
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards trans-dual deuterated cyclopropanes via photoredox synergistic deuteration with D2O†
Yuanqing
Wu‡
a,
Chuxiong
Peng‡
a,
Qichen
Zhan‡
 a,
Xudong
Lou
a,
Shijie
Liu
a,
Xiaofeng
Lin
a,
Yulin
Han
*e,
Peng
Cao
*abcd and
Tao
Cao
*a
a,
Xudong
Lou
a,
Shijie
Liu
a,
Xiaofeng
Lin
a,
Yulin
Han
*e,
Peng
Cao
*abcd and
Tao
Cao
*a
aState Key Laboratory of Technologies for Chinese Medicine Pharmaceutical Process Control and Intelligent Manufacture, School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, Jiangsu 210023, China. E-mail: cao_peng@njucm.edu.cn; caot@njucm.edu.cn
bThe Quzhou Affiliated Hospital of Wenzhou Medical University, Quzhou People's Hospital, Quzhou, Zhejiang 324000, China
cJiangsu Provincial Medical Innovation Center, Affiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu 210028, China
dGaoyou Hospital of Traditional Chinese Medicine, Yangzhou, Jiangsu 225600, China
eKey Laboratory of Pollution Exposure and Health Intervention of Zhejiang Province, Interdisciplinary Research Academy, Zhejiang Shuren University, Hangzhou, Zhejiang 310015, China. E-mail: yulin.han@zjsru.edu.cn
First published on 22nd April 2025
Abstract
As the demand for deuterated compounds continues to rise in medicinal chemistry, various methods have been developed to incorporate deuterium atoms. Among these, achieving consecutive trans-dual deuteration remains a challenging task. We have designed a novel strategy to synthesize trans-dual deuterated cyclopropanes at adjacent carbon positions. This approach involves H/D exchange followed by a photocatalyzed deuteroaminomethylation of cyclopropenes, with deuterium oxide serving as the sole deuterium source. The reaction is carried out under mild conditions and exhibits a broad substrate scope, high diastereoselectivity, and promising potential for further applications, making it an attractive transformation for future studies.
Due to the pronounced H/D isotope effect, deuterated compounds often exhibit significantly different thermodynamic and kinetic properties compared to their non-deuterated counterparts.1 Substituting hydrogen atoms with deuterium in biologically active compounds can alter their pharmacological profiles, offering a straightforward yet effective approach to improving pharmacokinetics and metabolic stability (Fig. 1a).2 Since the FDA's approval of deutetrabenazine in 2017 (ref. 3)—the first deuterated drug, used to treat Huntington's chorea and tardive dyskinesia—an increasing number of deuterated drugs have been developed for various diseases.4 Examples include donafenib (for non-small-cell lung cancer),5 deucravacitinib (for psoriasis and other autoimmune disorders),6 and VV-116 (for COVID-19 (ref. 7) and RSV8).
