
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA unified approach to meta-selective methylation, mono-, di- and trifluoromethylation of arenes†
Elisa Y.
Lai
 ab,
Lutz
Ackermann
*b and
Magnus J.
Johansson
*a
ab,
Lutz
Ackermann
*b and
Magnus J.
Johansson
*a
aMedicinal Chemistry, Research and Early Development, Cardiovascular, Renal and Metabolism (CVRM), Biopharmaceuticals R&D, AstraZeneca, Gothenburg, Pepparedsleden 1, 431 50 Mölndal, Sweden. E-mail: Magnus.J.Johansson2@astrazeneca.com
bInstitut für Organische und Biomolekulare Chemie, Wöhler Research Institute for Sustainable Chemistry (WISCh), Georg-August-Universität Göttingen, Tammannstraße 2, 37077, Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 10th April 2025
Abstract
Matched molecular series (MMS) are series of molecules that differ only by a single modification at a specific site. The synthesis of MMS is a desirable strategy in drug discovery campaigns. Small aliphatic motifs, notably methyl, mono-, di- and trifluoromethyl substituents (C1 units), are known to have profound effects on the physiochemical properties and/or potency of drug candidates. In this context, we herein report a unique strategy for achieving direct meta-selective methylation, mono-, di-, and trifluoromethylation from the same parent compound. This approach takes advantage of a highly meta-selective ruthenium(II)-catalyzed alkylation, followed by a subsequent photocatalyzed protodecarboxylation or silver-mediated fluorodecarboxylation to reveal the (fluoro)methyl moiety. This method enables the late-stage access to MMS in small molecules bearing a variety of orienting groups as well as bio-relevant molecules containing complex functionalities, bypassing the need for de novo synthesis to access individual compounds in a series. Moreover, key physiochemical properties of drug candidates were successfully modulated, highlighting opportunities to accelerate medicinal chemistry programs in a sustainable fashion.
Introduction
In medicinal chemistry, incorporating small C1 aliphatic substituents, including methyl, monofluoromethyl, difluoromethyl and trifluoromethyl groups, has proven to be a valuable strategy for enhancing the pharmacokinetic or pharmacodynamic properties of parent compounds (Fig. 1A). These derivatives are referred to as matched molecular series (MMS) as an extension of the concept of matched molecular pairs (MMP), where a series of compounds share a common scaffold but structurally differ from one another at a single site.1–4 The addition of a methyl group to a target molecule has the potential to enhance the potency or efficacy of a compound, widely recognized as the “magic methyl effect”.5,6 On a different note, fluoromethyl groups, while similar in size to methyl substituents, exert different and unique effects on drug candidate properties.7–10 The presence of fluorine atoms is reported to improve stability by replacing metabolically labile hydrogen atom(s),11–13 to modulate conformation,14–16 or to influence membrane permeability via lipophilicity modulation.17 In addition, mono- and difluoromethyl groups may serve as bioisosteres of hydroxyl and thiol functional groups due to their ability to behave as hydrogen bond donors.18–20 As a result, methyl, mono-, di- and trifluoromethyl analogues of drug candidates can bring immense value in the drug development process, as important biological properties can be positively influenced while leaving core structures intact.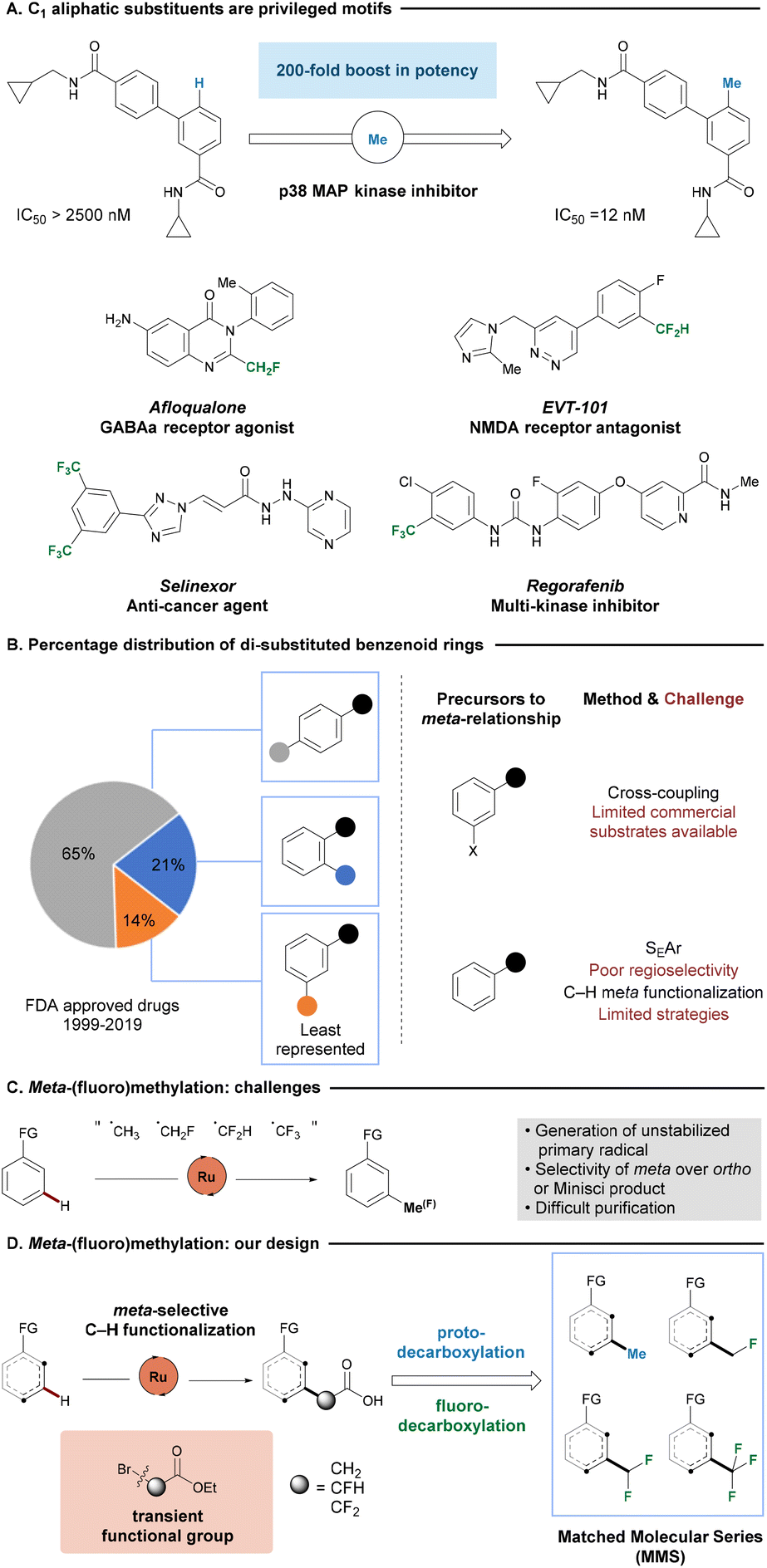 | ||
| Fig. 1 Access to di-substituted arenes. | ||
Benzenoid rings are by far the most privileged ring system present in small molecule drugs.21–24 Considering substitution patterns of arenes, para-substituted phenyl rings significantly outnumber analogues bearing ortho- or meta-substitution patterns, with the latter being the least represented scaffold (Fig. 1B).25 The scarcity of the meta-substitution pattern underscores a considerable unmet need for efficient synthetic methodologies. To access a di-substituted benzenoid without the need for reconstructing the ring, chemists typically either perform functional group interconversions on one of the existing substituents of a di-functionalized ring or introduce a new group to a mono-functionalized ring. The former strategy, typically employed in C–C cross-coupling reactions,26–28 is heavily dependent on