
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible excited state electron transfer in an acceptor–acceptor hetero dyad†
Jesper Dahl
Jensen
a,
Shayan
Louie
b,
Yanmei
He
ac,
Junsheng
Chen
 a,
Colin
Nuckolls
*b and
Bo W.
Laursen
*a
a,
Colin
Nuckolls
*b and
Bo W.
Laursen
*a
aNano-Science Center & Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100, Copenhagen Ø, Denmark. E-mail: bwl@chem.ku.dk
bDepartment of Chemistry, Columbia University, New York, New York 10027, USA
cDivision of Chemical Physics and NanoLund, Lund University, P. O. Box 124, 22100 Lund, Sweden
First published on 23rd April 2025
Abstract
In this manuscript, we create a new hetero dyad consisting of two electron acceptors with nearly isoenergetic HOMO and LUMO levels, namely perylene diimide (PDI) and aza dioxa triangulenium (ADOTA). This dyad system displays an unusual and reversible excited state electron transfer process. Upon excitation, the dyad shows complete energy transfer from the locally excited PDI to the ADOTA moiety in ∼1 ps, followed by photoinduced electron transfer (PET), forming oxidized PDI and reduced ADOTA. While this PET process is fast (kPET≈ 150 ps), the reversibility establishes an equilibrium between fluorescent locally excited ADOTA and the dark charge shifted PET state. We investigate the formation of and decay from this unusual reversible excited state electron transfer system by fs transient absorption and time-resolved fluorescence spectroscopy in different solvent mixtures because the solvent modulates the deactivation rate of the PET state. Electrochemistry confirms that both the local HOMOs and LUMOs of PDI and ADOTA are nearly isoenergetic but can be shifted by solvent polarity, which elucidates the reason for the unusual reversible electron transfer process and its sensitivity to the solvent. We further investigate near degeneracy of the LUMOs through spectroscopy of the chemically reduced dyad. We find that there is an equilibrium between the reduction of the cationic ADOTA to a neutral dyad, which is favored in DCM. However, in DMF, we find reduction of the PDI leads to formation of the zwitterionic dyad.
Introduction
Hetero dyads and multi chromophoric systems are interesting constructs in which supramolecular properties such as energy transfer and photoinduced electron transfer (PET) can be utilized for energy harvesting and charge separation.1–4 Such constructs usually consist of well-defined energy/electron donor and acceptor units supporting the efficient directional flow energy and charge. In contrast to this design paradigm, we here investigate a perylene diimide–aza dioxa triangulenium hetero dyad (PDI–ADOTA) deliberately designed with nearly isoenergetic local HOMO and LUMO levels on the two units to explore the possibilities of reversible electron and energy transfer.PDI is a highly versatile molecular building block in functional optoelectronic materials and supramolecular nanostructures.5–9 PDI acts as an efficient light absorber and antenna system for light harvesting due to its high molar absorption coefficient or as the energy acceptor and emitter in emissive materials due to its high photostability and rate of emission. The relatively low LUMO energy of PDI also makes it an excellent electron acceptor material in photovoltaics (PV) and in fluorescent probes based on reductive PET.10–12 It is well established that supramolecular systems are designed with clear rationale for which parts are energy or electron donors and which are acceptors, with S1 energies and spectral overlaps being key properties for energy transfer, and the relative HOMO/LUMO energies being the key for electron transfer.13 While PDI can serve both as donor and acceptor for energy transfer, it almost always serves as the electron acceptor in PVs and fluorescent probes due to its relatively low reduction potential.
Previously we designed and created a dyad system with PDI as the light absorbing antenna to amplify the brightness of a DAOTA triangulenium emitter with long fluorescence lifetime to allow for the first time-gated fluorescence imaging with single molecule sensitivity (Fig. 1a).14 This dyad system showed 100% energy transfer from locally excited PDI to locally excited DAOTA in less than 1 ps, in agreement with the more redshifted absorption and emission of the DAOTA. However, inspection of the relative HOMO and LUMO energies reveals that electron transfer from the excited DAOTA to PDI is energetically favorable by a large margin – yet we did not see this (despite the 18 ns long lifetime of the excited DAOTA). The dyad worked perfectly with properties of the expected supramolecular sum of the components according to their roles as energy donors and acceptors. In this case PET apparently is kinetically prohibited due to weak electronic coupling of both DAOTA and PDI through the phenyl-imide linker separating them.17,18
 | ||
| Fig. 1 (a) Structure of PDI–DAOTA dyad from ref. 14, and (b) new PDI–ADOTA dyad, along with their expected local HOMO and LUMO orbital energies, based on reduction potentials and optical E00 values of the individual components of PDI and DAOTA and on the measurements on PDI–ADOTA. Reduction potentials of DAOTA from ref. 15, and PDI from ref. 16, E00 values for DAOTA and PDI from ref. 14. | ||
While the PDI–DAOTA dyad perfectly fulfills its intended function as a bright long fluorescence lifetime fluorophore and demonstrates the power of supramolecular design of dyes, it also makes us speculate what properties will emerge from dyad systems when the roles as energy/electron donor and acceptor are ambiguous. When orbital and excited state energies are close to being degenerate between the molecular building blocks, we hypothesize that the role as donor or acceptor will be decided by details like conformation or local environment, providing novel emergent properties or sensor responses. To explore this avenue of near degenerate hetero dyads, we designed a dyad using the more electron deficient triangulenium derivative, ADOTA and a phenoxy-substituted PDI (Fig. 1b). Hereby bringing both HOMO and LUMO energies of the two units in close proximity of each other. By using a phenoxy linker in the bay position of the PDI we furthermore increase electronic contact between PDI and linker, thus enabling electron transfer processes between PDI and ADOTA.
For this nearly degenerated acceptor–acceptor hetero dyad, we find that the small energy separation between the local orbitals on PDI and ADOTA enables unusual reversible electron transfer in both the ground and excited states. By employing changes in solvent polarity, we can fine tune orbital energies and electron transfer rates and thus impose large changes in both photophysical and redox properties.