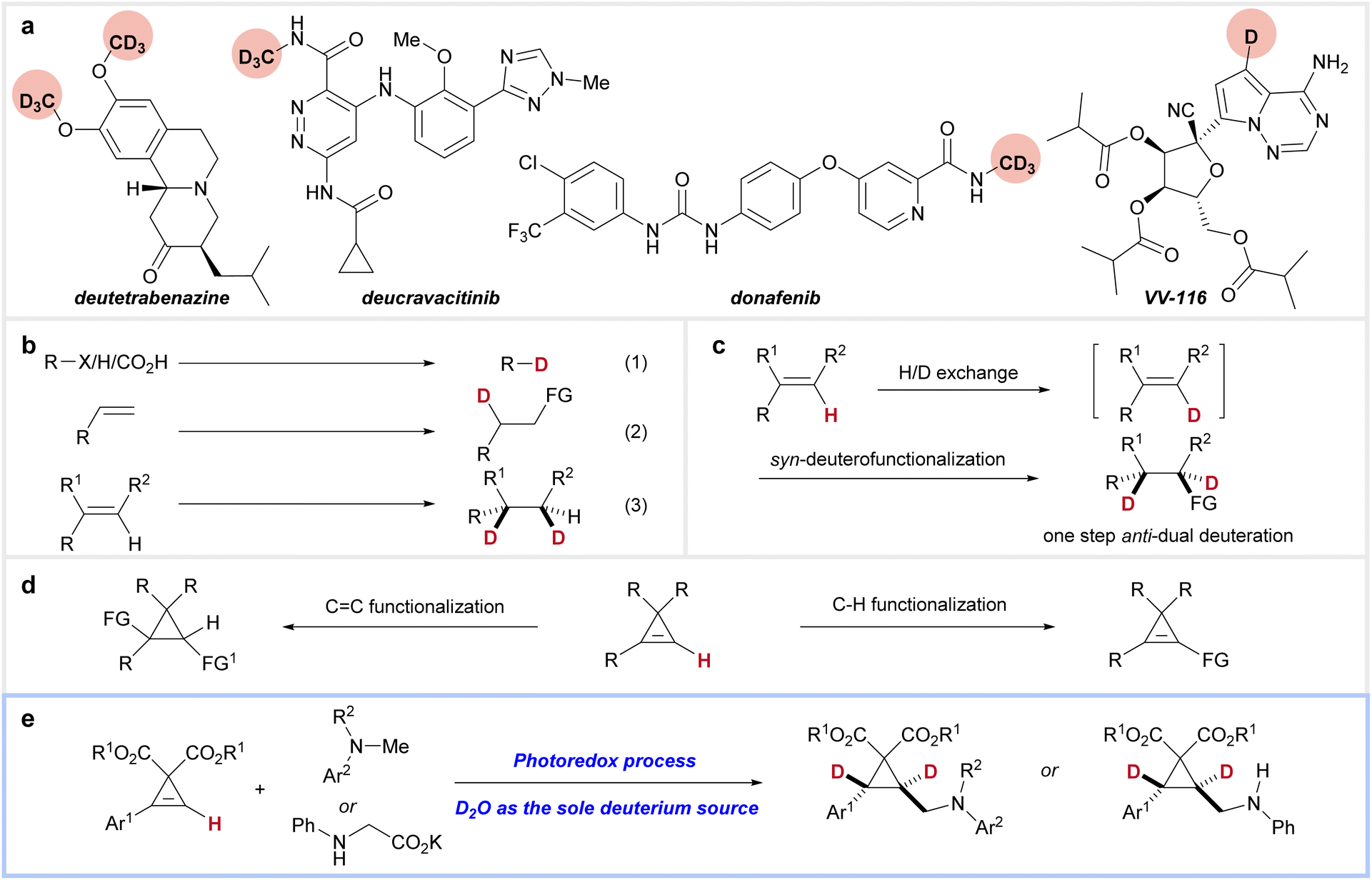 | ||
| Fig. 1 Background of this study. (a) Examples of deuterated drugs. (b) Reported strategies for synthesizing deuterated compounds. (c) Our strategy for trans-dual deuteration. (d) Functionalization pathways of cyclopropenes. (e) This work for the synthesis of trans-dual deuterated cyclopropanes. | ||
Given the significance of deuterated compounds, strategies for their synthesis have garnered considerable attention from synthetic chemists.9 Approaches such as dehalogenative deuteration,10 H/D exchange (hydrogen isotope exchange),11 and decarboxylative deuteration12 offer straightforward methods for introducing a deuterium atom (Fig. 1b, eqn (1)). An alternative strategy involves the deuterofunctionalization of alkenes, a versatile method for synthesizing functionalized aliphatic chains (Fig. 1b, eqn (2)).13 While these methods typically incorporate only one deuterium atom into the products, reductive deuteration of alkenes provides a direct route to dual-deuterated compounds at adjacent carbon atoms with either syn-selectivity or a lack of stereoselectivity (Fig. 1b, eqn (3)).11e,14 However, to the best of our knowledge, highly selective anti-dual deuteration of alkenes remains a significant challenge.
We envisioned that a synergistic combination of H/D exchange at a reactive C–H bond in alkenes and syn-deuterofunctionalization could enable dual deuteration at consecutive carbons, yielding trans-selective dual deuteration that is unattainable by existing methods (Fig. 1c). This approach provides highly regio- and stereoselective dual deuteration while avoiding scrambling to undesired positions. Due to their strained structures, readily available cyclopropenes exhibit high reactivity toward both transition-metal-catalyzed and radical-mediated additions (Fig. 1d).15 Additionally, the alkenyl C–H bond in cyclopropenes is relatively reactive, making them excellent substrates for our strategy.16 Herein, we report our recent results, achieving the synthesis of trans-dual deuterated cyclopropanes via synergistic H/D exchange-photocatalyzed deuteroaminomethylation of cyclopropenes using deuterium oxide as the sole deuterium source (Fig. 1e).