the availability of the starting material. However, meta-substituted aryl halides, the most widely used precursors for such transformations, have the poorest commercial availability.29 Alternatively, electrophilic aromatic substitution (SEAr) is one of the most conventional reactions to introduce a new substituent on a phenyl ring. However, the success of SEAr relies on the electronic and steric nature of the existing substituents on the phenyl ring. Electron withdrawing substituents can direct SEAr to meta-functionalization, but often suffer from sluggish reaction rates.30,31 More recently, researchers achieved direct remote C–H functionalization with carefully designed template systems. However, the installation and removal of templates added additional synthetic steps and challenges.32
Various synthetic methods have been developed to incorporate C1-alkyl moieties onto an arene through direct C–H bond functionalization, with the aim of minimizing the number of synthetic steps to the synthesis of desired analogues.33–39 Thus, strategies allowing for ortho-methylations40–43 and trifluoromethylation44–47 have been developed. In sharp contrast, direct C–H ortho-mono- or difluoromethylation, to the best of our knowledge, has not yet been reported. Furthermore, meta-C1-alkylation across all analogues continues to be challenging. Meta-methylation has been elegantly accomplished by Yu.48–50 Yet, di-functionalization remains a largely unsolved issue. Hence, there is a continued strong demand to develop a unified protocol that allows access to all four possible C1 aliphatic substituents to efficiently diversify the parent compound, thereby avoiding the need for lengthy de novo syntheses.
Ruthenium(II/III) catalysis in combination with a carboxylate ligand was reported to be able to direct XAT-substrates towards overall meta-functionalization of an arene bearing an intrinsic Lewis-basic motif.51–60 The initial project design relied on generating small aliphatic radicals directly through a single electron transfer (SET/XAT) process from a ruthenium intermediate.61–63 However, screening a variety of radical precursors proved the transformation to be challenging, mainly due to the instability and short lifetime of the primary radical (Fig. 1C). In addition, the similarity in structure and lipophilicity between the desired product, substrate and side products often resulted in difficult purification.64 Therefore, we considered the (fluoro)methylation, through initial C–H functionalization of an alkylating agent, along with a C–C cleavage would lead to our desired products.
Here in, we report an expedient method to achieve methylation, mono-, di-, and trifluoromethylation from the same parent compound through a common intermediate (Fig. 1D). Our strategy explores the excellent remote selectivity of ruthenium catalysis, in combination with a photocatalyzed protodecarboxylation or silver-mediated fluorodecarboxylation to furnish the desired protonated or fluorinated product.65–67 Our approach proved compatible with a range of different coordinating groups bearing an N(sp2) Lewis-basic motif, which is omnipresent in active pharmaceutical ingredients (API) and related drug-like compounds. This generally applicable protocol also allows for the expedient access of MMS via the late-stage functionalization (LSF) of complex bioactive structures,68,69 and provides opportunities for investigating structure–activity relationships (SAR) of potential drug candidates without lengthy de novo syntheses of individual compound of interest. In addition, a new restriction proposal from the European Union legislation attempts to address the issue of pollution from undegradable per- and polyfluoroalkyl substances (PFAS) by banning the manufacture, supply and use of such substances, with the exception of API.70,71 The restriction prompts scientists to develop alternative routes to synthesize the API without relying on precursors falling into the category of PFAS.72 Our method offers opportunities to address the problem by decorating the target compound with trifluoromethyl group in the final step of synthesis. Finally, our strategy offers complementarity in site-selectivity to small aliphatic substituent incorporation at either proximal ortho-C–H or remote para-C–H bonds on arenes, to expand the accessibility of largely unexplored chemical space.