Results and discussion
We synthesized the PDI–ADOTA dyad and studied its photophysical properties in multiple solvents and different redox states. We assemble the dyad by a convergent synthesis from the functional building blocks in Scheme 1 (Experimental details in ESI†). Phenol-ADOTA (ADOTA-OH) and Br-PDI were each synthesized according to literature procedures.19,20 We introduce ADOTA-OH in the final step to the “bay” position of PDI via a SNAr reaction to yield the desired PDI–ADOTA dyad. As reference compounds for the dyad we also synthesized PDI-OPh and ADOTA-Ref (Scheme 1). | ||
| Scheme 1 Synthesis of PDI–ADOTA dyad by base catalyzed SNAr reaction in N-methyl-2-pyrrolidone (NMP), and structures of the reference compounds PDI-OPh and ADOTA-Ref. | ||
Fig. 2 display the UV-vis absorption spectra of the dyad and its reference compounds in dichloromethane (DCM). The lowest energy transition in the dyad absorption spectrum is observed as a shoulder at 545 nm with intensity and position corresponding to the first transition in ADOTA-Ref. The three following intense peaks at 523, 487, and 455 nm correspond to typical PDI vibronic peaks. Compared to the reference compound, PDI-OPh, the peaks are sharper with a slight blue shift of ∼7 nm. We assign the redshift and broadening in PDI-OPh to the electron donating effect and flexibility of the phenoxy group.21–25 However, when the phenoxy linker is connected to the electron deficient ADOTA cation the typical sharp vibronic bands of PDI are reinstated.25 Beside this effect, the dyad spectrum resembles a superposition of the two building blocks (see overlay in Fig. S1†), suggesting only a weak electronic coupling between the two dye systems.
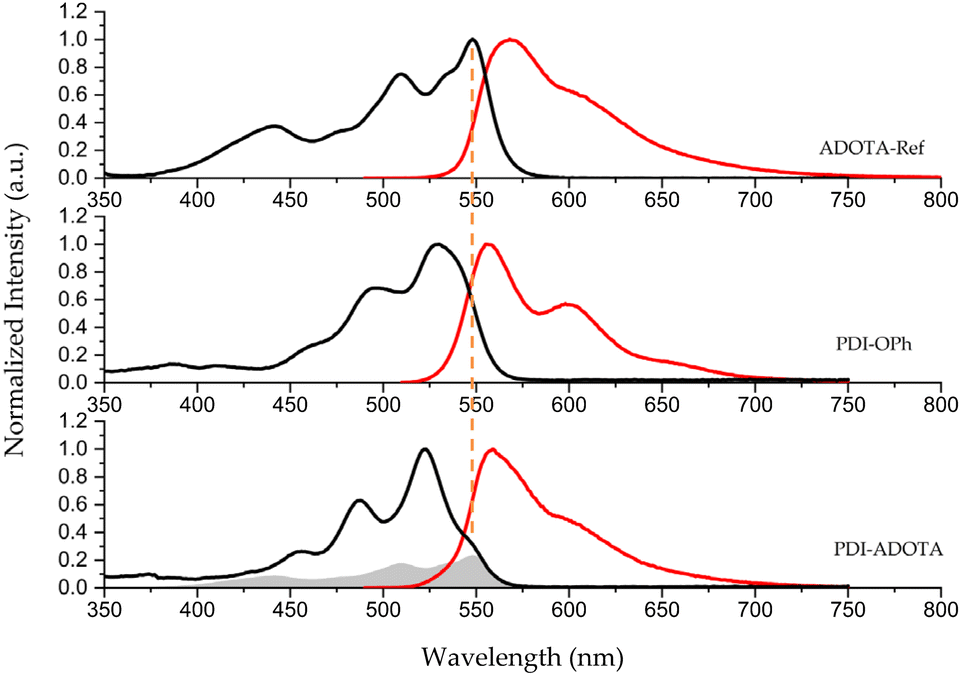 | ||
| Fig. 2 Normalized absorption (black), and emission (red) spectra for ADOTA-Ref, PDI-OPh and PDI–ADOTA in DCM solution. Grey trace is ADOTA-Ref scaled to relative molar absorption coefficients. | ||
The lowest energy absorption in the dyad, highlighted with an orange line, is thus assigned to a local excitation in the less intense absorbing ADOTA chromophore (ε ≈ 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 s−1, Table 1). An overview of optical properties and HOMO–LUMO energies are given in Table S1.†
900 M−1 s−1, Table 1). An overview of optical properties and HOMO–LUMO energies are given in Table S1.†
| λ abs (nm) | λ emi (nm) | ε (M−1 cm−1) | τ (ns) | Φ f (%) | k f (106 s−1) | k nr (106 s−1) | |
|---|---|---|---|---|---|---|---|
| a Intensity weighted average fluorescence lifetime. b Measured using pr-ADOTA as the reference (Φf = 83%).26 c k f = Φf/τ. d k nr = (1/τ) − kf. | |||||||
| ADOTA-Ref | 548 | 568 | 14900 |
19.4 | 73 | 38 | 14 |
| PDI-OPh | 530 | 555 | 46000 |
4.6 | 86 | 186 | 31 |
| PDI–ADOTA (sh) | 545 | 559 | 22700 |
11.2 | 31 | 28 | 62 |
| PDI–ADOTA | 523 | — | 62900 |
— | — | — | — |
| PDI–ADOTA with 50 v% ACN | 522 | 560 | 65600 |
1.0 | 3.1 | 31 | 970 |
The emission spectrum of PDI–ADOTA shows no dependency on excitation wavelength and the excitation spectra match the absorption spectra (Fig. S2†), supporting that the dyad is the only emissive species present. The similarity of the emission spectra of PDI and ADOTA however makes it impossible to assign the nature of the emitting state just based on spectral shape. Fluorescence lifetimes and quantum yields for the dyad and reference compounds measured in DCM are compiled in Table 1. The fluorescence lifetime of the dyad is found to be 11.2 ns while PDI-OPh and ADOTA-Ref display lifetimes of 4.6 and 19.4 ns, respectively. It is also found that the fluorescence quantum yield of the dyad is significantly reduced, Φ = 0.31, compared to quantum yields above 0.7 for each of the dyad building blocks.