Cyclopropene S1 was initially selected as the starting material to react with diphenylmethylamine (S2) under the catalysis of 4CzIPN and blue LED irradiation. Using potassium carbonate as the base and acetonitrile as the solvent, the addition product (1) was obtained in 40% yield (Table 1, entry 1). Optimization of the reaction conditions revealed that K2HPO4 and NMP were more effective as the base and solvent, respectively (Table 1, entries 2 and 3). Interestingly, the addition of a small amount of water slightly improved the reaction yield (Table 1, entry 4). Inspired by the beneficial effect of water, we hypothesized that replacing it with deuterium oxide (D2O) might yield deuterated products. Indeed, dual deuteration of 1 was observed, but the non-benzylic position exhibited only 44% deuterium incorporation (Table 1, entry 5). We attributed this low incorporation to the hydrogen atoms in K2HPO4. Consequently, we screened other hydrogen-free bases and found that potassium carbonate significantly improved deuterium incorporation (Table 1, entries 6–8). With potassium carbonate as the base and increasing the D2O content to 10%, we achieved 94% and 95% deuterium incorporation at the benzylic and non-benzylic positions, respectively, with a yield of 77% (Table 1, entry 9). However, increasing the D2O content further to 15% did not enhance deuterium incorporation and led to a significant decrease in yield (Table 1, entry 10). The reaction was completely suppressed when conducted under an air atmosphere or using pure D2O as the solvent (Table 1, entries 11 and 12). Alternative photocatalysts such as eosin Y and g-C3N4 proved ineffective (Table 1, entries 13 and 14). However, when Ir(ppy)2(dtbbpy)PF6 was used as the photocatalyst, the reaction proceeded smoothly, albeit with a slightly decreased yield of 1 and lower deuteration at the D2 position (Table 1, entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Yield of 1b (%) | D1 (%) | D2 (%) |
| a Reaction conditions: S1 (0.1 mmol), S2 (0.3 mmol), photocatalyst (0.005 mmol), and base (0.3 mmol) in solvent (1 mL) irradiated with blue LEDs at room temperature overnight. b Isolated yield. c Not detected. d Under an air atmosphere. e Eosin Y (5 mol%) as the photocatalyst. f g-C3N4 (5 mg) as the photocatalyst. g Ir(ppy)2(dtbbpy)PF6 (1 mol%) as the photocatalyst. | |||||
| 1 | K2CO3 | CH3CN | 40 | N.D.c | |
| 2 | K2HPO4 | CH3CN | 66 | N.D. | |
| 3 | K2HPO4 | NMP | 79 | N.D. | |
| 4 | K2HPO4 | NMP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (95:5) :H2O (95:5) |
85 | N.D. | |
| 5 | K2HPO4 | NMP:D2O (95:5) |
86 | 87 | 44 |
| 6 | K2CO3 | NMP:D2O (95:5) |
83 | 88 | 86 |
| 7 | Cs2CO3 | NMP:D2O (95:5) |
79 | 85 | 86 |
| 8 | K3PO4 | NMP:D2O (95:5) |
76 | 84 | 85 |
| 9 | K 2 CO 3 |
NMP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :D
2
O (90:10) :D
2
O (90:10)
|
77 | 94 | 95 |
| 10 | K2CO3 | NMP:D2O (85:15) |
61 | 95 | 95 |
| 11d | K2CO3 | NMP:D2O (90:10) |
0 | N.D. | |
| 12 | K2CO3 | D2O | 0 | N.D. | |
| 13e | K2CO3 | NMP:D2O (90:10) |
0 | N.D. | |
| 14f | K2CO3 | NMP:D2O (90:10) |
0 | N.D. | |
| 15g | K 2 CO 3 |
NMP:D
2
O (90:10)
|
75 | 96 | 93 |

With the optimized conditions in hand, we investigated the scope of this reaction (Fig. 2). For phenyl-substituted cyclopropenes reacting with diphenylmethylamine, halogen substituents such as bromine (2) and fluorine (3) were well tolerated, and chlorine atoms at the para- (4), meta- (5), and ortho-positions (6) did not affect the reaction. Other electron-withdrawing groups, including trifluoromethyl (7) and ester groups (8), were also compatible with this transformation. Cyclopropenes bearing electron-donating substituents produced the corresponding dual-deuterated cyclopropanes efficiently (9–11). Additionally, substrates featuring ethyl, tert-butyl, and benzyl ester groups were suitable for this reaction (12–14). It should be noted that when 3 mmol of S1 was used, the yield remained unaffected, allowing for a gram-scale synthesis of 1.