Results and discussion
We commenced our studies by exploring a range of alkyl halides bearing a transient radical stabilizing group (Scheme S1 in ESI†). BPin, nitrile and BF3K methyl halides were shown to be ineffective in Ru-catalyzed alkylation while BrCH2TMS produced the desired meta product in 9% NMR yield. However, further optimization did not improve the yield and switching to ICH2TMS altered the selectivity to, in this case, the ortho-position. After careful consideration, substrates bearing an ester stabilizing group were chosen to be the ideal candidates. First, the ester group offers excellent site-selectivity to the desired meta position. Second, all the non-, mono- and di-fluorinated substates are commercially available, which could greatly reduce the cost of an industrial application. Third, an acid functional group can be revealed easily from the ester protection group through saponification, which could then be utilized in a subsequent decarboxylative protonation or fluorination step.Next, we explored the scope and limitations of the ruthenium-medi ated alkylation (Scheme 1). We were pleased to observe that the protocol can be applied to a range of different medicinally relevant heteroarenes including pyridine (1a), pyrimidine (1b), quinoline (1c), pyrazole (1d) and thiazole (1e). To our delight, bioactive moieties such as purine nucleobase derivative (1i, 1j), including those bearing sensitive protecting groups, were well-tolerated under the optimized conditions. In addition, para-substituents bearing different electronic properties, including methoxy (1f), methyl (1g) and trifluoromethyl (1h) groups, proved feasible. Surprisingly, when switching from 2-phenylthiazole to 2-phenyloxazole, the mechanism was completely overturned from meta-C–H functionalisation to conventional Friedel Crafts-type reaction.73,74 Disappointingly, dihydrooxazole and imidazopyridine did not furnish any desired products, with only unreacted substrate observed.
 | ||
| Scheme 1 Ruthenium-catalyzed meta-selective alkylation of arenes. aReactions were carried out using [RuCl2(p-cymene)]2 (10 mol%) and P(4-(CF3)C6H4)3 (30 mol%). | ||
We then turned our attention to investigate the one pot saponification-protodecarboxylation, which would lead to our final meta-methylation goal. Conventionally, decarboxylation is performed under harsh reaction conditions, typically requiring heating to over 200 °C, which would pose a challenge to molecules bearing sensitive functional groups. Inspired by a photochemical 12CO2 to 13/14CO2 isotope exchange strategy reported by the Audisio group, we tested our ester substrates under 450 nm blue LED irradiation.75 The generation of alpha fluoro-radicals through photo-decarboxylation was also reported.76,77 A high-throughput experimentation (HTE) optimization campaign was conducted with three different esters bearing zero, one or two fluorine atoms as substrates (ESI, Scheme S2†). The substrates were subjected to three different bases for the saponification step. Then, solvents were removed under vacuum followed by addition of photocatalyst and a new set of solvents. Photocatalyzed decarboxylation was shown to suffer from increasing difficulty going from esters intermediates bearing zero, one, to two fluorine atoms. The presence of more electron-withdrawing fluorine atoms could potentially make the electropotential of the carboxylate ion more positive, thus more difficult to oxidize, hindering the radical decarboxylation. Therefore, we decided to adopt this strategy only to access the privileged methyl substituent. Of note, it is surprising that all three solvents gave satisfying conversion. DMSO was eventually selected for carrying out the scope exploration since it is classified as a greener solvent compared to DMF or DMA. With optimized conditions in hand, we explored the versatility of photodecarboxylation (Scheme 2). All the ester intermediates obtained from the ruthenium(II/III)-catalyzed alkylation step successfully furnished the desired methylation product in satisfactory yields. Deacetylation occurred when acetate intermediate 2j was subjected to saponification, but to our delight, the presence of free hydroxyl group was well tolerated in the subsequent protodecarboxylation. When 10 equivalents of D2O were added in the photocatalyzed reaction, a final CH2D group was successfully formed to yield 6ga, offering opportunities for the late-stage selective deuterium labelling of drug molecules. The success of deuteration opens the door for potential tritiation of compounds of interest.78 In order to obtain mono- and difluoromethylated compounds, a silver-catalyzed fluorodecarboxylation methodology was applied. This strategy also offers a unique opportunity to access trifluoromethylated products, which were inaccessible via protodecarboxylation, and was exemplified in the LSF of bioactive molecules. The non-fluorinated and monofluorinated intermediates that were obtained from either conditions A or conditions B (Scheme 1) were subjected to the same set of conditions for fluorodecarboxylation (Scheme 3). The ester intermediates (2&3) were first hydrolyzed by K2CO3 to furnish the acetates, then a solvent switch in the same reaction vial was performed, followed by the subsequent silver-mediated fluorodecarboxylation. The intermediates from Scheme 1 successfully underwent the saponification-fluorodecarboxylation sequence to furnish the corresponding mono- or difluoromethylated product with the exception of purine derivatives (2i, 2j, 3j). These set of compounds proved to be challenging in the fluorodecarboxylation step. Instead, we opted for protodecarboxylation strategy described in Scheme 2, yet still managed to furnish moderate to good yield of the desired products (7i, 7j, 8j), which highlighted the versatility of the two-step protocol. Of note, all the respective mono- and difluromethylated products were obtained in moderate to good yields. Interestingly, fluorodecarboxylation of the monofluorinated intermediate (8a–8i) generally resulted in higher yields compared to the non-fluorinated counterpart (7a–7i). Li's group reported a detailed study of such transformation, indicating that the reactivity of carboxylic acids drops in the order tertiary > secondary > primary,79 which serves in part to explain the discrepancy in yields we observed from the two sets of intermediates.