From the fluorescence lifetimes and quantum yields, it is possible to calculate the rate of fluorescence (kf) and the rate of non-radiative decay (knr), see Table 1. We find that kf for PDI–ADOTA is similar to ADOTA-Ref, thereby confirming that the emissive state in the dyad is a local excited state on the ADOTA unit. This is the case, no matter if the PDI or the ADOTA component is excited, proving that quantitative excitation energy transfer (EET) from PDI to ADOTA takes place in the dyad. The lower fluorescence quantum yield of the dyad is due to a fourfold increase in the total rate on non-radiative deactivation (knr), suggesting the appearing of a new deactivation pathway in the dyad.
Considering the dyad design, we suspect this new deactivation process to be a result of PET. To further elucidate this process, we conducted fluorescence measurements of the dyad in DCM with increasing amounts of the more polar acetonitrile (ACN). While the reference systems only show minor changes in properties upon addition of ACN (Tables S2, S3 and Fig. S3†) the dyad shows significant drop in both fluorescence lifetime and quantum yield (Table S2 and Fig. S5†). For a solvent mixture with 50% ACN, the lifetime is reduced to 1 ns and the quantum yield to 3.1% compared to 31% in pure DCM (Table 1). While kf is practically unaffected, knr is enhanced by a factor of 15. To arrive at a detailed quantitative model for energy and electron transfer in the PDI–ADOTA dyad, a more comprehensive analysis of the fluorescence data as well as electrochemistry and femtosecond transient absorption (fs-TA) is required, as will be presented in the following sections.
To facilitate the analysis and discussion of these data we here first present a model of the PDI–ADOTA dyad. Fig. 3 summarizes the important states, and the processes connecting these: that is, the locally excited (LE) states on the PDI and ADOTA units, and the charge shifted PET state formed by electron transfer from PDI to LE ADOTA.
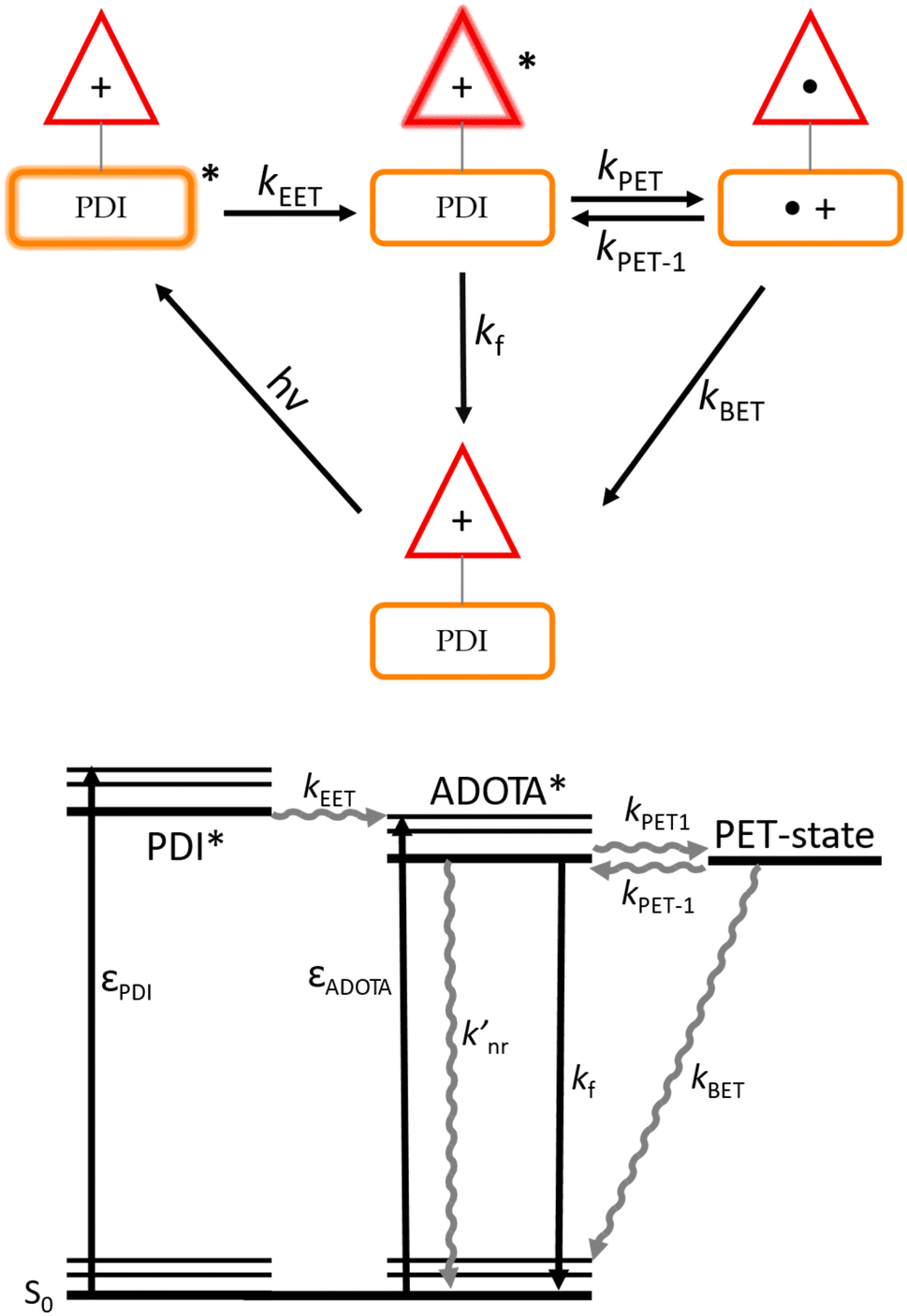 | ||
| Fig. 3 Top: model of various excited and charge shifted states of the PDI–ADOTA dyad and the processes connecting these states. Bottom: corresponding Jablonski diagram. | ||
As discussed above, the emissive state is, in all solvent mixtures, the LE ADOTA. As a consequence of this, and in agreement with the match between absorption and excitation spectra (Fig. S2†), 100% EET from LE PDI to LE ADOTA must take place, similar to what was found for the previous reported PDI–DAOTA dyad.14 We find that the LE ADOTA is in fast equilibrium with a charge shifted PET state where the cationic charge is shifted from ADOTA to PDI, forming the oxidized PDI and reduced ADOTA (Fig. 3). The lower fluorescence quantum yield and lifetime is assigned to deactivation of the PET state by non-radiative back electron transfer (BET) yielding the dyad ground state. The derived non-radiative rates knr for the dyad are thus a weighted sum of the “intrinsic” non-radiative rate of the LE ADOTA  and kBET from the PET state. Before we derive this model, we first study the driving force for the PET process, dictated by the relative HOMO energies, which are found by cyclic voltammetry (CV) and optical spectroscopy (see Fig. S7–11† for reproduced CVs).