 | ||
| Fig. 2 Substrate scope for the synthesis of trans-dual deuterated cyclopropanes. Reaction conditions: cyclopropene (0.1 mmol), amine (0.3 mmol), 4CzIPN (0.005 mmol), and K2CO3 (0.3 mmol) in a mixture of NMP and D2O (90:10, 1 mL) irradiated with blue LEDs at room temperature overnight. aReaction on a 3 mmol scale to afford 0.96 g of 1. bIr(ppy)2(dtbbpy)PF6 (1 mol%) was used. c4CzIPN (10 mol%) was used. | ||
Beyond diphenylmethylamine, other tertiary amines such as phenyldimethylamine (15), phenylethylmethylamine (16), and phenylisopropylmethylamine (17) also performed well under these conditions. Substituted phenyldimethylamines were further explored, with substrates bearing chlorine atoms at the ortho- and meta-positions (18 and 19) or a fluorine atom at the para-position (20) showing no adverse effects. Substrates with electron-donating groups such as methoxy (21), dimethyl (22), and methyl (23) groups, as well as electron-withdrawing ester groups (24), were also tolerated. Notably, an ethynyl group (25) on the substrate retained the C–C triple bond, demonstrating the higher reactivity of the cyclopropene moiety compared to the alkyne moiety. Notably, the alkynyl C–H bond was deuterated at 68% during the reaction. Additionally, a carbazole derivative (26) was successfully synthesized, further highlighting the versatility of this transformation.
Interestingly, under the standard conditions, potassium phenylglycinate was also found to be suitable for the anti-selective dual deuteration reaction via a photoredox decarboxylation process (27).12,17 This approach provides dual-deuterated cyclopropanes bearing a secondary aminomethyl substituent. Furthermore, cyclopropenes bearing alkyl groups (28–30) or halogen atoms (31–34) were successfully employed in this process, affording the corresponding products in good yields. The trans-selectivity of this transformation was unambiguously confirmed by the single-crystal X-ray diffraction analysis of compound 27.
When benzylmethylphenylamine S3 was used, product 35 was obtained smoothly. However, the deuteration content at D1 was only 65%, while the benzyl position (D3) exhibited 43% deuteration (eqn (4)). This outcome was attributed to the scrambling of reactive benzylic hydrogens. Nevertheless, substrates S4, S5, and S6 did not undergo the desired reaction (eqn (5)).

The applicability of this transformation was demonstrated through the modification of functional molecules (Fig. 3a and b). By introducing a phenylmethylamino group, several biologically active moieties were incorporated into the reaction, expanding its potential applications (Fig. 3a). Consequently, products derived from ibuprofen (36), gemfibrozil (37), adamantane (38), and dihydrocholesterol (39) were successfully synthesized. Furthermore, hydrolysis of compound 1 yielded the diacid intermediate 1-diacid, which subsequently underwent condensation with biologically active alcohols to afford compounds 40 and 41 (Fig. 3b). These transformations enabled the installation of a dual-deuterated cyclopropyl warhead.