 | ||
| Scheme 2 One pot saponification-protodecarboxylation sequence. aTraceless removal of acetyl group during saponification. b10 equiv. D2O were added along with 4CzIPN. cPercentage deuterium incorporation. | ||
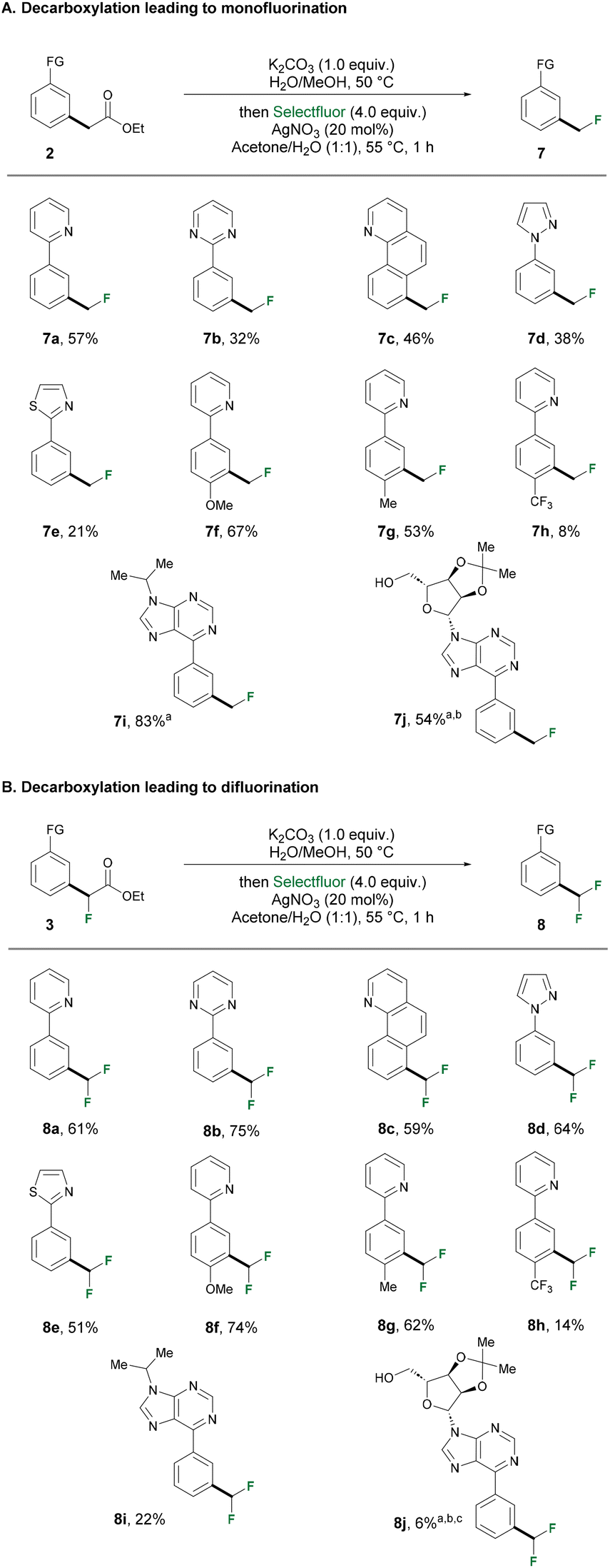 | ||
| Scheme 3 One pot saponification-fluorodecarboxylation sequence. aReaction was carried out with the corresponding ethyl acetate and K2CO3 (2.0 equiv.) in MeOH/MeOH for saponification, followed by irradiation under 450 nm blue light with (Ir[dF(CF3)ppy]2(dtbpy))PF6 (6.0 mol%) in DMF (0.1 M) for 24 h at 35 °C. bTraceless removal of acetyl group during saponification. cConversion determined by LC-MS. | ||
Our newly developed (fluoro)methylation strategy was then applied to more complex bioactive molecules bearing multiple sites for potential functionalization. We were pleased to find that the ruthenium(II/III)-catalyzed alkylation successfully incorporated a masked (fluoro)methyl radical into the desired site of functionalization with traces amount (less than 5% by LC-MS) of ortho-product observed when ethyl bromoacetate was used as the radical precursor (Fig. 2A). In all cases, bis-functionalization was not observed. The targeted meta-product and undesired ortho-product were easily separated owing to their differences in lipophilicity brought about by the transient ester functional group, highlighting an important advantage of the indirect (fluoro)methylation. With emapunil and diazepam as starting material, all four homologues bearing one extra C1 unit at the meta position could be synthesized. In addition, 86% deuterium incorporation was achieved when 10 equivalents of D2O was added during the protodecarboxylation of diazepam-ethyl acetate intermediate to furnish 11aa in 12% yield over 2 steps. Ruthenium-catalyzed alkylation was shown to be compatible with pritelivir to yield the unfluorinated and monofluorinated ester intermediate. Yet, fluorodecarboxylation did not yield the desired product with the latter. Nonetheless, an overall meta-methylation and monofluoromethylation was still achieved producing two analogues of the parent compound. Of note, trimethyltin hydroxide was used to selectively hydrolyze the transient ester functional group since saponification would also hydrolyze the tertiary amide present in the core structure. In any medicinal chemistry campaign, time is one of the most important factors. As a result, LSF is evolving to be one of the most appealing strategies for accessing synthetically challenging target compounds. We compared the preparation of derivatives of pharmaceuticals (9a–9d, 11a–11d, 13a–13b) with traditional de novo synthesis to produce identical compounds. In this context, expedient access to these unique sets of MMS brings significant value to the study of physiochemical properties and potency of drug candidates, thus speeding up the DMTA cycle in drug discovery.