and kBET from the PET state. Before we derive this model, we first study the driving force for the PET process, dictated by the relative HOMO energies, which are found by cyclic voltammetry (CV) and optical spectroscopy (see Fig. S7–11† for reproduced CVs).
The formal reduction potentials (E0′) found by CV are reported in Table 2. For ADOTA-Ref the first one-electron reduction forming the neutral ADOTA radical is reversible as expected and occurs at potentials matching with previous reported values.15,27,28 Likewise, the first two one-electron reductions of PDI-OPh are reversible and follow the standard scenario for PDI compounds.16,24,29
When examining the PDI–ADOTA three sequential one-electron reductions is observed, in good agreement with the reference compounds and confirming that the coupling between the chromophores is weak (Fig. S8†). The first reduction for PDI–ADOTA matches well with the reduction of ADOTA and the next two reductions matches with the reduction of the PDI moiety (Table 2). Since the oxidation potentials are not accessible by CV, the HOMO energies are estimated from the reduction potentials and the optical transitions. Taking the reduction potential as the energy of the LUMO and subtracting the energy of the S1 state (E00) results in the energy of the corresponding HOMO. E00 for ADOTA in the dyad is found as the average between the red shoulder in the absorption (546 nm), and λmax of the emission (559 nm). The HOMO energy of the PDI in the dyad is found by adding half of the Stokes shift found for PDI-OPh to the vibronic (0,0′) peak at 523 nm in the dyad spectrum (Fig. 2 and Table S1†). The estimated energies of local HOMOs and LUMOs are summarized in Fig. 4. It is clear that PDI-OPh and the PDI unit in the dyad have higher lying HOMO energy levels compared to ADOTA, thereby making the reductive PET favorable. However, the driving force for PET is quite small since the difference in HOMO energy is only 70 meV. The small energy difference between the local HOMO levels of the PDI and ADOTA units is why reversible PET (kPET-1) is possible.30
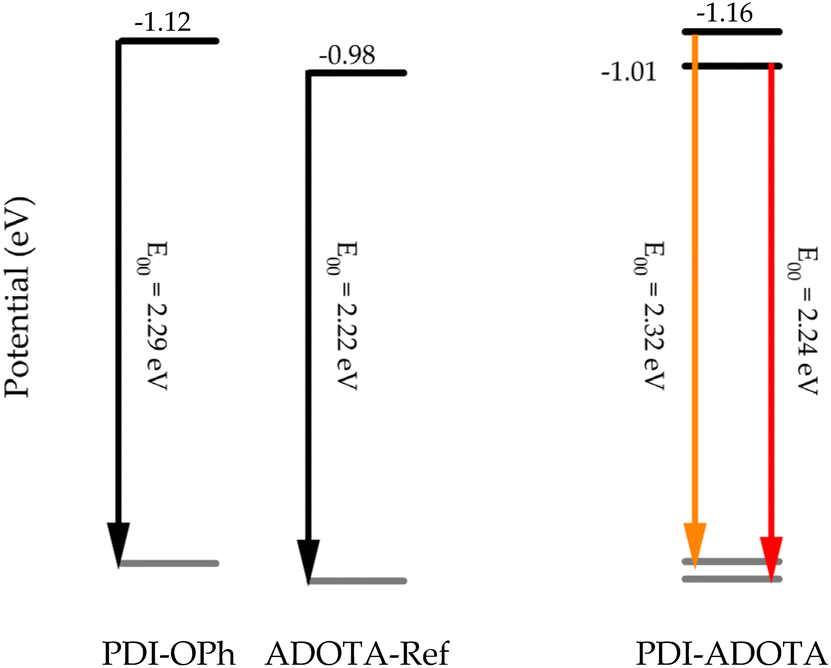 | ||
| Fig. 4 Relative energies in eV of HOMOs and LUMOs of the references and the dyad based on reduction potentials and E00 energies of the chromophores in DCM. | ||
To further understand and map both the excited state energy transfer and the PET process in the dyad, we measured fs-TA in both DCM and 50 v% ACN in DCM. Fig. 5a shows the TA results for 50 v% ACN while TA measurements for PDI–ADOTA in pure DCM are shown in Fig. S16.† Pumping the PDI–ADOTA dyad at 535 nm should, according to the absorption spectra, result in preferential excitation of the PDI chromophore (by a factor of 2.5, Fig. S1†). The PDI-OPh (and PDI in general) shows very distinct excited state absorption (ESA) signals at 700 and 900 nm where ADOTA only has a very weak ESA signal at 660 nm (Fig. S12†). When comparing the TA spectra of PDI-OPh and PDI–ADOTA at early times (100–800 fs) in both solvent mixtures, it is clear the spectral shape is the same (black trace, Fig. 5b and S13†), confirming the presence of the LE PDI state shortly after excitation of the dyad. The LE PDI state in the dyad however decays in ∼1 ps (Fig. 5c), and a new excited state is formed, which is supported by the appearance of a new blue shifted ESA signal (Fig. 5b, blue trace). The new state is similar to that of LE ADOTA (Fig. S14†). We take this as a sign of fast and efficient EET process from PDI to ADOTA, as outlined in our model (Fig. 3). The lifetime of the LE PDI state was found by carrying out a global fitting with a sequential model (details in ESI†), resulting in τPDI* = 0.8 ± 0.2 ps, for both solvent mixtures, showing that the energy transfer process is not influenced by the solvent (lifetimes of states from TA are compiled in Table S4†).