 | ||
| Fig. 3 Synthetic application of this reaction. (a) Modification of complex molecules. (b) Transformation of 1 into compounds 40 and 41. (c) Synthesis of compound 42. (d) AIE properties of S7 and 42. (e) DLS analysis of S7 and 42. aReaction conditions: cyclopropene (0.1 mmol), amine (0.3 mmol), 4CzIPN (0.005 mmol), and K2CO3 (0.3 mmol) in a mixture of NMP and D2O (90:10, 1 mL) irradiated with blue LEDs at room temperature overnight. b4CzIPN (10 mol%) was used. | ||
Additionally, compound 42, incorporating a tetraphenylethylene moiety, was synthesized via the reaction of S1 with S7 under standard conditions (Fig. 3c). Given the widespread use of tetraphenylethylene in constructing materials with aggregation-induced emission (AIE) properties,18 we compared the AIE behavior of S7 and 42 (Fig. 3d). Notably, under identical conditions, 42 exhibited a stronger AIE effect than its precursor S7. The aggregation of both compounds was confirmed by DLS (Fig. 3e) and TEM (Fig. S1 in the ESI†) analyses, revealing that the average size of aggregated 42 (986.6 nm) was larger than that of S7 (746.9 nm). This increased aggregation size may contribute to the enhanced AIE effect, as larger aggregates often lead to a more restricted molecular environment. Additionally, we hypothesize that the rigid cyclopropane core in 42 reduces molecular flexibility and restricts intramolecular rotation or vibration in the aggregated state. This enhanced rigidity likely suppresses non-radiative decay pathways, which typically quench emission, thereby further amplifying the AIE effect compared to the more flexible S7.19
To investigate the possible mechanism of this reaction, several control experiments were conducted. The addition of TEMPO under standard conditions completely suppressed the reaction; instead, the adduct product S2-TEMPO was detected by HRMS, indicating that the transformation proceeds via a radical mechanism (Fig. 4a). In a separate experiment, the reaction of S1 with potassium carbonate in the absence of S2 and without blue LED irradiation led to the formation of S1-D, with deuteration occurring at the olefinic hydrogen at 96% (Fig. 4b). This result demonstrates that the acidic olefinic hydrogen can undergo H/D exchange during the reaction.16a A light on/off experiment (Fig. 4c), along with a measured quantum yield of 0.30 (see the ESI† for details), ruled out the possibility of a chain process in this reaction. Furthermore, Stern–Volmer quenching experiments were performed. As shown in Fig. 4d, the fluorescence of the photocatalyst 4CzIPN was quenched by the addition of various concentrations of S2, while no quenching was observed upon the addition of S1. These results suggest that electron transfer occurs between the photocatalyst 4CzIPN and S2.
 | ||
| Fig. 4 Mechanistic studies. (a) Trap of the radical intermediate by TEMPO. (b) Deuteration of S1. (c) A light on/off experiment. (d) Stern–Volmer quenching experiments. (e) A proposed mechanism. | ||
Based on the above results, a plausible mechanism has been proposed (Fig. 4e). Potassium carbonate deprotonates the olefinic hydrogen of S1, generating the S1-anion, which is then deuterated by D2O to form S1-D. Concurrently, upon irradiation, the photocatalyst (PC) is activated to its excited state (PC*), where it undergoes a single electron transfer (SET) process with S2, followed by deprotonation, generating intermediate Int 1 and a radical anion (PC˙−). Int 1 then undergoes a radical addition to S1-D, forming a more thermodynamically stable trans-cyclopropyl radical intermediate (Int 2vs.Int 2′). Int 2 reacts with PC˙−via a SET process to yield the cyclopropyl anion intermediate (Int 3) and regenerate the ground-state PC. Finally, deuteration of Int 3 leads to the formation of product 1, with the thermodynamically favoured trans-selective product being formed during the process.
Conclusions
In conclusion, we have developed a trans-selective dual deuteration strategy for cyclopropenes by combining H/D exchange and syn-deuterofunctionalization. The mild reaction conditions, broad substrate scope, high diastereoselectivity, and significant deuterium incorporation make this transformation highly attractive. Post-modification of biologically active molecules and the synthesis of an AIE compound further highlight its potential applications in related fields. Ongoing work in this laboratory focuses on extending this strategy to other unsaturated compounds and conducting more detailed mechanistic studies.Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
Y. W., C. P., and Q. Z.: investigation, methodology, and writing – review and editing. X. Lou, S. L., and X. Lin: investigation and methodology. Y. H.: investigation, methodology, and project administration. P. C.: project administration, funding acquisition, and writing – review and editing. T. C.: project administration, funding acquisition, and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Key R&D Program of China (2023YFC2308200), the National Natural Science Foundation of China (22107052), and the Nanjing University of Chinese Medicine (XPT22107052) is greatly acknowledged.Notes and references
- J. Yang, Deuterium: Discovery and Applications in Organic Chemistry, Elsevier, Amsterdam, 2016 Search PubMed.