 | ||
| Fig. 2 Late-stage functionalization of bioactive molecules and physiochemical properties modulation. aDetailed reaction conditions are provided in ESI.†bPercentage deuterium incorporation. cUnstable in plasma. dReported in literature.42 | ||
Access to MMS of marketed pharmaceuticals enabled the generation of key DMPK data including solubility, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D and protein binding (Fig. 2B). A general increase in logD was observed when a methyl group was installed. Further replacing the hydrogen atoms by fluorine atoms reduced lipophilicity, resulting in a drop in logD (CH2F and CF2H compared to CH3), agreeing with previous studies.80–82 The trifluoromethylated analogue presented a noticeable increase in lipophilicity compared to the methylated compound, which was consistent with previous report that trifluoromethyl group increased the hydrophilicity when there is no solubilizing group in close proximity.83 Monofluoromethylated and difluoromethylated compounds are in general less lipophilic than the corresponding methylated and trifluoromethylated analogues due to the present of larger dipole moments.84 As the parent compound became more lipophilic (Emapunil > Diazepam > Pritelivir), the boost in logD from –CH3 to –CF3 also became more prominent.85 It is also interesting to note that installation of CH2F group may have a dramatic impact on the solubility of the target compound. Finally, the site of functionalization can modulate the properties of the parent compound differently. The difference in the medicinal chemistry relevant data of methylation occurring at ortho vs. meta position of diazepam could be explained by the perturbation of the conformation of the phenyl ring.42Ortho-methylation has a higher probability of twisting the phenyl ring out of plane from the 7-membered ring due to higher steric congestion.
D and protein binding (Fig. 2B). A general increase in logD was observed when a methyl group was installed. Further replacing the hydrogen atoms by fluorine atoms reduced lipophilicity, resulting in a drop in logD (CH2F and CF2H compared to CH3), agreeing with previous studies.80–82 The trifluoromethylated analogue presented a noticeable increase in lipophilicity compared to the methylated compound, which was consistent with previous report that trifluoromethyl group increased the hydrophilicity when there is no solubilizing group in close proximity.83 Monofluoromethylated and difluoromethylated compounds are in general less lipophilic than the corresponding methylated and trifluoromethylated analogues due to the present of larger dipole moments.84 As the parent compound became more lipophilic (Emapunil > Diazepam > Pritelivir), the boost in logD from –CH3 to –CF3 also became more prominent.85 It is also interesting to note that installation of CH2F group may have a dramatic impact on the solubility of the target compound. Finally, the site of functionalization can modulate the properties of the parent compound differently. The difference in the medicinal chemistry relevant data of methylation occurring at ortho vs. meta position of diazepam could be explained by the perturbation of the conformation of the phenyl ring.42Ortho-methylation has a higher probability of twisting the phenyl ring out of plane from the 7-membered ring due to higher steric congestion.
Conclusion
In summary, we have devised a strategy allowing direct access to a meta-selective C–H methylation and mono-, di-, and trifluoromethylation protocol of arene to quickly access a range of C1-substituted compounds from the same parent molecule. The site-selectivity was tuned by exploiting the unique reactivity of ruthenium catalysis, and the molecular diversity was achieved by either photocatalyzed protodecarboxylation or silver-mediated fluorodecarboxylation. Moreover, this approach was successfully applied to marketed pharmaceuticals to produce MMS of the parent drug. Specifically, trifluoromethylation was achieved in the last step of synthesizing a potential API, thereby bypassing the generation of intermediate that could be classified as PFAS. An array of pharmaceutically relevant analogues was generated quickly, thus could speed up substantially the drug development campaign. In the meantime, the sustainable strategy reduces the CO2 footprint by significantly reducing the number of step counts compared to de novo synthesis. Lastly, the remote incorporation of C1 unit provided opportunities to modulate physiochemical properties of bioactive molecules, presenting the potential of this methodology to accelerate optimization towards suitable drug candidates.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: E. Y. L., M. J. J. and L. A.; methodology: E. Y. L.; writing – original draft: E. Y. L.; writing – review & editing: E. Y. L., M. J. J. and L. A.; funding acquisition: M. J. J. and L. A.; supervision: M. J. J. and L. A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank European Union's Horizon 2020 research and innovation program (Marie Skłodowska-Curie Grant Agreement No. 860762) for financial support, Martina Apell for HRMS analysis, Ileana Guzzetti and Richard Lewis for NMR analysis, Lucas Guillemard for project discussion, Robert Sheppard for helpful comments, Martin Hayes and Rong Zhao for proof-reading and Malin Lemurell for project support. M. J. J. like to thank the Swedish Foundation for Strategic Environmental Research (Mistra SafeChem, project number 2018/11).Notes and references
- M. Awale, S. Riniker and C. Kramer, Matched Molecular Series Analysis for ADME Property Prediction, J. Chem. Inf. Model., 2020, 60, 2903–2914 CrossRef CAS PubMed.
- C. E. Keefer and G. Chang, The use of matched molecular series networks for cross target structure activity relationship translation and potency prediction, MedChemComm, 2017, 8, 2067–2078 RSC.
- C. Tyrchan and E. Evertsson, Matched Molecular Pair Analysis in Short: Algorithms, Applications and Limitations, Comput. Struct. Biotechnol. J., 2017, 15, 86–90 CrossRef CAS PubMed.
- M. Wawer and J. Bajorath, Local Structural Changes, Global Data Views: Graphical Substructure–Activity Relationship Trailing, J. Med. Chem., 2011, 54, 2944–2951 CrossRef CAS PubMed.
- D. Aynetdinova, M. C. Callens, H. B. Hicks, C. Y. X. Poh, B. D. A. Shennan, A. M. Boyd, Z. H. Lim, J. A. Leitch and D. J. Dixon, Installing the “magic methyl” –C–H methylation in synthesis, Chem. Soc. Rev., 2021, 50, 5517–5563 RSC.