 | ||
| Fig. 5 (a) Transient absorption spectra of PDI–ADOTA in 50 v% ACN. (b) TA spectral traces of the three states found in PDI–ADOTA at times equal to dotted lines in (a) with the same color. (c) fs-TA decay traces at 900 nm (black dot-dash line in (a)), assigned to LE PDI for PDI–ADOTA in DCM and 50 v% ACN. (d) fs-TA decay trace at 585 nm (black dot-dash line in (a)), assigned to the PDI radical cation, showing the grow-in and subsequent decay of the PET-state in DCM and 50 v% ACN. The solid lines in (c) and (d) are obtained from global analysis. | ||
The short lifetime of the initial LE PDI state also means that no emission in the dyad will occur from PDI since the rate of energy transfer (kEET) is in the range of 1012 s−1, which is 104 times faster than the radiative rate of PDI (kf ≈ 108 s−1, Table 1). Within several hundred ps after LE ADOTA is formed, a new characteristic peak at 585 nm appears in the TA spectrum and becomes dominant at around t = 0.5 ns (red line in Fig. 5a and red trace Fig. 5b). Based on the relative PDI and ADOTA HOMO energies we speculated that this could be a signature of electron transfer from PDI to ADOTA. Indeed, this 585 nm peak matches perfectly with the reported absorption for an oxidized radical cation of PDI by Salbeck et al. and has a similar excited state absorption to that reported by R. J. Lindquist et al. for a photooxidized radical cation of a perylene monoimide.16,31 In the context of the dyad, this corresponds to formation of the charge shifted PET state illustrated in Fig. 3. Interestingly, the grow-in of the 585 nm signal is independent of solvent with a time constant of 150 ± 10 ps (τADOTA*) based on global fitting (Fig. 5d), corresponding to a forward PET rate kPET1 of 6 × 109 s−1. Only the subsequent decay back to the ground state is strongly dependent on solvent, with time constants of τPET-ACN = 0.9 ns in 50% ACN/DCM and τPET-DCM ≫ 3 ns in pure DCM. These ground state recovery time constants agree well with the observed fluorescence lifetimes of 1.0 ns and 11 ns (Table 1), showing that both the emissive LE ADOTA state and the PET state are present and decays with the same time constants. Since the TA spectral profile is not changing after equilibrium is established (t ∼300 ps, Fig. S15†) the LE ADOTA and the PET state must be present in a constant ratio, and thus be connected by an equilibrium that is faster than the reactions back to the ground state. That is, kPET and kPET-1 ≫  and kBET. By comparison of the evolution-associated spectra of the LE ADOTA to PET state equilibrium obtained in DCM and with 50 v% ACN (Fig. S16†), we find very little difference, proving that the equilibrium distribution is nearly the same in both solvents. Based on the TA and fluorescence data, we thus find that the solvent polarity only affects the non-radiative decay of the PET state (kBET), which is responsible for the observed fluorescence quenching. Polarity dependence of electron transfer rates in donor acceptor systems are most often rationalized according to changes in driving force due to the large change in when neutral donor and acceptors form charged PET states.32 This is not the case due to the ground state charge of ADOTA which leads to a charge shifted PET state with the overall same +1 charge (Fig. 3). In agreement with this, no solvent effect is observed for the forward electron transfer to the PET state. While the kinetic data (from TA and fluorescence) clearly show that polar ACN accelerate back electron transfer we have found no prior studies exploring this effect for comparable systems.
and kBET. By comparison of the evolution-associated spectra of the LE ADOTA to PET state equilibrium obtained in DCM and with 50 v% ACN (Fig. S16†), we find very little difference, proving that the equilibrium distribution is nearly the same in both solvents. Based on the TA and fluorescence data, we thus find that the solvent polarity only affects the non-radiative decay of the PET state (kBET), which is responsible for the observed fluorescence quenching. Polarity dependence of electron transfer rates in donor acceptor systems are most often rationalized according to changes in driving force due to the large change in when neutral donor and acceptors form charged PET states.32 This is not the case due to the ground state charge of ADOTA which leads to a charge shifted PET state with the overall same +1 charge (Fig. 3). In agreement with this, no solvent effect is observed for the forward electron transfer to the PET state. While the kinetic data (from TA and fluorescence) clearly show that polar ACN accelerate back electron transfer we have found no prior studies exploring this effect for comparable systems.