- (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed; (b) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed; (c) T. G. Gant, J. Med. Chem., 2013, 57, 3595–3611 CrossRef PubMed.
- Y.-A. Heo and L. J. Scott, Drugs, 2017, 77, 1857–1864 CrossRef CAS PubMed.
- (a) R. M. C. Di Martino, B. D. Maxwell and T. Pirali, Nat. Rev. Drug Discovery, 2023, 22, 562–584 CrossRef CAS PubMed; (b) H. M. Chandra Mouli, A. Vinod, S. Kumari, A. K. Tiwari, M. K. Kathiravan, V. Ravichandiran and R. Peraman, Bioorg. Chem., 2023, 135, 106490 CrossRef CAS PubMed.
- S. J. Keam and S. Duggan, Drugs, 2021, 81, 1915–1920 CrossRef CAS PubMed.
- S. T. Wrobleski, R. Moslin, S. Lin, Y. Zhang, S. Spergel, J. Kempson, J. S. Tokarski, J. Strnad, A. Zupa-Fernandez, L. Cheng, D. Shuster, K. Gillooly, X. Yang, E. Heimrich, K. W. McIntyre, C. Chaudhry, J. Khan, M. Ruzanov, J. Tredup, D. Mulligan, D. Xie, H. Sun, C. Huang, C. D'Arienzo, N. Aranibar, M. Chiney, A. Chimalakonda, W. J. Pitts, L. Lombardo, P. H. Carter, J. R. Burke and D. S. Weinstein, J. Med. Chem., 2019, 62, 8973–8995 CrossRef CAS PubMed.
- (a) H.-j. Qian, Y. Wang, M.-q. Zhang, Y.-c. Xie, Q.-q. Wu, L.-y. Liang, Y. Cao, H.-q. Duan, G.-h. Tian, J. Ma, Z.-b. Zhang, N. Li, J.-y. Jia, J. Zhang, H. A. Aisa, J.-s. Shen, C. Yu, H.-l. Jiang, W.-h. Zhang, Z. Wang and G.-y. Liu, Acta Pharmacol. Sin., 2022, 43, 3130–3138 CrossRef CAS PubMed; (b) Y. Xie, W. Yin, Y. Zhang, W. Shang, Z. Wang, X. Luan, G. Tian, H. A. Aisa, Y. Xu, G. Xiao, J. Li, H. Jiang, S. Zhang, L. Zhang, H. E. Xu and J. Shen, Cell Res., 2021, 31, 1212–1214 CrossRef CAS PubMed.
- R. Zhang, Y. Zhang, W. Zheng, W. Shang, Y. Wu, N. Li, J. Xiong, H. Jiang, J. Shen, G. Xiao, Y. Xie and L. Zhang, Signal Transduction Targeted Ther., 2022, 7, 123 CrossRef CAS PubMed.
- (a) G. Prakash, N. Paul, G. A. Oliver, D. B. Werz and D. Maiti, Chem. Soc. Rev., 2022, 51, 3123–3163 RSC; (b) S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed; (c) N. Li, Y. Li, X. Wu, C. Zhu and J. Xie, Chem. Soc. Rev., 2022, 51, 6291–6306 RSC; (d) P. L. Norcott, Chem. Commun., 2022, 58, 2944–2953 RSC.
