For selected examples, see: ; (f) R. B. Gupta and R. W. Franck, J. Am. Chem. Soc., 1987, 1, 5393–5403 CrossRef; (g) J. Llaveria, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10958–10961 CrossRef CAS PubMed; (h) J. H. Xu, S. C. Zheng, J. W. Zhang, X. Y. Liu and B. Tan, Angew. Chem., Int. Ed., 2016, 55, 11834–11839 CrossRef CAS PubMed; (i) B. M. Reeves, H. B. Hepburn, A. Grozavu, P. J. Lindsay-scott and T. J. Donohoe, Angew. Chem., Int. Ed., 2019, 58, 15697–15701 CrossRef CAS PubMed; (j) D. Zhang, Z. Su, Q. He, Z. Wu, Y. Zhou, C. Pan, X. Liu and X. Feng, J. Am. Chem. Soc., 2020, 142, 15975–15985 CrossRef CAS PubMed; (k) C. McLaughlin, J. Bitai, L. J. Barber, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2021, 12, 12001–12011 RSC; (l) Y. Lu, P. N. Dey and C. M. Beaudry, Chem.–Eur. J., 2021, 27, 4028–4032 CrossRef CAS PubMed; (m) M. Kischkewitz, B. Marinic, N. Kratena, Y. Lai, H. B. Hepburn, M. Dow, K. E. Christensen and T. J. Donohoe, Angew. Chem., Int. Ed., 2022, 61, e202204682 CrossRef CAS PubMed; (n) J. C. Abell, C. P. Bold, L. Vicens, T. Jentsch, N. Velasco, J. L. Tyler, R. N. Straker, A. Noble and V. K. Aggarwal, Org. Lett., 2023, 25, 400–404 CAS; (o) A. J. N. Lamhauge, D. A. Mcleod, L. Casper, G. A. Oliver, L. Viborg, T. Warburg and K. A. Jørgensen, Chem.–Eur. J., 2023, e202301830 Search PubMed.
- For reviews, see: (a) W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996–8017 CAS; (b) J. M. Saya, E. Ruijter and R. V. A. Orru, Chem.–Eur. J., 2019, 25, 8916–8935 CAS; (c) C. J. Huck, Y. D. Boyko and D. Sarlah, Nat. Prod. Rep., 2022, 39, 2231–2291 CAS; (d) M. Zhu, X. Zhang, C. Zheng and S. L. You, Acc. Chem. Res., 2022, 55, 2510–2525 CAS . For a selected example, see: ; (e) J. Ma, F. Strieth-Kalthoff, T. Dalton, M. Freitag, J. L. Schwarz, K. Bergander, C. Daniliuc and F. Glorius, Chem, 2019, 5, 2854–2864 CAS; (f) H. J. Miao, L. Le Wang, H. Bin Han, Y. De Zhao, Q. L. Wang and Z. W. Bu, Chem. Sci., 2020, 11, 1418–1424 CAS; (g) J. K. Kerkovius, A. Stegner, A. Turlik, P. H. Lam, K. N. Houk and S. E. Reisman, J. Am. Chem. Soc., 2022, 144, 15938–15943 CAS; (h) B. Singh, A. J. Ansari, N. Malik and S. S. V. Ramasastry, Chem. Sci., 2023, 11, 6963–6969 Search PubMed.
- (a) K. M. Peese and D. Y. Gin, J. Am. Chem. Soc., 2006, 128, 8734–8735 CAS; (b) K. M. Peese and D. Y. Gin, Chem.–Eur. J., 2008, 14, 1654–1665 CAS . For intermolecular reports, but not explored toward caged-amine scaffolds, see: ; (c) A. R. Katritzky and Y. Takeuchi, J. Am. Chem. Soc., 1970, 92, 4134–4136 CAS; (d) A. R. Katritzky and N. Dennis, Chem. Rev., 1989, 89, 827–861 CAS; (e) M. E. Jung, L. M. Zeng, T. S. Peng, H. Y. Zeng, Y. Le and J. Y. Su, J. Org. Chem., 1992, 57, 3528–3530 CrossRef CAS; (f) V. C. Pham and J. L. Charlton, J. Org. Chem., 1995, 60, 8051–8055 CAS; (g) W. Sliwa, Heterocycles, 1996, 43, 2005–2029 CAS; (h) M. J. Sung, H. I. Lee, Y. Chong and J. K. Cha, Org. Lett., 1999, 1, 2017–2019 CAS; (i) H. I. Lee, M. J. Sung, H. B. Lee and J. K. Cha, Heterocycles, 2004, 62, 407–422 CAS.
- For a book, see: (a) Domino Reactions: Concepts for Efficient Organic Synthesis, ed. L. F. Tietze, Wiley-VCH, Weinheim, 2014. For Reviews, see: Search PubMed; (b) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 3, 551–564 Search PubMed; (c) E. C. Cherney and P. S. Baran, Isr. J. Chem., 2011, 51, 391–405 CAS; (d) R. Ardkhean, D. F. J. Caputo, S. M. Morrow, H. Shi, Y. Xiong and E. A. Anderson, Chem. Soc. Rev., 2016, 45, 1557–1569 CAS.
- Y. Hayashi, Acc. Chem. Res., 2021, 54, 1385–1398 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2190610. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01527h |
| This journal is © The Royal Society of Chemistry 2025 |