- H. Schönherr and T. Cernak, Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of Fluorine in Medicinal Chemistry, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- W. K. Hagmann, The Many Roles for Fluorine in Medicinal Chemistry, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, Fluorine in Medicinal Chemistry, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- G. Rong, C. Wang, L. Chen, Y. Yan and Y. Cheng, Fluoroalkylation promotes cytosolic peptide delivery, Sci. Adv., 2020, 6, eaaz1774 CrossRef CAS PubMed.
- B. M. Johnson, Y.-Z. Shu, X. Zhuo and N. A. Meanwell, Metabolic and Pharmaceutical Aspects of Fluorinated Compounds, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- B. K. Park and N. R. Kitteringham, Effects of Fluorine Substitution on Drug Metabolism: Pharmacological and Toxicological Implications, Drug Metab. Rev., 1994, 26, 605–643 CrossRef CAS PubMed.
- L. E. Zimmer, C. Sparr and R. Gilmour, Fluorine Conformational Effects in Organocatalysis: An Emerging Strategy for Molecular Design, Angew. Chem., Int. Ed., 2011, 50, 11860–11871 CrossRef CAS PubMed.
- L. Hunter, The C–F bond as a conformational tool in organic and biological chemistry, Beilstein J. Org. Chem., 2010, 6, 38 Search PubMed.
- D. Y. Buissonneaud, T. van Mourik and D. O'Hagan, A DFT study on the origin of the fluorine gauche effect in substituted fluoroethanes, Tetrahedron, 2010, 66, 2196–2202 CrossRef CAS.
- Z. Wang, H. R. Felstead, R. I. Troup, B. Linclau and P. T. F. Williamson, Lipophilicity Modulations by Fluorination Correlate with Membrane Partitioning, Angew. Chem., Int. Ed., 2023, 62, e202301077 CrossRef CAS PubMed.
- J. Yang, S. Ponra, X. Li, B. B. C. Peters, L. Massaro, T. Zhou and P. G. Andersson, Catalytic enantioselective synthesis of fluoromethylated stereocenters by asymmetric hydrogenation, Chem. Sci., 2022, 13, 8590–8596 RSC.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed.
- J. A. Erickson and J. I. McLoughlin, Hydrogen Bond Donor Properties of the Difluoromethyl Group, J. Org. Chem., 1995, 60, 1626–1631 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, Rings in Drugs, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- J. Wang and T. Hou, Drug and Drug Candidate Building Block Analysis, J. Chem. Inf. Model., 2010, 50, 55–67 CrossRef CAS PubMed.
- P. Ertl, S. Jelfs, J. Mühlbacher, A. Schuffenhauer and P. Selzer, Quest for the Rings. In Silico Exploration of Ring Universe To Identify Novel Bioactive Heteroaromatic Scaffolds, J. Med. Chem., 2006, 49, 4568–4573 CrossRef CAS PubMed.
- X. Q. Lewell, A. C. Jones, C. L. Bruce, G. Harper, M. M. Jones, I. M. McLay and J. Bradshaw, Drug Rings Database with Web Interface. A Tool for Identifying Alternative Chemical Rings in Lead Discovery Programs, J. Med. Chem., 2003, 46, 3257–3274 CrossRef CAS PubMed.
- A. Nilova, L.-C. Campeau, E. C. Sherer and D. R. Stuart, Analysis of Benzenoid Substitution Patterns in Small Molecule Active Pharmaceutical Ingredients, J. Med. Chem., 2020, 63, 13389–13396 CrossRef CAS PubMed.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- For the purposes of this manuscript, commercial availability for each aryl halides bearing a halide (X) and a R group with either ortho-, meta-, or para-substitution pattern was assessed using the Reaxys database. The reported counts represent compounds with molecular weight of 408 or less. Using these criteria, searches via Reaxys produces the following number of commercial compounds for each substitution pattern: ortho-aryl halide: 67346; meta-aryl halide: 65605; para-aryl halide, 198, 131, Reaxys, Elsevier, n.d., https://www.reaxys.com/#/search/quick, accessed 2024-10-22.
- A. D. Nguyen, R. J. Zanolini and J. L. Gustafson, in Handbook of CH- Functionalization, 2022, pp. 1–26, DOI:10.1002/9783527834242.chf0184.
- G. A. Olah, Aromatic substitution. XXVIII. Mechanism of electrophilic aromatic substitutions, Acc. Chem. Res., 1971, 4, 240–248 CrossRef CAS.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, Achieving Site-Selectivity for C–H Activation Processes Based on Distance and Geometry: A Carpenter's Approach, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS PubMed.
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, C–H activation, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- T. W. Lyons and M. S. Sanford, Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- J. F. Hartwig, Evolution of C–H Bond Functionalization from Methane to Methodology, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed.
- J. F. Hartwig and M. A. Larsen, Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed.
- E. Weis, M. A. Hayes, M. J. Johansson and B. Martín-Matute, Iridium-catalyzed C–H methylation and d3-methylation of benzoic acids with application to late-stage functionalizations, iScience, 2021, 24, 102467 CrossRef CAS PubMed.
- S. Ni, M. Hribersek, S. K. Baddigam, F. J. L. Ingner, A. Orthaber, P. J. Gates and L. T. Pilarski, Mechanochemical Solvent-Free Catalytic C–H Methylation, Angew. Chem., Int. Ed., 2021, 60, 6660–6666 CrossRef CAS PubMed.
- S. D. Friis, M. J. Johansson and L. Ackermann, Cobalt-catalysed C–H methylation for late-stage drug diversification, Nat. Chem., 2020, 12, 511–519 CrossRef CAS PubMed.
- X.-Y. Chen and E. J. Sorensen, Pd-Catalyzed, ortho C–H Methylation and Fluorination of Benzaldehydes Using Orthanilic Acids as Transient Directing Groups, J. Am. Chem. Soc., 2018, 140, 2789–2792 CrossRef CAS PubMed.
- L. Hu, X. Chen, Q. Gui, Z. Tan and G. Zhu, Highly mono-selective ortho-trifluoromethylation of benzamides via 8-aminoquinoline assisted Cu-promoted C–H activations, Chem. Commun., 2016, 52, 6845–6848 RSC.
- M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Exceedingly Fast Copper(II)-Promoted ortho C-H Trifluoromethylation of Arenes using TMSCF3, Angew. Chem., Int. Ed., 2014, 53, 10439–10442 CrossRef CAS PubMed.
- L.-S. Zhang, K. Chen, G. Chen, B.-J. Li, S. Luo, Q.-Y. Guo, J.-B. Wei and Z.-J. Shi, Palladium-Catalyzed Trifluoromethylation of Aromatic C–H Bond Directed by an Acetamino Group, Org. Lett., 2013, 15, 10–13 CrossRef CAS PubMed.
- X.-G. Zhang, H.-X. Dai, M. Wasa and J.-Q. Yu, Pd(II)-Catalyzed Ortho Trifluoromethylation of Arenes and Insights into the Coordination Mode of Acidic Amide Directing Groups, J. Am. Chem. Soc., 2012, 134, 11948–11951 CrossRef CAS PubMed.
- H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator, Nature, 2018, 558, 581–585 CrossRef CAS PubMed.
- G. Cheng, P. Wang and J.-Q. Yu, meta-C–H Arylation and Alkylation of Benzylsulfonamide Enabled by a Palladium(II)/Isoquinoline Catalyst, Angew. Chem., Int. Ed., 2017, 56, 8183–8186 CrossRef CAS PubMed.
- X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Ligand-enabled meta-C–H activation using a transient mediator, Nature, 2015, 519, 334–338 CrossRef CAS PubMed.
- L. Guillemard, L. Ackermann and M. J. Johansson, Late-stage meta-C–H alkylation of pharmaceuticals to modulate biological properties and expedite molecular optimisation in a single step, Nat. Commun., 2024, 15, 3349 CrossRef CAS PubMed.
- G. Li, C. Jia, X. Cai, L. Zhong, L. Zou and X. Cui, Ruthenium-catalyzed meta-C–H bond alkylation of aryl 2-pyridyl ketones, Chem. Commun., 2020, 56, 293–296 RSC.
- X.-G. Wang, Y. Li, H.-C. Liu, B.-S. Zhang, X.-Y. Gou, Q. Wang, J.-W. Ma and Y.-M. Liang, Three-Component Ruthenium-Catalyzed Direct Meta-Selective C–H Activation of Arenes: A New Approach to the Alkylarylation of Alkenes, J. Am. Chem. Soc., 2019, 141, 13914–13922 CrossRef CAS PubMed.
- J. Li, K. Korvorapun, S. De Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Ruthenium(II)-catalysed remote C–H alkylations as a versatile platform to meta-decorated arenes, Nat. Commun., 2017, 8, 15430 CrossRef PubMed.
- P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Visible-Light-Enabled Ruthenium-Catalyzed meta-C–H Alkylation at Room Temperature, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS PubMed.
- A. Sagadevan and M. F. Greaney, meta-Selective C–H Activation of Arenes at Room Temperature Using Visible Light: Dual-Function Ruthenium Catalysis, Angew. Chem., Int. Ed., 2019, 58, 9826–9830 CrossRef CAS PubMed.
- J. A. Leitch and C. G. Frost, Ruthenium-catalysed σ-activation for remote meta-selective C–H functionalisation, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC.
- A. J. Paterson, C. J. Heron, C. L. McMullin, M. F. Mahon, N. J. Press and C. G. Frost, α-Halo carbonyls enable meta selective primary, secondary and tertiary C–H alkylations by ruthenium catalysis, Org. Biomol. Chem., 2017, 15, 5993–6000 RSC.
- J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, N-Acyl Amino Acid Ligands for Ruthenium(II)-Catalyzed meta-C–H tert-Alkylation with Removable Auxiliaries, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed.
- N. Hofmann and L. Ackermann, meta-Selective C–H Bond Alkylation with Secondary Alkyl Halides, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed.
- P. J. Deneny, R. Kumar and M. J. Gaunt, Visible light-mediated radical fluoromethylation via halogen atom transfer activation of fluoroiodomethane, Chem. Sci., 2021, 12, 12812–12818 RSC.
- S. M. Hell, C. F. Meyer, S. Ortalli, J. B. I. Sap, X. Chen and V. Gouverneur, Hydrofluoromethylation of alkenes with fluoroiodomethane and beyond, Chem. Sci., 2021, 12, 12149–12155 RSC.
- K. Korvorapun, M. Moselage, J. Struwe, T. Rogge, A. M. Messinis and L. Ackermann, Regiodivergent C–H and Decarboxylative C–C Alkylation by Ruthenium Catalysis: ortho versus meta Position-Selectivity, Angew. Chem., Int. Ed., 2020, 59, 18795–18803 CrossRef CAS PubMed.
- Y. Chen, Advances in the Synthesis of Methylated Products through Indirect Approaches, Adv. Synth. Catal., 2020, 362, 998–1014 CrossRef CAS.
- K. Korvorapun, N. Kaplaneris, T. Rogge, S. Warratz, A. C. Stückl and L. Ackermann, Sequential meta-/ortho-C–H Functionalizations by One-Pot Ruthenium(II/III) Catalysis, ACS Catal., 2018, 8, 886–892 CrossRef CAS.
- Z. Ruan, S.-K. Zhang, C. Zhu, P. N. Ruth, D. Stalke and L. Ackermann, Ruthenium(II)-Catalyzed meta C–H Mono- and Difluoromethylations by Phosphine/Carboxylate Cooperation, Angew. Chem., Int. Ed., 2017, 56, 2045–2049 CrossRef CAS PubMed.
- C. C. Yuan, X. L. Chen, J. Y. Zhang and Y. S. Zhao, meta-Selective C–H difluoromethylation of various arenes with a versatile ruthenium catalyst, Org. Chem. Front., 2017, 4, 1867–1871 RSC.
- L. Zhang and T. Ritter, A Perspective on Late-Stage Aromatic C–H Bond Functionalization, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Late-stage C–H functionalization offers new opportunities in drug discovery, Nat. Rev. Chem, 2021, 5, 522–545 CrossRef CAS PubMed.
- E. C. A. (ECHA), Per- and polyfluoroalkyl substances (PFAS), https://echa.europa.eu/hot-topics/perfluoroalkyl-chemicals-pfas, accessed 2024-10-02, 2024.
- N. D. Tyrrell, A Proposal That Would Ban Manufacture, Supply, and Use of All Fluoropolymers and Most Fluorinated Reagents within the Entire EU, Org. Process Res. Dev., 2023, 27, 1422–1426 CrossRef CAS.
- M. Spennacchio, M. Bernús, J. Stanić, D. Mazzarella, M. Colella, J. J. Douglas, O. Boutureira and T. Noël, A unified flow strategy for the preparation and use of trifluoromethyl-heteroatom anions, Science, 2024, 385, 991–996 CrossRef CAS PubMed.
- A. E. Obukhov and L. I. Belen'kii, Reactivity and electronic structure of the ground and excited states of oxazole, 2-phenyloxazole, and 2-phenylthiazole, Chem. Heterocycl. Compd., 1999, 35, 832–854 CrossRef CAS.
- L. I. Belen'kii, Effect of the acid-base properties of heteroaromatic compounds on their electrophilic substitution reactions (review), Chem. Heterocycl. Compd., 1986, 22, 587–608 CrossRef.
- V. Babin, A. Talbot, A. Labiche, G. Destro, A. Del Vecchio, C. S. Elmore, F. Taran, A. Sallustrau and D. Audisio, Photochemical Strategy for Carbon Isotope Exchange with CO2, ACS Catal., 2021, 11, 2968–2976 CrossRef CAS.
- F. Pasca, Y. Gelato, M. Andresini, D. Serbetci, P. Natho, G. Romanazzi, L. Degennaro, M. Colella and R. Luisi, Continuous Flow Decarboxylative Monofluoroalkylation Enabled by Photoredox Catalysis, JACS Au, 2025, 5, 684–692 CrossRef CAS PubMed.
- Á. Gutiérrez-Bonet and W. Liu, Synthesis of Alkyl Fluorides and Fluorinated Unnatural Amino Acids via Photochemical Decarboxylation of α-Fluorinated Carboxylic Acids, Org. Lett., 2023, 25, 483–487 CrossRef PubMed.
- S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Recent Developments for the Deuterium and Tritium Labeling of Organic Molecules, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed.
- F. Yin, Z. Wang, Z. Li and C. Li, Silver-Catalyzed Decarboxylative Fluorination of Aliphatic Carboxylic Acids in Aqueous Solution, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed.
- B. Jeffries, Z. Wang, R. I. Troup, A. Goupille, J. Y. Le Questel, C. Fallan, J. S. Scott, E. Chiarparin, J. Graton and B. Linclau, Lipophilicity trends upon fluorination of isopropyl, cyclopropyl and 3-oxetanyl groups, Beilstein J. Org. Chem., 2020, 16, 2141–2150 CrossRef CAS PubMed.
- A. P. Truong, G. Tóth, G. D. Probst, J. M. Sealy, S. Bowers, D. W. G. Wone, D. Dressen, R. K. Hom, A. W. Konradi, H. L. Sham, J. Wu, B. T. Peterson, L. Ruslim, M. P. Bova, D. Kholodenko, R. N. Motter, F. Bard, P. Santiago, H. Ni, D. Chian, F. Soriano, T. Cole, E. F. Brigham, K. Wong, W. Zmolek, E. Goldbach, B. Samant, L. Chen, H. Zhang, D. F. Nakamura, K. P. Quinn, T. A. Yednock and J.-M. Sauer, Design of an orally efficacious hydroxyethylamine (HEA) BACE-1 inhibitor in a preclinical animal model, Bioorg. Med. Chem. Lett., 2010, 20, 6231–6236 CrossRef CAS PubMed.
- C. G. Wermuth, P. Ciapetti, B. Giethlen and P. Bazzini, in Comprehensive Medicinal Chemistry II, ed. J. B. Taylor and D. J. Triggle, Elsevier, Oxford, 2007, vol. 2, pp. 649–711 Search PubMed.
- N. Muller, When Is a Trifluoromethyl Group More Lipophilic than a Methyl Group? Partition Coefficients and Selected Chemical Shifts of Aliphatic Alcohols and Trifluoroalcohols, J. Pharm. Sci., 1986, 75, 987–991 CrossRef CAS PubMed.
- Q. A. Huchet, B. Kuhn, B. Wagner, H. Fischer, M. Kansy, D. Zimmerli, E. M. Carreira and K. Müller, On the polarity of partially fluorinated methyl groups, J. Fluorine Chem., 2013, 152, 119–128 CrossRef CAS.
- B. Jeffries, Z. Wang, H. R. Felstead, J.-Y. Le Questel, J. S. Scott, E. Chiarparin, J. Graton and B. Linclau, Systematic Investigation of Lipophilicity Modulation by Aliphatic Fluorination Motifs, J. Med. Chem., 2020, 63, 1002–1031 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01367d |
| This journal is © The Royal Society of Chemistry 2025 |