- (a) P. Li, C. Guo, S. Wang, D. Ma, T. Feng, Y. Wang and Y. Qiu, Nat. Commun., 2022, 13, 3774 CrossRef CAS PubMed; (b) X. Wang, M.-H. Zhu, D. P. Schuman, D. Zhong, W.-Y. Wang, L.-Y. Wu, W. Liu, B. M. Stoltz and W.-B. Liu, J. Am. Chem. Soc., 2018, 140, 10970–10974 CrossRef CAS PubMed; (c) C. Liu, Z. Chen, C. Su, X. Zhao, Q. Gao, G.-H. Ning, H. Zhu, W. Tang, K. Leng, W. Fu, B. Tian, X. Peng, J. Li, Q.-H. Xu, W. Zhou and K. P. Loh, Nat. Commun., 2018, 9, 80 CrossRef PubMed; (d) Z.-Z. Zhou, J.-H. Zhao, X.-Y. Gou, X.-M. Chen and Y.-M. Liang, Org. Chem. Front., 2019, 6, 1649–1654 RSC; (e) V. Soulard, G. Villa, D. P. Vollmar and P. Renaud, J. Am. Chem. Soc., 2017, 140, 155–158 CrossRef PubMed; (f) Z. Song, J. Zeng, T. Li, X. Zhao, J. Fang, L. Meng and Q. Wan, Org. Lett., 2020, 22, 1736–1741 CrossRef CAS PubMed; (g) Y. Li, Z. Ye, Y.-M. Lin, Y. Liu, Y. Zhang and L. Gong, Nat. Commun., 2021, 12, 2894 CrossRef CAS PubMed; (h) X. Ling, Y. Xu, S. Wu, M. Liu, P. Yang, C. Qiu, G. Zhang, H. Zhou and C. Su, Sci. China:Chem., 2020, 63, 386–392 CrossRef CAS; (i) D. Wood and S. Lin, Angew. Chem., Int. Ed., 2023, 62, e202218858 CrossRef CAS PubMed.
- (a) C. Teja, S. Kolb, P. Colonna, J. Grover, S. Garcia-Argote, G. K. Lahiri, G. Pieters, D. B. Werz and D. Maiti, Angew. Chem., Int. Ed., 2024, 63, e202410162 CrossRef CAS PubMed; (b) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed; (c) A. Sattler, ACS Catal., 2018, 8, 2296–2312 CrossRef CAS; (d) R. Zhou, L. Ma, X. Yang and J. Cao, Org. Chem. Front., 2021, 8, 426–444 RSC; (e) P. Li, Y. Wang, H. Zhao and Y. Qiu, Acc. Chem. Res., 2024, 58, 113–129 CrossRef PubMed; (f) M. Kramer, D. Watts and A. N. Vedernikov, Organometallics, 2020, 39, 4102–4114 CrossRef CAS; (g) J. Corpas, P. Viereck and P. J. Chirik, ACS Catal., 2020, 10, 8640–8647 CrossRef CAS; (h) T. R. Puleo, A. J. Strong and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 1467–1472 CrossRef CAS PubMed; (i) C. Zarate, H. Yang, M. J. Bezdek, D. Hesk and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 5034–5044 CrossRef CAS PubMed; (j) Z. Zhao, R. Zhang, Y. Liu, Z. Zhu, Q. Wang and Y. Qiu, Nat. Commun., 2024, 15, 3832 CrossRef CAS PubMed; (k) Y. Kuang, H. Cao, H. Tang, J. Chew, W. Chen, X. Shi and J. Wu, Chem. Sci., 2020, 11, 8912–8918 RSC; (l) Q. K. Kang, Y. Li, K. Chen, H. Zhu, W. Q. Wu, Y. Lin and H. Shi, Angew. Chem., Int. Ed., 2022, 61, e202117381 CrossRef CAS PubMed; (m) R. Ogasahara, K. Ban, M. Mae, S. Akai and Y. Sawama, ChemMedChem, 2024, 19, e202400201 CrossRef CAS PubMed; (n) X. Yan, Y. Pang, Y. Zhou, R. Chang and J. Ye, J. Am. Chem. Soc., 2024, 147, 1186–1196 CrossRef PubMed; (o) N. Li, J. Li, M. Qin, J. Li, J. Han, C. Zhu, W. Li and J. Xie, Nat. Commun., 2022, 13, 4224 CrossRef CAS PubMed; (p) A. Uttry, S. Mal and M. van Gemmeren, J. Am. Chem. Soc., 2021, 143, 10895–10901 CrossRef CAS PubMed; (q) J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2020, 11, 1026–1031 RSC.
- (a) N. Li, Y. Ning, X. Wu, J. Xie, W. Li and C. Zhu, Chem. Sci., 2021, 12, 5505–5510 RSC; (b) T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 CrossRef CAS PubMed.
- (a) J. Guo, Z. Cheng, J. Chen, X. Chen and Z. Lu, Acc. Chem. Res., 2021, 54, 2701–2716 CrossRef CAS PubMed; (b) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 CrossRef CAS PubMed; (c) Q. Shi, M. Xu, R. Chang, D. Ramanathan, B. Peñin, I. Funes-Ardoiz and J. Ye, Nat. Commun., 2022, 13, 4453 CrossRef CAS PubMed; (d) D. Ramanathan, Q. Shi, M. Xu, R. Chang, B. Peñín, I. Funes-Ardoiz and J. Ye, Org. Chem. Front., 2023, 10, 1182–1190 RSC.