With the three subsequent situations established by the fs-TA, we return to the details of the time-resolved fluorescence data. Fluorescence lifetimes of ADOTA-Ref and PDI-OPh are both mono-exponential and match well with previous reported values for the two fluorophores (19.3 ns and 4.5 ns respectively, Table S2†). In all solvent mixtures, the observed fluorescence decays of the dyad contain three components (Table S2†): the dominant component is in all cases τ1 that varies from 11.4 ns in DCM to 0.7 ns in 50 v% ACN and is assigned to the LE ADOTA state in equilibrium with the PET state, in agreement with the ground state recovery of the fs-TA. In all solvent mixtures, a fast component of τ2 = 0.2 ns is observed, which we now assign to emission from initially formed population of LE ADOTA states prior to formation of the PET state. This agrees with the fs-TA time constant of τADOTA* = 150 ps for this situation. A third very small and constant lifetime component τ3 ≈ 5 ns is observed in all samples. We have not yet been able to assign this component and it is discussed further in the ESI.† The time dependent spectra of the three states shown in Fig. 5b matches well with evolution associated spectra obtained through global analysis of all the TA data shown in Fig. S16,† which also confirms the time constants (Table S4†). Summing up the TA and fluorescence data, we find that LE ADOTA is the lowest excited state in the system and that ultra-fast EET lead to formation of this state from LE PDI with 100% efficiency in less than 1 ps after excitation (Fig. 3). In a reductive PET process, LE ADOTA is reduced to the neutral radical state by receiving an electron from PDI. This PET process happens with a rate of kPET ≈ 6 × 109 s−1 which is quite slow for covalently linked donor–acceptor systems, but agrees well with the small driving force ΔGPET ≈ 70 mV, as defined by the small energy difference between the local HOMO levels on PDI and ADOTA in the dyed (Table 2). This small energy difference also explains the very unusual fast equilibrium between the LE ADOTA state and the PET state where an electron is shuffling back and forth between PDI and ADOTA. Assuming a one-to-one equilibrium, this corresponds to more than 50 times within the lifetime of the excited state (in DCM).
In the PDI–ADOTA dyad PDI takes on the role of electron donor for the more electron deficient ADOTA. This is contrary to most other dyad systems containing PDI but has been reported for some photo voltage systems.31,33,34
The local LUMO levels in the dyad are not as well-aligned as the HOMO levels (Table 2). However, we speculated that we might be able to tune the LUMO levels to reduce the 150 meV difference in energy. The first indication of this was the slight broadening of the absorption of PDI–ADOTA in 50 v% ACN in DCM, which resulted in the disappearance of the shoulder at 545 nm in the spectra (Fig. S4†). From the fs-TA we know that the HOMO energy levels are unaffected by the solvent (no change in PET equilibrium) hence we speculate that it is the LUMO which is shifted when adding ACN. We therefore hypothesized that by increasing the polarity even further we might be able to bring the LUMOs closer in energy. We probed this by measuring reduction potentials in DCM and DMF, and by characterizing the optical properties in DMF and THF as function of titration with varying equivalents of cobaltocene, a chemical reductant. In DMF, a slight redshift for the absorption and emission is found, with λabs = 528 nm and λemi = 564 nm, although the emission is quenched to a large extend (Fig. S6†). The CV of the dyad in DCM (Fig. 6c and Table 2) clearly shows three distinct one electron reductions assigned to; first a one elctron reduction of ADOTA and secondly two sequential one electron reductions of PDI, as discussed above and illustrated in Fig. 6a. However, in DFM the first reduction wave is broad and contains both one elctron reduction of the PDI and the ADOTA units of the dyad (Fig. 6c). This is confirmed by the CVs of the reference compounds that clearly show the first one-electron reduction in DMF occur at almost the same potential for ADOTA and PDI (Fig. S8† and Table 3). This means that in DMF we have lowered the LUMO of the PDI sufficiently for it to be degenerate in energy with the LUMO of ADOTA, resulting in simultaneous reduction of the two unites, as illustrated in Fig. 6b.
 | ||
| Fig. 6 (a and b) Graphic illustrations of the sequential reduction processes in DCM and THF. (c) Cyclic voltammograms of PDI–ADOTA in DCM and DMF, top and bottom, respectively. (d) Absorption spectra of dyad with 0 (black), 1 (red), 2 (blue), and 3 (green) equivalents of Cp2Co in THF and DMF, top and bottom, respectively. | ||
| E 0′ (1st) | E 0′ (2nd) | |
|---|---|---|
| a Values reported in V vs. Fc/Fc+. Electrochemical methods are found in the ESI. | ||
| ADOTA-Ref | −0.93 | — |
| PDI-OPh | −0.95 | −1.22 |
| PDI–ADOTA | −0.93 | −1.22 |
We further probed this degeneracy by measuring the absorption of reduced ADOTA and PDI in the dyad in both polar and non-polar solvent. Here we speculate that using 1 equivalent of chemical reductant in non-polar solvent will reduce the ADOTA chromophore forming the neutral radical, while in highly polar solvent we expect some reduction occurring on the PDI unit, resulting in the formation of its radical anion as illustrated in Fig. 6a and b respectively. For stability reason, we had to change the non-polar solvent from DCM to THF, but the polarity is very similar.35 As a chemical reductant, we chose cobaltocene (CoCp2) with a reduction potential of −1.33 V vs. Fc/Fc+.36 The reduced PDI-OPh has a clear absorption signal around 700 nm, with multiple other peaks in the range of 800–1000 nm, in agreement with the reported spectra of the radical anion of PDI (Fig. S25†).16 For the ADOTA radical no absorption is observed, except in the UV-region (Fig. S26†), this is most likely due to the radical being unstable at the longer time scale and forming products that only have absorption in the UV-range. The radical species are however expected to have weak absorption based on previous reports on absorption of reduced related triangulenium compounds.37,38 When looking at the absorption of PDI–ADOTA with 0–3 equivalents of CoCp2 in THF it is clear that after addition of 1 equivalent the only change is a drop in the absorption at 525 nm, and no signal appears in the 600–1000 nm region (Fig. 6d, top, red trace). This agrees with reduction of ADOTA since no PDI radical peak at above 600 nm appears. The drop observed in the main absorption peak at 525 nm is ∼20%, which matches well with the relative molar absorption coefficients of PDI and ADOTA (εPDI = 46000 M−1 cm−1 and εADOTA = 10000 M−1 cm−1). Upon addition of further one equivalent CoCp2 the clear peaks of reduced PDI appears (Fig. 6d, top, blue trace, and overlay in Fig. S27†) a further increase of this signal is found when adding a third equivalent of reductant (Fig. 6d). The titration was repeated in DMF and interestingly upon addition the first equivalent of CoCp2 clear PDI radical peaks appears, proving the formation of reduced PDI (Fig. 6d, bottom, red trace). This observation proves that in DMF the LUMOs are so close in energy that a ground state equilibrium between the neutral radical on ADOTA and the radical anion on PDI is established. This solvent dependency agrees with the reduction potentials found from CV (Fig. 6b, Tables 2 and 3). We rationalize that these solvent dependent redox properties are rooted in the large difference in polarity of the dyad depending on which unit is reduced: one-electron reduction of ADOTA yields an overall neutral dyad while reduction of the PDI unit results in a polar zwitterionic dyad, the former being favored in low polarity conditions (THF and DCM) and the latter in DMF (Fig. 6a and b). The possibility of toggling between different reduction sites depending on solvent polarity is a characteristic feature of this new hetero dyads with almost degenerate orbital energies.
Conclusions
By combining neutral (PDI) and cationic (ADOTA) electron deficient π-systems with aligned orbital energies, we obtained an acceptor–acceptor dyad with ambiguous electron transfer properties. Despite the alignment of orbital energies, we find that electronic coupling is weak enough that the individual π-systems are nearly unperturbed in the ground and local excited states, however transfer of energy and electrons is highly efficient and gives rise to a not previously reported feature of dyad systems: fast reversible electron transfer in the excited state. This establishes an equilibrium between the emissive LE ADOTA state and a non-emissive charge shifted PET state comprising the neutral ADOTA radical and the cationic PDI radical. Non-radiative deactivation of this PET state (kBET) is highly sensitive to solvent polarity and acts, due to the fast equilibrium, as a fluorescence quenching mechanism for the dyad in polar solvents.In a more polar solvent (DMF) we also managed to tune the local LUMO energies into being close to degenerate. This results in simultaneous partial reduction of both units using a chemical reductant. Our study shows that hetero dyads with nearly isoenergetic local HOMOs and LUMOs can produce emergent hybrid characteristics with large changes in photophysical and redox properties in response to minor changes in the local environment and is therefore an interesting design strategy for the development of sensors and responsive materials.
Data availability
Experimental data are available from corresponding author upon reasonable request.Author contributions
JDJ, CN and BWL conceived the study. JDJ carried out the synthesis, steady-state spectroscopy, fluorescence lifetime measurements and electrochemical measurements. JC carried out the TA measurement and related data analysis with YH assisting. SL handled the chemical reduction measurements and data analysis. JDJ and BWL wrote the original manuscript. All other authors commented and reviewed the manuscript and figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. J. and B. W. L. acknowledges funding support from the Novo Nordisk Foundation (NNF20OC0062176). This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 871124 Laserlab-Europe. J. C. acknowledges funding support from the Novo Nordisk Foundation (NNF22OC0073582). Y. H. acknowledges the support from the China Scholarship Council (202006150002).Notes and references
- H.-Q. Peng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Biological Applications of Supramolecular Assemblies Designed for Excitation Energy Transfer, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS PubMed.
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Förster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents, Chem. Soc. Rev., 2020, 49, 5110–5139 RSC.
- P. A. Liddell, D. Kuciauskas, J. P. Sumida, B. Nash, D. Nguyen, A. L. Moore, T. A. Moore and D. Gust, Photoinduced Charge Separation and Charge Recombination to a Triplet State in a Carotene−Porphyrin−Fullerene Triad, J. Am. Chem. Soc., 1997, 119, 1400–1405 CrossRef CAS.
- Y. Hou, X. Zhang, K. Chen, D. Liu, Z. Wang, Q. Liu, J. Zhao and A. Barbon, Charge separation, charge recombination, long-lived charge transfer state formation and intersystem crossing in organic electron donor/acceptor dyads, J. Mater. Chem. C, 2019, 7, 12048–12074 RSC.
- S. R. Peurifoy, E. Castro, F. Liu, X. Y. Zhu, F. Ng, S. Jockusch, M. L. Steigerwald, L. Echegoyen, C. Nuckolls and T. J. Sisto, Three-Dimensional Graphene Nanostructures, J. Am. Chem. Soc., 2018, 140, 9341–9345 CrossRef CAS.
- M. Mayländer, K. Kopp, O. Nolden, M. Franz, P. Thielert, A. Vargas Jentzsch, P. Gilch, O. Schiemann and S. Richert, PDI–trityl dyads as photogenerated molecular spin qubit candidates, Chem. Sci., 2023, 14, 10727–10735 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Perylene Bisimide Dye Assemblies as Archetype Functional Supramolecular Materials, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- J. M. Bradley, A. F. Coleman, P. J. Brown, Y. Huang, R. M. Young and M. R. Wasielewski, Harvesting electrons and holes from photodriven symmetry-breaking charge separation within a perylenediimide photosynthetic model dimer, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2313575120 CrossRef CAS PubMed.
- H. Langhals, A. J. Esterbauer, A. Walter, E. Riedle and I. Pugliesi, Förster Resonant Energy Transfer in Orthogonally Arranged Chromophores, J. Am. Chem. Soc., 2010, 132, 16777–16782 CrossRef CAS PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Non-fullerene acceptors for organic solar cells, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Molecular helices as electron acceptors in high-performance bulk heterojunction solar cells, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- D. Aigner, S. A. Freunberger, M. Wilkening, R. Saf, S. M. Borisov and I. Klimant, Enhancing photoinduced electron transfer efficiency of fluorescent pH-probes with halogenated phenols, Anal. Chem., 2014, 86, 9293–9300 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer-Verlag New York Inc., New York, 3 Revised edn, 2006 Search PubMed.
- L. Kacenauskaite, N. Bisballe, R. Mucci, M. Santella, T. Pullerits, J. S. Chen, T. Vosch and B. W. Laursen, Rational Design of Bright Long Fluorescence Lifetime Dyad Fluorophores for Single Molecule Imaging and Detection, J. Am. Chem. Soc., 2021, 143, 1377–1385 CrossRef CAS PubMed.
- J. D. Jensen, N. Bisballe, L. Kacenauskaite, M. S. Thomsen, J. Chen, O. Hammerich and B. W. Laursen, Utilizing Selective Chlorination to Synthesize New Triangulenium Dyes, J. Org. Chem., 2021, 86, 17002–17010 CrossRef CAS PubMed.
- J. Salbeck, H. Kunkely, H. Langhals, W. Saalfrank and J. Daub, Elektronentransfer (ET)-Verhalten von Fluoreszenzfarbstoffen, Chimia, 1989, 6–9 CrossRef CAS.
- H. Langhals, Cyclic carboxylic imide structures as structure elements of high stability. Novel developments in perylene dye chemistry, Heterocycles, 1995, 1, 477–500 CrossRef.
- C. Li and H. Wonneberger, Perylene Imides for Organic Photovoltaics: Yesterday, Today, and Tomorrow, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- M. Rosenberg, A. K. R. Junker, T. J. Sørensen and B. W. Laursen, Fluorescence pH Probes Based on Photoinduced Electron Transfer Quenching of Long Fluorescence Lifetime Triangulenium Dyes, Chemphotochem, 2019, 3, 233–242 CrossRef CAS.
- P. Rajasingh, R. Cohen, E. Shirman, L. J. W. Shimon and B. Rybtchinski, Selective Bromination of Perylene Diimides under Mild Conditions, J. Org. Chem., 2007, 72, 5973–5979 CrossRef CAS PubMed.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, A recyclable supramolecular membrane for size-selective separation of nanoparticles, Nat. Nanotechnol., 2011, 6, 141–146 CrossRef CAS PubMed.
- M.-J. Lin, Á. J. Jiménez, C. Burschka and F. Würthner, Bay-substituted perylene bisimide dye with an undistorted planar scaffold and outstanding solid state fluorescence properties, Chem. Commun., 2012, 48, 12050 RSC.
- R. F. Kelley, W. S. Shin, B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, Photoinitiated Charge Transport in Supramolecular Assemblies of a 1,7,N,N‘-Tetrakis(zinc porphyrin)-perylene-3,4:9,10-bis(dicarboximide), J. Am. Chem. Soc., 2007, 129, 3173–3181 CrossRef CAS PubMed.
- D. Inan, R. K. Dubey, N. Westerveld, J. Bleeker, W. F. Jager and F. C. Grozema, Substitution Effects on the Photoinduced Charge-Transfer Properties of Novel Perylene-3,4,9,10-tetracarboxylic Acid Derivatives, J. Phys. Chem. A, 2017, 121, 4633–4644 CrossRef CAS PubMed.
- D. Ke, C. Zhan, S. Xu, X. Ding, A. Peng, J. Sun, S. He, A. D. Q. Li and J. Yao, Self-Assembled Hollow Nanospheres Strongly Enhance Photoluminescence, J. Am. Chem. Soc., 2011, 133, 11022–11025 CrossRef CAS PubMed.
- S. A. Bogh, M. Simmermacher, M. Westberg, M. Bregnhoj, M. Rosenberg, L. De Vico, M. Veiga, B. W. Laursen, P. R. Ogilby, S. P. A. Sauer and T. J. Sørensen, Azadioxatriangulenium and Diazaoxatriangulenium: Quantum Yields and Fundamental Photophysical Properties, ACS Omega, 2017, 2, 193–203 CrossRef CAS PubMed.
- J. D. Jensen, R. K. Jakobsen, Z. Yao and B. W. Laursen, Investigating Design Rules for Photoinduced Electron Transfer Quenching in Triangulenium Probes, Chem.–Eur. J., 2023, e202301077 CrossRef.
- S. Dileesh and K. R. Gopidas, Photoinduced electron transfer in azatriangulenium salts, J. Photochem. Photobiol., A, 2004, 162, 115–120 CrossRef CAS.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, Electrochemistry, Spectroscopy and Electrogenerated Chemiluminescence of Perylene, Terrylene, and Quaterrylene Diimides in Aprotic Solution, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- D. Rehm and A. Weller, Kinetics Of Fluorescence Quenching By Electron And H-Atom Transfer, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- R. J. Lindquist, B. T. Phelan, A. Reynal, E. A. Margulies, L. E. Shoer, J. R. Durrant and M. R. Wasielewski, Strongly oxidizing perylene-3,4-dicarboximides for use in water oxidation photoelectrochemical cells, J. Mater. Chem. A, 2016, 4, 2880–2893 RSC.
- M. R. Wasielewski, Photoinduced electron transfer in supramolecular systems for artificial photosynthesis, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, Intramolecular Charge-Transfer Tuning of Perylenes: Spectroscopic Features and Performance in Dye-Sensitized Solar Cells, J. Phys. Chem. C, 2007, 111, 15137–15140 CrossRef CAS.
- D. K. Panda, F. S. Goodson, S. Ray and S. Saha, Dye-sensitized solar cells based on multichromophoric supramolecular light-harvesting materials, Chem. Commun., 2014, 50, 5358–5360 RSC.
- J. P. Cerón-Carrasco, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraïdi, Solvent polarity scales: determination of new ET(30) values for 84 organic solvents, J. Phys. Org. Chem., 2014, 27, 512–518 CrossRef.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- T. J. Sørensen, M. F. Nielsen and B. W. Laursen, Synthesis and Stability of N,N '-Dialkyl-1,13-dimethoxyquinacridinium (DMQA(+)): A [4]Helicene with Multiple Redox States, ChemPlusChem, 2014, 79, 1030–1035 CrossRef.
- R. Gueret, L. Poulard, M. Oshinowo, J. Chauvin, M. Dahmane, G. Dupeyre, P. P. Lainé, J. Fortage and M.-N. Collomb, Challenging the [Ru(bpy)3]2+ Photosensitizer with a Triazatriangulenium Robust Organic Dye for Visible-Light-Driven Hydrogen Production in Water, ACS Catal., 2018, 8, 3792–3802 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01397f |
| This journal is © The Royal Society of Chemistry 2025 |