- (a) H. Li, B. Zhang, Y. Dong, T. Liu, Y. Zhang, H. Nie, R. Yang, X. Ma, Y. Ling and J. An, Tetrahedron Lett., 2017, 58, 2757–2760 CrossRef CAS; (b) S. Guo, X. Wang and J. S. Zhou, Org. Lett., 2020, 22, 1204–1207 CrossRef CAS PubMed; (c) Y. Wang, Q. Wang, L. Wu, K. Jia, M. Wang and Y. Qiu, Nat. Commun., 2024, 15, 2780 CrossRef CAS PubMed; (d) K. Yang, T. Feng and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202312803 CrossRef CAS PubMed; (e) S. Kolb and D. B. Werz, Chem.–Eur. J., 2023, 29, e202300849 CrossRef CAS PubMed; (f) X. Liu, R. Liu, J. Qiu, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2020, 59, 13962–13967 CrossRef CAS PubMed; (g) J. Zhang, C. Mück-Lichtenfeld and A. Studer, Nature, 2023, 619, 506–513 CrossRef CAS PubMed.
- (a) Y. Cohen, A. Cohen and I. Marek, Chem. Rev., 2020, 121, 140–161 CrossRef PubMed; (b) R. Vicente, Chem. Rev., 2020, 121, 162–226 CrossRef PubMed; (c) P. Li, X. Zhang and M. Shi, Chem. Commun., 2020, 56, 5457–5471 RSC; (d) Z.-B. Zhu, Y. Wei and M. Shi, Chem. Soc. Rev., 2011, 40, 5534–5563 RSC; (e) L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415–8434 CrossRef CAS PubMed; (f) G. Lefebvre, O. Charron, J. Cossy and C. Meyer, Org. Lett., 2021, 23, 5491–5495 CrossRef CAS PubMed; (g) N. S. Dange, F. Robert and Y. Landais, Org. Lett., 2016, 18, 6156–6159 CrossRef CAS PubMed; (h) M. Ueda, N. Doi, H. Miyagawa, S. Sugita, N. Takeda, T. Shinada and O. Miyata, Chem. Commun., 2015, 51, 4204–4207 RSC; (i) C. Peng, F. Gu, X. Lin, N. Ding, Q. Zhan, P. Cao and T. Cao, Green Chem., 2023, 25, 671–677 RSC; (j) F. Gu, B. Lin, Z. H. Peng, S. Liu, Y. Wu, M. Luo, N. Ding, Q. Zhan, P. Cao, Z. Zhou and T. Cao, Adv. Sci., 2024, 11, 2407931 CrossRef CAS PubMed; (k) P. Chandu, S. Biswas, S. Garai and D. Sureshkumar, Green Chem., 2023, 25, 9086–9091 RSC; (l) S. Biswas, M. Mallick, G. Nanda K S, P. Chandu and D. Sureshkumar, Org. Lett., 2024, 26, 10207–10212 CrossRef CAS PubMed; (m) Q. Huang, Y. Chen, X. Zhou, L. Dai and Y. Lu, Angew. Chem., Int. Ed., 2022, 61, e202210560 CrossRef CAS PubMed; (n) X.-K. He, L.-Q. Lu, B.-R. Yuan, J.-L. Luo, Y. Cheng and W.-J. Xiao, J. Am. Chem. Soc., 2024, 146, 18892–18898 CrossRef CAS PubMed.
- (a) K. Liu, T. Li, D.-Y. Liu, W. Li, J. Han, C. Zhu and J. Xie, Sci. China:Chem., 2021, 64, 1958–1963 CrossRef CAS; (b) S. Chuprakov, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2005, 127, 3714–3715 CrossRef CAS PubMed.
- (a) J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed; (b) C. L. Ji, Y. N. Lu, S. Xia, C. Zhu, C. Zhu, W. Li and J. Xie, Angew. Chem., Int. Ed., 2025, 64, e202423113 CrossRef CAS PubMed.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (c) H.-T. Feng, Y.-X. Yuan, J.-B. Xiong, Y.-S. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2414708. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00350d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |