
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterlayer engineering-induced charge redistribution in Bi2Te3 toward efficient Zn2+ and NH4+ storage†
Xiaojie
Liang
a,
Fangzhong
Liu
a,
Haonan
Yue
a,
Yaoyong
Dong
a,
Lijuan
Chen
e,
Ting
Song
 a,
Yong
Pei
a,
Xianyou
Wang
a,
Bei
Long
*a,
Yao
Xiao
*cf and
Xiongwei
Wu
*bd
a,
Yong
Pei
a,
Xianyou
Wang
a,
Bei
Long
*a,
Yao
Xiao
*cf and
Xiongwei
Wu
*bd
aSchool of Chemistry, Xiangtan University, Xiangtan, 411105 Hunan, P. R. China. E-mail: longbei@xtu.edu.cn
bSchool of Chemistry and Materials Science, Hunan Agricultural University, Changsha, 410128 Hunan, P. R. China. E-mail: wxw@hunau.edu.cn
cCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, 325035 Zhejiang, P. R. China. E-mail: xiaoyao@wzu.edu.cn
dCollege of Chemistry and Chemical Engineering, National & Local Joint Engineering Laboratory for New Petro-chemical Materials and Fine Utilization of Resources, Hunan Normal University, Changsha, 410081 Hunan, P. R. China
eCollege of Intelligent Science and Engineering, Hunan Institute of Engineering, Xiangtan 411104 Hunan, P. R. China
fKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, P. R. China
First published on 5th April 2025
Abstract
Bismuth-based materials show promise for aqueous energy storage systems due to their unique layered structures and high storage capacity. Some bismuth-based materials have been applied to store Zn2+ or NH4+, indicating that one bismuth-based compound may be innovatively used in both zinc-ion and ammonium-ion batteries (ZIBs and AIBs). Herein, we successfully design a poly(3,4-ethylenedioxythiophene) (PEDOT) coated and embedded Bi2Te3 (Bi2Te3@PEDOT). Theoretical calculations and experimental studies demonstrate that the PEDOT coating and its intercalation into the interlayer enhance the structural stability of Bi2Te3 and significantly improve the storage capacities for Zn2+ and NH4+. The PEDOT intercalation results in an increased interlayer spacing and a charge redistribution in the interlayer, facilitating charge transfer. Additionally, the insertion-type mechanism of Zn2+ and NH4+ in Bi2Te3@PEDOT is revealed through ex situ tests. The optimized electrode (5 mg cm−2) exhibits high discharge capacities of 385 mA h g−1 in ZIBs and 235 mA h g−1 in AIBs at 0.2 A g−1 and long-term cycle stability. Bi2Te3@PEDOT performs robustly even at a high mass loading of 10 mg cm−2. Bi2Te3@PEDOT//MnO2 (ZIBs) and Bi2Te3@PEDOT//ZnMn2O4 (AIBs) full cells offer high reversible capacities. This work provides a reference for designing bifunctional energy storage materials.
Introduction
Aqueous batteries (ABs) are a new type of promising energy storage device due to their abundant raw materials, inherent safety, and high cost-effectiveness.1–5 Various types of ABs have been developed such as Na+, K+, Al3+, Zn2+, Ca2+, H+ and NH4+ ion batteries.6–9 Among them, zinc-ion and ammonium-ion batteries (ZIBs and AIBs) have gained significant attention owing to the high theoretical specific capacity (5855 mA h cm−3) of Zn anodes and the small hydrated ion size (0.331 nm) of NH4+.10–13 Though some materials have been reported to show good electrochemical performances in ZIBs and AIBs, such as MoO3 and CuS, two different modification methods are essential to separately obtain high storage capacities for Zn2+ and NH4+.14–16Recently, the design of bifunctional materials has been drawing more and more attention. For instance, Yu et al. synthesized a heterostructural VS2/VOx by an in situ electrochemical method; the VS2/VOx cathode displayed a high specific capacity of 156 mA h g−1 at 10 A g−1 in ZIBs and maintained a reversible capacity of 150 mA h g−1 over more than 1000 cycles in AIBs.17 Li and coworkers designed a layered NH4V4O10 cathode with an adjustable interlayer distance through an in situ electrochemical strategy, and the optimized electrode exhibited good electrochemical performances for both Zn2+ and NH4+ storage.18 However, there are few reports on bifunctional anodes for both ZIBs and AIBs.
In previous works, layered bismuth-based materials such as BiOXs, Bi2O2CO3, and Bi2SeO5 exhibited good Zn2+ or NH4+ storage properties due to their large interlayer spacings and robust crystal structures.19–21 Therefore, bismuth-based materials show great potential for both Zn2+ and NH4+ storage. To achieve excellent electrochemical properties in bismuth-based materials, it is very important to improve electronic/ionic conductivity.22,23 Doping/defect engineering is often used to promote electronic rearrangement and therefore enhances ion/electron transfer, such as Co-doped BiOBr and Se vacancy-activated Bi2Se3.20,24 However, localized or uneven doping and defects result in their restricted enhancement. For instance, excessive Co doping increases the diffusion barrier in a local area.20 Moreover, the designs of the coating and composite can only improve electron or ion transfer. Fortunately, interlayer engineering is a good choice for layered materials. Benefiting from the existence of structural pillars (cations, graphene, polymers and so on), the interlamellar spacing is increased and the electrostatic interaction between layers is decreased, enabling fast reversible ion diffusion/intercalation and high structural stability.25–27
In this study, we prepare a PEDOT-coated/embedded Bi2Te3 (Bi2Te3@PEDOT) as a bifunctional anode material for efficient Zn2+ and NH4+ storage. The coating of PEDOT improves the stability and electronic conductivity of Bi2Te3, and the embedded PEDOT changes the interlayer spacing and charge distribution of Bi2Te3 and thus enhances Zn2+/NH4+ transport kinetics. The Bi2Te3@PEDOT electrode shows superior capacity for Zn2+ and NH4+ storage at mass loadings of 5 and 10 mg cm−2. Remarkably, Zn2+ and NH4+ full cells also display satisfactory discharge capacity and cycling stability.
Results and discussion
The preparation processes of Bi2Te3 and Bi2Te3@PEDOT are schematically displayed in Fig. 1a. Firstly, hexagonal Bi2Te3 nanosheets are synthesized via a solvothermal method, and then PEDOT is coated on and embedded in Bi2Te3 during the polymerization process of EDOT. The compositions and distinctions of Bi2Te3 and Bi2Te3@PEDOT are characterized by various tests. The X-ray diffraction (XRD) patterns reveal that both samples exhibit similar characteristic peaks which are consistent with Bi2Te3 (JCPDS no. 15-0863), suggesting that the crystal structure of Bi2Te3 remains unchanged after the incorporation of PEDOT (Fig. 1b). Significantly, the (006) crystal plane in Bi2Te3@PEDOT (17.22°) shifts to a low angle compared to Bi2Te3 (17.48°), indicating that the lattice spacing increases from 0.507 to 0.515 nm due to the insertion of PEDOT (inset of Fig. 1b). The pure PEDOT is characterized by means of multiple methods (Fig. S1†). Raman spectra further confirm the presence of both Bi2Te3 and PEDOT in Bi2Te3@PEDOT (Fig. 1c). The peaks at 62, 102, and 134 cm−1 arise from the A11g, E2g, and A21g modes of Bi2Te3, while the characteristic peaks of PEDOT are located at 990, 1263, 1365, 1435, 1510, and 1566 cm−1.27–29 Additionally, the peaks in Bi2Te3@PEDOT exhibit a visible red shift (inset in Fig. 1c). X-ray photoelectron spectroscopy (XPS) is employed to analyze the chemical state of Bi2Te3 and Bi2Te3@PEDOT. In the Te 3d XPS spectra (Fig. 1d), the peaks at 571.8 and 582.2 eV in Bi2Te3 originate from the Te–Bi bonds, and the Te–O bonds at 572.5 and 586.0 eV are attributed to surface oxidation.30 Similarly, the peaks at 157.2/162.5 eV and 158.6/164.0 eV in the Bi 4f XPS spectra correspond to the Te–Bi and O–Bi bonds, respectively (Fig. S2a†).31,32 However, those peaks in Bi2Te3@PEDOT shift towards higher binding energies, indicating the change in the chemical environment due to the introduction of PEDOT. Further evidence for the successful synthesis of Bi2Te3@PEDOT is provided by the C 1s and S 2p XPS spectra and the Fourier-transform infrared (FT-IR) spectroscopy spectra (Fig. S2b–d†).33,34 Nitrogen adsorption/desorption isotherms and pore size distribution curves reveal that the cumulative pore volume of Bi2Te3 rises from 0.0395 to 0.1201 cm3 g−1, and the specific surface area grows from 7.54 to 19.66 m2 g−1 after the introduction of PEDOT (Fig. S3†).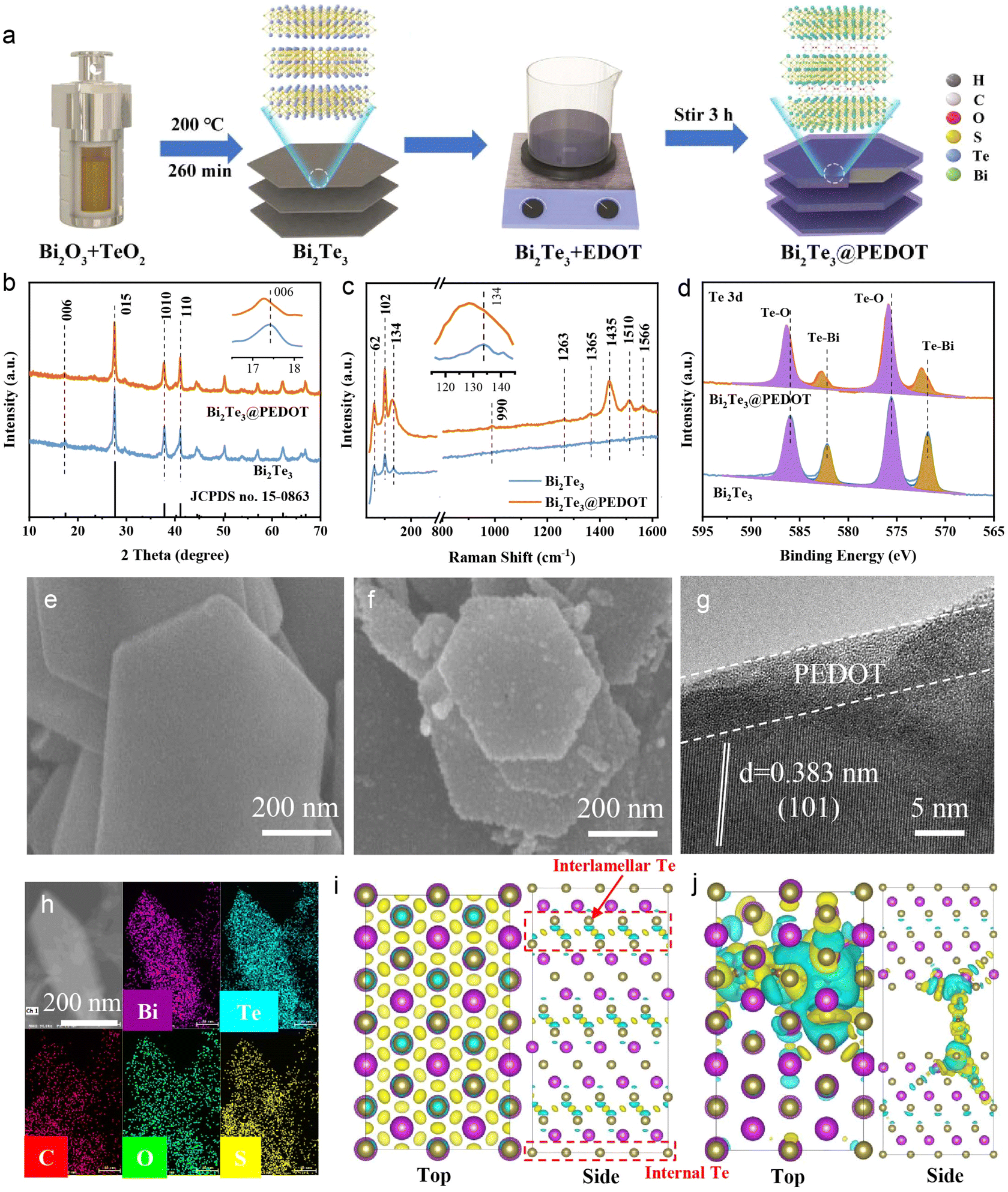 | ||
| Fig. 1 (a) Schematic diagram of the preparation of Bi2Te3 and Bi2Te3@PEDOT. (b) XRD patterns, (c) Raman spectra, and (d) Te 3d XPS spectra of Bi2Te3 and Bi2Te3@PEDOT. SEM images of (e) Bi2Te3 and (f) Bi2Te3@PEDOT. (g) TEM images of Bi2Te3@PEDOT. (h) Elemental mapping of Bi2Te3@PEDOT. Charge density distributions of (i) Bi2Te3 and (j) PEDOT-Bi2Te3 (the purple and brown colors represent Bi and Te atoms, and the yellow and blue regions reflect electron accumulation and electron depletion, respectively). | ||
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images provide insights into the microstructure and morphology of the samples. The SEM image reveals that Bi2Te3 exhibits a smooth hexagonal nanosheet structure (Fig. 1e). Conversely, Bi2Te3@PEDOT displays a rough surface, due to the PEDOT coating layer (Fig. 1f). This explains the increased specific surface area in Bi2Te3@PEDOT and enhances the charge transfer between Bi2Te3@PEDOT and electrolyte. The TEM images reveal a PEDOT coating layer with a thickness of approximately 10 nm in Bi2Te3@PEDOT (Fig. S4a and b†). The high-resolution TEM (HRTEM) image exhibits the lattice fringes (0.383 nm) which corresponds to the (101) crystal plane of Bi2Te3 (greater than 0.377 nm for pure Bi2Te3) (Fig. 1g).35 This result corresponds to the shift in the XRD test. Simultaneously, the polycrystalline nature of Bi2Te3@PEDOT is confirmed by the selected area electron diffraction (SAED) test (Fig. S4c†). Elemental mapping illustrates that Bi, Te, S, O, and C elements are evenly distributed across the nanosheet (Fig. 1h). The above results demonstrate the successful synthesis of Bi2Te3 and Bi2Te3@PEDOT.
Density functional theory (DFT) calculations are used to investigate the role of the embedded PEDOT in the Bi2Te3 structure. The optimized crystal structures of Bi2Te3 (8.87 × 15.38 × 30.55 Å, α = 90.15°, β = 90°, γ = 90°) and PEDOT-Bi2Te3 (8.94 × 15.44 × 37.96 Å, α = 90.09°, β = 90.03°, γ = 89.98°) are presented in Fig. S5.† The insertion of PEDOT results in an expanded interlayer distance and a slight lattice distortion, which is consistent with the experimental results. Differential charge distribution diagrams reveal significant interlamellar electron exchange in the Bi2Te3, reflecting a strong interaction in the interlayer (Fig. 1i). PEDOT intercalation reduces such electron exchange, weakening the interlayer interaction (Fig. 1j). Bader charge analysis further supports this finding. Specifically, the average electron count of Te atoms adjacent to PEDOT decreases from 0.37 (without PEDOT) to 0.06 e, indicating that more electron transfer occurs between PEDOT and its neighboring Te atoms. The density of states (DOS) is simulated for both Bi2Te3 and PEDOT-Bi2Te3 (Fig. S6†). The Bi2Te3 model exhibits typical semiconductor properties. The downward shift of the DOS peaks in the PEDOT-Bi2Te3 model along with the significant DOS at the Fermi level indicates a transformation from semiconductor to conductor, suggesting that the insertion of PEDOT significantly improves the electrical conductivity of PEDOT-Bi2Te3.36 The reduced interlayer electronic interactions and improved electrical conductivity are expected to facilitate ion diffusion in the interlayer, therefore enhancing the electrochemical performances of Bi2Te3@PEDOT.
The electrochemical properties of Bi2Te3 and Bi2Te3@PEDOT in ZIBs are evaluated firstly. In cyclic voltammetry (CV) tests, Bi2Te3 exhibits two redox peaks at 0.62, 0.68, 1.15, and 1.20 V, suggesting the multi-step insertion/deinsertion of Zn2+ in Bi2Te3. Meanwhile, Bi2Te3@PEDOT shows a pair of redox peaks at ∼0.6/1.2 V, reflecting a variation in the insertion/deinsertion process of Zn2+ after the introduction of PEDOT (Fig. S7†).37 This change is explored in the subsequent studies. Meanwhile, the first three CV curves of Bi2Te3@PEDOT overlap better than those of Bi2Te3, indicating its improved reversibility. The first three galvanostatic charge–discharge (GCD) curves show an average discharge plateau at about 0.69 V in Bi2Te3 and 0.67 V in Bi2Te3@PEDOT, implying their suitability as anodes in full cells (Fig. S8†). The rate performances and cycling properties are further explored. At current densities of 0.2, 0.5, 1.0, 2.0, 3.0, 5.0, and 10 A g−1, Bi2Te3@PEDOT offers specific capacities of 378, 340, 320, 312, 310, 300, and 253 mA h g−1, significantly higher than those of Bi2Te3 (195, 180, 152, 123, 110, 97, and 80 mA h g−1) and PEDOT (Fig. 2a and S9†). Fig. 2b and c shows the GCD curves at different current densities, in which Bi2Te3@PEDOT exhibits more stable charging and discharging plateaus. In the cycle life test, Bi2Te3@PEDOT maintains a discharge capacity of 295 mA h g−1 after 200 cycles at 1 A g−1, which is significantly higher than that of Bi2Te3 (96 mA h g−1) (Fig. 2d). Significantly, PEDOT exhibits a negligible Zn2+ storage capacity (18 mA h g−1), reflecting its low capacity contribution to Bi2Te3@PEDOT. After 1000 cycles at 10 A g−1, Bi2Te3@PEDOT retains 82% of its initial capacity, while Bi2Te3 retains only 40% (Fig. 2e). Additionally, the high mass loading Bi2Te3@PEDOT electrodes (10 mg cm−2) are further tested. The discharge capacities of Bi2Te3@PEDOT are 325, 290, 263, 257, 253, 251, and 219 mA h g−1 at current densities from 0.2 to 10 A g−1, much higher than those of Bi2Te3 (125, 99, 85, 75, 64, 56, and 49 mA h g−1) (Fig. 2f). After 500 cycles at 5 A g−1, Bi2Te3@PEDOT maintains a discharge capacity of 187 mA h g−1, while Bi2Te3 retains only 68 mA h g−1 (Fig. 2g). The rate and cycling performances of Bi2Te3@PEDOT-x are compared (Fig. 2h and S10†). It is clear that Bi2Te3@PEDOT-2 (Bi2Te3@PEDOT) outperforms Bi2Te3@PEDOT-1 and Bi2Te3@PEDOT-3, reflecting that a suitable amount of PEDOT is critical to battery performance.
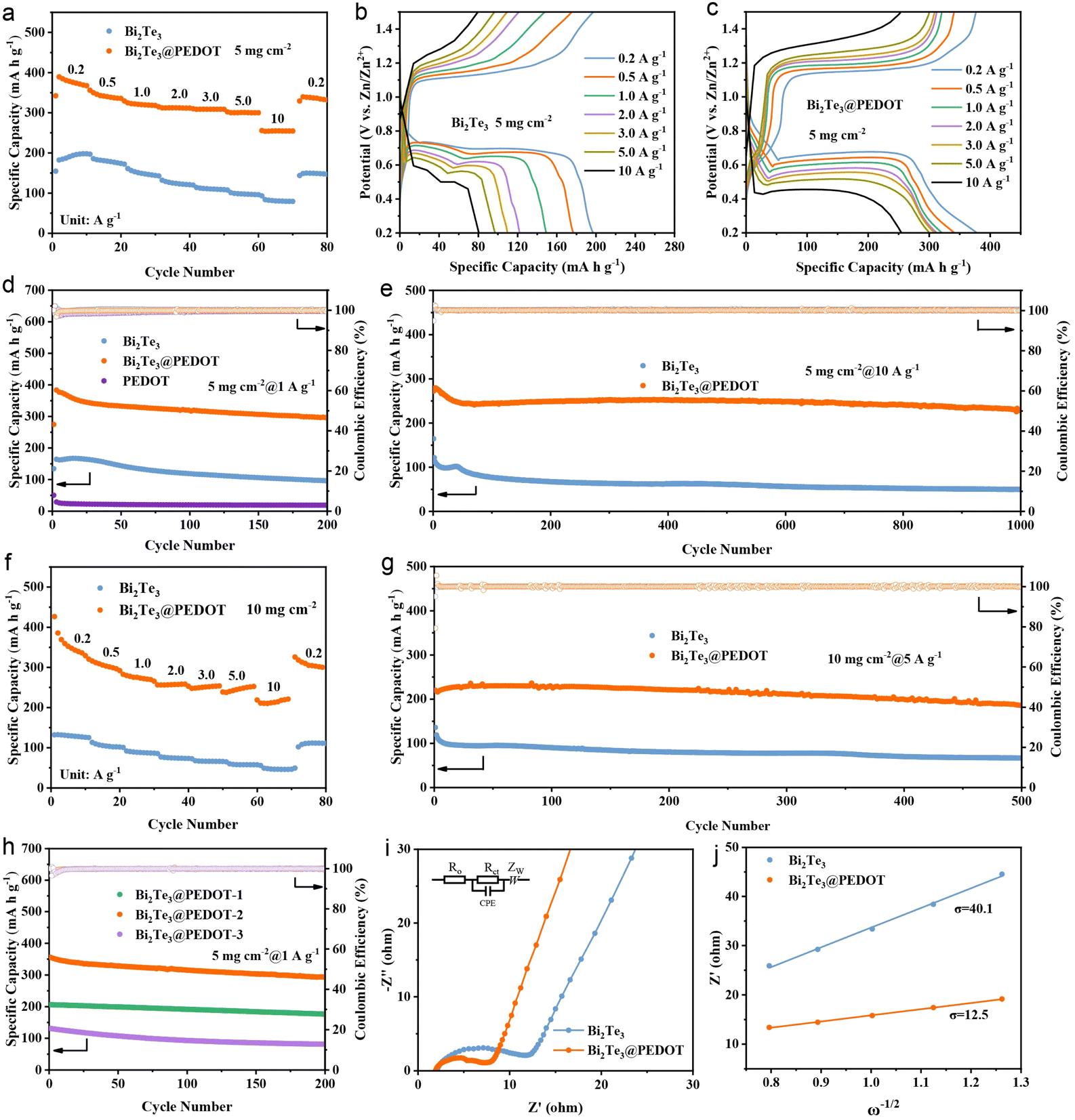 | ||
| Fig. 2 (a) Rate performances and (b and c) corresponding GCD curves of Bi2Te3 and Bi2Te3@PEDOT. (d) Cycle life of Bi2Te3, Bi2Te3@PEDOT, and PEDOT at 1 A g−1. (e) Cyclic performances of Bi2Te3 and Bi2Te3@PEDOT at 10 A g−1 (5 mg cm−2). (f) Rate capabilities and (g) cyclic performances of Bi2Te3 and Bi2Te3@PEDOT at 5 A g−1 (10 mg cm−2). (h) Cycle life of Bi2Te3@PEDOT-x at 1 A g−1. (i) EIS spectra and (j) relationship between ω−1/2 and Z′ of Bi2Te3 and Bi2Te3@PEDOT. | ||
The electrochemical kinetics of Bi2Te3 and Bi2Te3@PEDOT is analyzed by CV tests at different sweep rates (Fig. S11a and b†). Fitting the slopes of the log(i) versus log(v) curves (Fig. S11c†), the b-values of the redox peaks R1, O1, R2, and O2 are 0.72, 0.98, 0.62, and 0.67, respectively. This suggests the enhanced diffusion-controlled behavior of Bi2Te3@PEDOT due to its fast ion diffusion.38,39 The electrochemical impedance spectra (EIS) show that the ohmic resistance (Ro = 2.13 Ω), charge transfer resistance (Rct = 9.9 Ω), and Warburg factor (σ = 40.1) of Bi2Te3 are larger than those of Bi2Te3@PEDOT (Ro = 1.94 Ω, Rct = 4.56 Ω, and σ = 16.8), thereby indicating an improved reaction kinetics in Bi2Te3@PEDOT (Fig. 2i and j). To further demonstrate the superiority of Bi2Te3@PEDOT, the Zn2+ diffusion coefficient is calculated by the galvanostatic intermittent titration technique (GITT) test and Fick's second law. As shown in Fig. S12,† the ion diffusion rate in Bi2Te3@PEDOT is larger than that in Bi2Te3. This result is attributed to the interlayer expansion and reduced electrostatic interaction of Bi2Te3 and thus the improved ion diffusion rate.40
To explore the storage mechanism in Bi2Te3 and Bi2Te3@PEDOT, ex situ XRD, XPS, and SEM are conducted. Ex situ XRD profiles of Bi2Te3 and Bi2Te3@PEDOT are displayed in Fig. 3a–d. Both materials maintain similar XRD patterns during the charge and discharge processes, indicating their intercalation/deintercalation mechanism. The (006) crystal plane in Bi2Te3 shifts to a higher angle after the electrode is discharged to 0.2 V, which results from strong electrostatic interactions between the embedded Zn2+ and Bi2Te3 layers (Fig. 3b).41 In contrast, the (006) crystal plane of Bi2Te3@PEDOT shifts to a lower angle at the fully discharged state. This opposite result is attributed to the insertion of PEDOT and the enlarged interlayer spacing which reduces the interaction forces between Zn2+ and Bi2Te3 layers (this is proved by DFT calculations).42 Meanwhile, the by-product of Zn4SO4(OH)6·4H2O (ZSH) is generated in both Bi2Te3 and Bi2Te3@PEDOT, though the intensity of ZSH peaks is weak in Bi2Te3@PEDOT. To assess the stability of the materials, Bi2Te3 and Bi2Te3@PEDOT are soaked in 2 mol L−1 ZnSO4 electrolyte for 2 h. As shown in Fig. S13a,† numerous bubbles are clearly observed on the surface of Bi2Te3, and some white by-products appear on Bi2Te3 after drying. In contrast, there is no obvious change for Bi2Te3@PEDOT. In addition, the XRD pattern of Bi2Te3 shows the peaks of Bi2TeO5 and Zn(SO4)(H2O)6, whereas no obvious new phase is detected in Bi2Te3@PEDOT (Fig. S13b†).
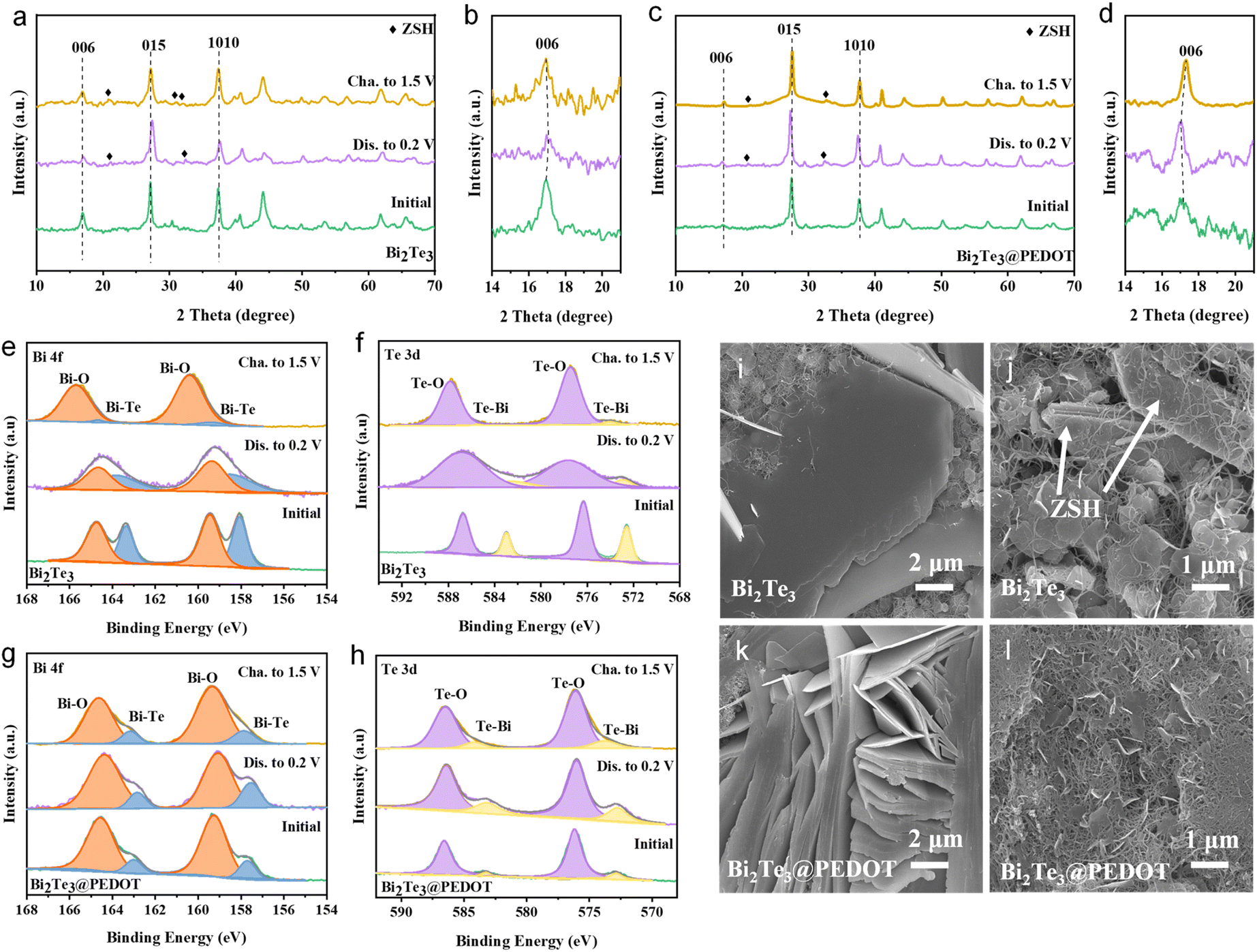 | ||
| Fig. 3 (a–d) Ex situ XRD patterns, (e and g) Bi 4f XPS spectra, and (f and h) Te 3d XPS spectra of Bi2Te3 and Bi2Te3@PEDOT. SEM images of (i and j) Bi2Te3 and (k and l) Bi2Te3@PEDOT at the fully discharged/charged states. | ||
The energy storage mechanism is further investigated using ex situ XPS spectra. In the Bi 4f spectra of Bi2Te3 (Fig. 3e), the peaks of the Bi–O and Bi–Te bonds irreversibly shift to high binding energy during the first cycle, and the Bi–Te peaks almost disappear at the fully charged state. Moreover, Te–O and Te–Bi peaks in the Te 3d XPS spectra also exhibit similar change, implying the poor reversibility and stability of Bi2Te3 (Fig. 3f). For comparison, those peaks in Bi2Te3@PEDOT do not change significantly during the reaction, highlighting its enhanced stability due to the coating and insertion of PEDOT (Fig. 3g and h). In the Zn 2p XPS spectra, no Zn signal is observed at the original state (Fig. S14†). Zn 2p peaks emerge when the electrode is discharged to 0.2 V and become weak at the fully charged state, indicating the Zn2+ intercalation/deintercalation.37,43 The O 1s XPS spectra can be divided into two peaks: the O–H peak at 532.0 eV and the O–Bi/O–Te peak at 530.5 eV (Fig. S15†).44 The area ratios of O–H and O–Bi/O–Te peaks increase and decrease in the discharge and charge processes, respectively, indicating the insertion/extraction of H+. The discharge capacities of H+ and Zn2+ in Bi2Te3@PEDOT are studied in ZnSO4/DMSO and H2SO4 (pH = 5) electrolytes, respectively (Fig. S16†). The specific discharge capacity of H+ (194 mA h g−1) is higher than that of Zn2+ (103 mA h g−1), indicating dominant H+ storage. Additionally, the morphology changes of the electrodes at different charge states are observed by ex situ SEM. Many large-sized ZSH flakes are observed in both Bi2Te3 and Bi2Te3@PEDOT at 0.2 V (Fig. 3i and k). The ZSH flakes are still visible in Bi2Te3 at the fully charged state (Fig. 3j). Meanwhile, the ZSH almost disappears in Bi2Te3@PEDOT, implying its enhanced reaction reversibility (Fig. 3l). The above study demonstrates the co-insertion mechanism of Zn2+ and H+ in both Bi2Te3 and Bi2Te3@PEDOT. Bi2Te3@PEDOT shows high stability and reversibility in the Zn2+/H+ insertion/deinsertion processes.
The remarkable performances of Bi2Te3@PEDOT in ZIBs prompt an exploration of its electrochemical properties in AIBs. Bi2Te3@PEDOT exhibits good rate performance with specific capacities of 266, 235, 200, 164, and 113 mA h g−1 at 0.1, 0.2, 0.5, 1.0, and 2.0 A g−1, respectively, which is higher than those of Bi2Te3 (245, 212, 168, 120, and 71 mA h g−1) (Fig. 4a). The corresponding GCD curves are displayed in Fig. S17,† and Bi2Te3@PEDOT exhibits more obvious discharge plateaus at all current densities. Bi2Te3@PEDOT and Bi2Te3 deliver the discharge capacities of 156 and 113 mA h g−1 with capacity retentions of 72% and 67%, respectively, after 700 cycles at 1 A g−1 (Fig. 4b). Notably, PEDOT shows a low NH4+ storage capacity, suggesting its low capacity contribution to Bi2Te3@PEDOT (Fig. S18†). Additionally, Bi2Te3 and Bi2Te3@PEDOT electrodes are tested under a high mass loading of 10 mg cm−2. The discharge capacities of Bi2Te3@PEDOT are 257, 219, 169, 112, and 50 mA h g−1 at current densities from 0.1 to 2.0 A g−1, while Bi2Te3 exhibits lower discharge capacities (Fig. 4c). After 300 cycles at 1.0 A g−1, Bi2Te3@PEDOT exhibits a significantly higher discharge capacity of 115 mA h g−1 than that of Bi2Te3 (66 mA h g−1) (Fig. 4d). Evidently, Bi2Te3@PEDOT is well suited for use as a host material for NH4+ storage.
 | ||
| Fig. 4 (a) Rate performances and (b) cycle life of Bi2Te3 and Bi2Te3@PEDOT (5 mg cm−2) at 1 A g−1. (c) Rate capabilities and (d) cycle life of Bi2Te3 and Bi2Te3@PEDOT (10 mg cm−2) at 1 A g−1. (e) EIS spectra and (f) relationship between Z′ and ω−1/2 of Bi2Te3 and Bi2Te3@PEDOT. (g and h) Ex situ XRD patterns, (i) Te 3d XPS spectra, and (j) N 1s XPS spectra of Bi2Te3@PEDOT. | ||
The reaction kinetics of Bi2Te3 and Bi2Te3@PEDOT is further investigated through CV, EIS, and GITT tests. In the first five CV curves, the redox peaks of Bi2Te3@PEDOT are more obvious, suggesting its higher capacity for NH4+ storage (Fig. S19†). The corresponding b-values for peaks R1, O1, R2, and O2 are 0.67, 0.77, 0.83, and 0.74 respectively, suggesting an increased capacitive contribution of Bi2Te3@PEDOT (Fig. S20†). The Rct and Warburg factor of Bi2Te3@PEDOT (68 Ω and 38.2) are significantly lower than those of Bi2Te3 (108 Ω and 57.6), implying its enhanced charge transport (Fig. 4e and f). The GITT results also reveal the fast insertion and extraction rates of NH4+ in Bi2Te3@PEDOT (Fig. S21†). The above results suggest the improved charge transfer of Bi2Te3 after the introduction of PEDOT.
To study the NH4+ storage mechanism in Bi2Te3@PEDOT, ex situ XRD and XPS tests are conducted. The XRD test reveals that no new phase appears after the electrode is fully discharged/charged (Fig. 4g). The partial enlargement of the XRD pattern is shown in Fig. 4h, in which the (006) crystal plane remains stable, implying that the insertion and extraction of NH4+ do not affect the structure of Bi2Te3@PEDOT. This good stability is due to the protective layer of PEDOT and the increased interlayer spacing in Bi2Te3@PEDOT. As exhibited in Bi 4f and Te 3d XPS spectra, the Bi–Te peaks shift to higher binding energies after discharging, while the peak positions remain unchanged after charging, indicating a degree of irreversible reaction in the first cycle (Fig. 4i and S22a†). In the N 1s spectra, a strong peak of –NH− appears at the fully discharged state, providing evidence that NH4+ inserts into Bi2Te3@PEDOT (Fig. 4j). However, a weakened peak can still be seen after charging, reflecting the incomplete extraction of NH4+.17,45 The intensity of O–H bonds in the O 1s spectra is enhanced after discharging and decreased after charging, demonstrating the co-insertion of H2O along with NH4+ (Fig. S22b†).45,46 The above results prove the insertion of NH4+ into Bi2Te3@PEDOT. Significantly, Bi2Te3@PEDOT outperforms most of the reported anodes in both ZIBs and AIBs (Tables S1 and S2†). Moreover, the high mass loading anode has been reported rarely.
The Zn2+and NH4+ storage properties of Bi2Te3, as well as the function of the embedded PEDOT, are further validated by DFT calculations. The ionic transport is affected by adsorption energy and lattice distortion. The charge density distributions are used to reveal the effect of PEDOT insertion on Zn2+ and NH4+ adsorption. In the PEDOT-Bi2Te3 model, the charge transfer between Zn2+ and surrounding Te atoms is reduced, leading to a change in adsorption energy from −0.547 (Bi2Te3) to −0.492 eV (PEDOT-Bi2Te3) (Fig. 5a and b). This is beneficial to Zn2+ diffusion. According to Bader charge analysis, when Zn2+ is inserted into Bi2Te3, the Zn transfers a charge of 0.428 e to Bi2Te3, and it drops to 0.089 e in PEDOT-Bi2Te3. Meanwhile, the three neighboring Te atoms obtain an average electron number of 0.046 e before PEDOT intercalation, and it decreases to 0.009 e after PEDOT intercalation. This is consistent with the charge density distributions. By contrast, the adsorption energy between NH4+ and Te atoms increases from −0.735 (Bi2Te3) to −2.204 eV (PEDOT-Bi2Te3) (Fig. 5c and d). Bader charge analysis shows that NH4+ transfers 0.53 e before PEDOT insertion and 0.73 e after PEDOT insertion, while the average electron numbers gained by the three neighboring Te atoms are 0.028 and 0.131 e, respectively, indicating the formation of strong covalent interactions between NH4+ and Bi2Te3 after the PEDOT insertion and the adverse NH4+ diffusion. On the other hand, the average bond length of Te–Bi near the adsorption site of Bi2Te3 is 3.08 Å and increases to 3.15 and 3.27 Å after Zn2+ and NH4+ insertion. This change shows a decrease from 3.07 Å to 3.07 and 3.10 Å with the help of interlayer engineering, facilitating Zn2+ and NH4+ diffusion. Further analysis of the Zn2+/NH4+ diffusion barriers in Bi2Te3 and PEDOT-Bi2Te3 reveals a significant reduction in diffusion barriers with the insertion of PEDOT. The diffusion pathways of Zn2+/NH4+ are illustrated in Fig. S23.† Specifically, the Zn2+/NH4+ diffusion barrier in Bi2Te3 (1.225/0.237 eV) declines to 0.261/0.112 eV with the help of PEDOT (Fig. 5e and f). This reflects that the insertion of PEDOT improves the diffusion of Zn2+/NH4+ due to the combined influence of adsorption energy and lattice distortion.
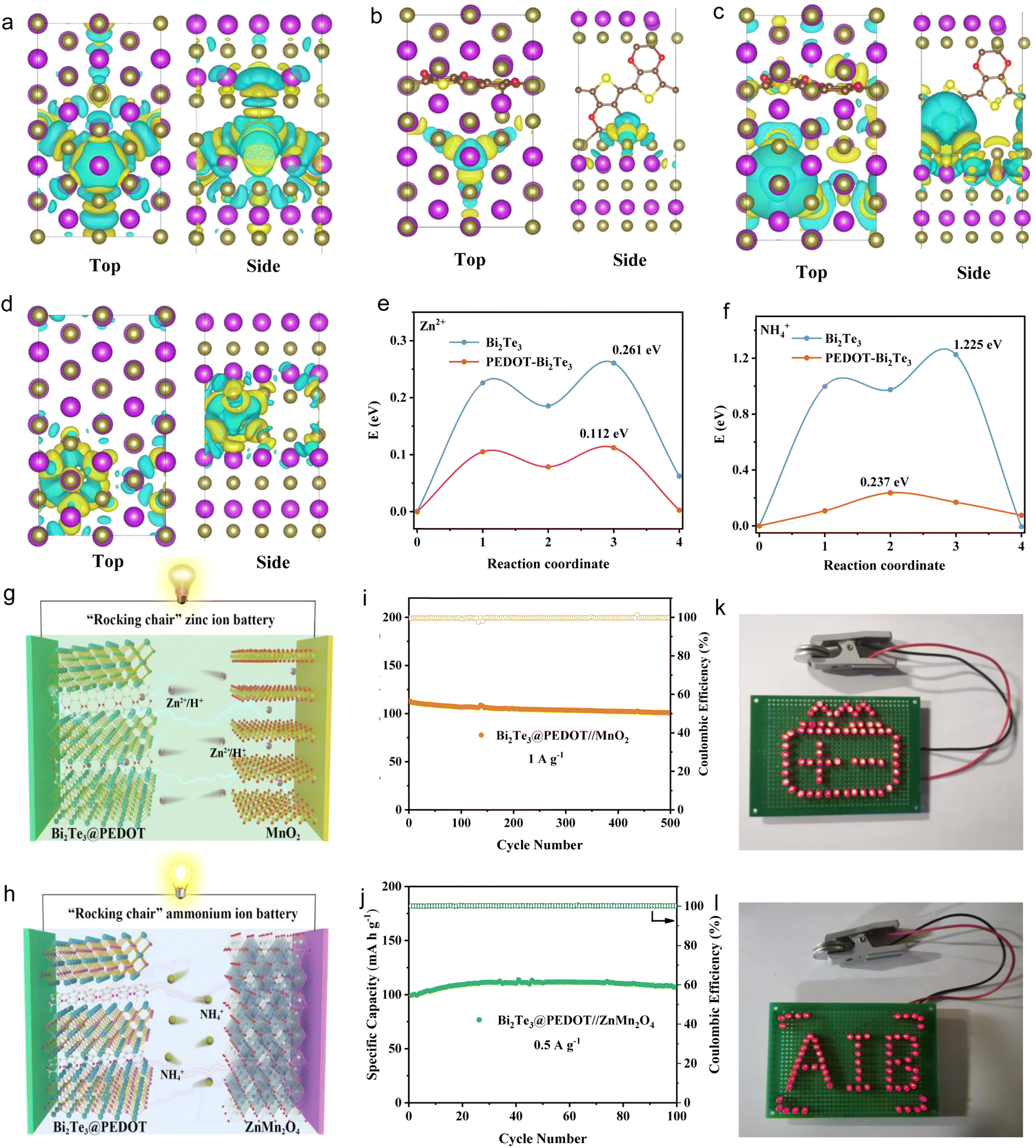 | ||
| Fig. 5 Charge density distributions of (a) Bi2Te3 + Zn2+, (b) PEDOT-Bi2Te3 + Zn2+, (c) Bi2Te3 + NH4+, and (d) PEDOT-Bi2Te3 + NH4+ (the purple and brown colors represent Bi and Te atoms, and the yellow and blue regions reflect electron accumulation and electron depletion, respectively). (e) Zn2+ and (f) NH4+ diffusion barriers of Bi2Te3 and PEDOT-Bi2Te3. (g and h) Structural illustration and (i and j) cycle life of Bi2Te3@PEDOT//MnO2 and Bi2Te3@PEDOT//ZnMn2O4 full cells. Digital photos of two (k) Bi2Te3@PEDOT//MnO2 and (l) Bi2Te3@PEDOT//ZnMn2O4 full cells lighting up red LEDs. | ||
Finally, full cells are assembled to investigate the practical application of Bi2Te3@PEDOT in both “rocking-chair” ZIBs and AIBs, respectively. Commercial MnO2 and ZnMn2O4 are used as cathodes due to their low cost and high operating voltages. The schematics of Bi2Te3@PEDOT//MnO2 and Bi2Te3@PEDOT//ZnMn2O4 full cells are shown in Fig. 5g and h, where Zn2+/H+ or NH4+ shuttles between the anode and cathode through the electrolyte. The zinc-ion full battery exhibits a high capacity of 110 mA h g−1 at 1 A g−1 and a capacity retention of 88% after 500 cycles (Fig. 5i). The ammonium-ion full battery delivers a discharge capacity of 110 mA h g−1 at 0.5 A g−1 and good cycling stability over 100 cycles (Fig. 5j). The GCD curves of the ZIB and AIB full cells are displayed in Fig. S24.† In addition, both full cells drive the self-assembled red LED logos (Fig. 5k and l).
Conclusions
In summary, a PEDOT-coated/embedded Bi2Te3 is successfully fabricated through a hydrothermal method followed by the polymerization of EDOT. Various characterizations and DFT calculations demonstrate that the embedded PEDOT enlarges the interlayer spacing and reduces the interlamellar interaction, thus improving the reaction kinetics of Bi2Te3. Meanwhile, the coating of PEDOT enhances structural stability, suppresses side reactions with the aqueous solution, and increases electronic conductivity. Ex situ tests demonstrate the insertion-type mechanism of Bi2Te3@PEDOT in both ZIBs and AIBs. The Bi2Te3@PEDOT electrode exhibits high discharge capacities of 385 and 235 mA h g−1 at 0.2 A g−1 in ZIBs and AIBs, respectively. Even at a high mass loading of 10 mg cm−2, Bi2Te3@PEDOT shows satisfactory rate performances and cycling stability. Furthermore, “rocking-chair” ZIBs and AIBs with the Bi2Te3@PEDOT anode offer high reversible capacities of 110 mA h g−1 at 1.0 A g−1 (ZIBs) and 110 mA h g−1 at 0.5 A g−1 (AIBs), respectively. This work extends the utilizing scope of layered bismuth-based materials and serves as a reference for bifunctional energy storage electrodes.Data availability
The data supporting this article have been included in the main text and ESI.†Author contributions
X. L.: writing – original draft, data curation; F. L.: writing – original draft, data curation; H. Y.: data curation, investigation; Y. D.: formal analysis, resources, software; L. C.: investigation; T. S.: investigation; Y. P.: resources, software; X. W.: project administration, funding acquisition; B. L.: writing – review & editing, conceptualization, supervision; Y. X.: writing – review & editing, conceptualization, supervision; X. W.: writing – review & editing, project administration, funding acquisition. X. L. and F. L. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (52102312), Scientific Research Project of Education Department of Hunan Province (23C0047), and National Key R & D Program of China (2021YFB2400400). The authors would like to thank Xiao Ming Li from SCI-GO (http://www.sci-go.com) for help with the XPS analysis.References
- M. Li, X. P. Wang, J. S. Meng, C. L. Zuo, B. K. Wu, C. Li, W. Sun and L. Q. Mai, Adv. Mater., 2024, 36, 2308628 CrossRef CAS PubMed.
- Z. N. Ju, Q. Zhao, D. L. Chao, Y. Hou, H. G. Pan, W. P. Sun, Z. Y. Yuan, H. Li, T. Y. Ma, D. W. Su and B. H. Jia, Adv. Energy Mater., 2022, 12, 2201074 Search PubMed.
- J. Liu, K. Wang, Y. Sun, H. Li, X. Han, X. Duan, Z. Huang and T. Ma, Nano Energy, 2025, 136, 110764 Search PubMed.
- C. Luo, H. Lei, Y. Xiao, X. Nie, Y. Li, Q. Wang, W. Cai, C. Dai, M. Yao, Y. Zhang and D. Yuan, Energy Mater., 2024, 4, 400036 Search PubMed.
- X. Liu, B. Xu, S. Deng, J. Han, Y. An, J. Zhao, Q. Yang, Y. Xiao and C. Fang, Carbon Energy, 2024, 6, e603 CrossRef CAS.
- G. Zeng, Q. Sun, S. Horta, P. R. Martínez-Alanis, P. Wu, J. Li, S. Wang, M. Ibáñez, Y. Tian, L. Ci and A. Cabot, Energy Environ. Sci., 2025, 18, 1683–1695 Search PubMed.
- W. Jiang, K. Zhu, W. Xie, Z. Wang, Z. Ou and W. Yang, Chem. Sci., 2024, 15, 2601–2611 Search PubMed.
- Y. Zhang, Z. Huang, L. Lei, H. Fan, X. Han, H. Li and T. Ma, Adv. Energy Mater., 2025, 15, 2404732 Search PubMed.
- F. Xiankai, X. Kaixiong, Z. Wei, D. Weina, Z. Hai, C. Liang and C. Han, Carbon Energy, 2024, 6, e536 Search PubMed.
- B.-H. Xiao, K. Xiao, J.-X. Li, C.-F. Xiao, S. Cao and Z.-Q. Liu, Chem. Sci., 2024, 15, 11229–11266 Search PubMed.
- G. Zeng, Q. Sun, S. Horta, S. Wang, X. Lu, C. Y. Zhang, J. Li, J. Li, L. Ci, Y. Tian, M. Ibáñez and A. Cabot, Adv. Mater., 2024, 36, 2305128 Search PubMed.
- B. Long, X. Ma, L. Chen, T. Song, Y. Pei, X. Wang and X. Wu, Adv. Funct. Mater., 2024, 34, 2411430 CrossRef CAS.
- S. Gong, M. Zhu, Y. Zhou, R. Li, J. Zhang, X. Jia, D. Chao and C. Wang, Chem. Sci., 2024, 15, 19870–19885 RSC.
- J. Shi, Y. Hou, Z. Liu, Y. Zheng, L. Wen, J. Su, L. Li, N. Liu, Z. Zhang and Y. Gao, Nano Energy, 2022, 91, 106651 Search PubMed.
- H. Liu, K. Zhang, S. Wang and X. Cai, Small, 2023, 20, 2310835 CrossRef PubMed.
- Q. Lei, J. Zhang, Z. Liang, Y. Yue, Z. Ren, Y. Sun, Z. Yao, J. Li, Y. Zhao, Y. Yin, P. Huai, Z. Lv, J. Li, Z. Jiang, W. Wen, X. Li, X. Zhou and D. Zhu, Adv. Energy Mater., 2022, 12, 2200547 CrossRef CAS.
- D. Yu, Z. Wei, X. Zhang, Y. Zeng, C. Wang, G. Chen, Z. X. Shen and F. Du, Adv. Funct. Mater., 2020, 31, 2008743 CrossRef.
- S. Li, D. Yu, J. Liu, N. Chen, Z. Shen, G. Chen, S. Yao and F. Du, Adv. Sci., 2023, 10, 2206836 CrossRef CAS PubMed.
- M. Han, Y. Qian, X. Li, N. Wang, T. Song, L. Liu, X. Wang, X. Wu, M.-K. Law and B. Long, J. Colloid Interface Sci., 2023, 645, 483–492 Search PubMed.
- B. Long, Q. Zhang, T. Duan, T. Song, Y. Pei, X. Wang, C. Zhi, X. Wu, Q. Zhang and Y. Wu, Adv. Sci., 2022, 9, 2204087 CrossRef CAS PubMed.
- D. Ling, Q. Wang, G. Tian, H. Yu, D. Zhang and Q. Wang, ACS Nano, 2023, 17, 25222–25233 CrossRef CAS PubMed.
- Q. Wang, M. Wang, L. Wen, W. Zeng, B. Ge, C. Zhang, Y. Yue and S. Wang, Adv. Funct. Mater., 2023, 34, 2214506 CrossRef.
- X. Wang, F. Zhou, Q. Liang, Q. Zhang, Y. Zhu, Z. Xiao and L. Wang, Adv. Funct. Mater., 2024, 34, 2408203 CrossRef CAS.
- B. Long, X. Ma, L. Chen, T. Song, Y. Pei, X. Wang and X. Wu, Adv. Funct. Mater., 2024, 34, 2411430 CrossRef CAS.
- S. Li, Y. Liu, X. Zhao, Q. Shen, W. Zhao, Q. Tan, N. Zhang, P. Li, L. Jiao and X. Qu, Adv. Mater., 2021, 33, e2007480 CrossRef PubMed.
- G. Zhang, T. Wu, H. Zhou, H. Jin, K. Liu, Y. Luo, H. Jiang, K. Huang, L. Huang and J. Zhou, ACS Energy Lett., 2021, 6, 2111–2120 CrossRef CAS.
- W. Bi, G. Gao, G. Wu, M. Atif, M. S. AlSalhi and G. Cao, Energy Storage Mater., 2021, 40, 209–218 CrossRef.
- J. Feng, Z. Zhuang, Y. Zhou and C. Li, Adv. Funct. Mater., 2024, 34, 2315188 Search PubMed.
- G. Wang, F. Meng, Y. Chen, A. Lotnyk and X. Shen, Adv. Sci., 2024, 11, e2308056 CrossRef PubMed.
- X. Liu, Y. Si, K. Li, Y. Xu, Z. Zhao, C. Li, Y. Fu and D. Li, Energy Storage Mater., 2021, 41, 255–263 Search PubMed.
- C. Zheng, Z. Guo, B. Jian, Z. Chen, J. Zhong, N. Li and S. Huang, Chem. Eng. J., 2023, 475, 146408 Search PubMed.
- S. Wang, Y. Qiao, X. Liu, S. Zhu, Y. Zheng, H. Jiang, Y. Zhang, J. Shen, Z. Li, Y. Liang, Z. Cui, P. K. Chu and S. Wu, Adv. Funct. Mater., 2022, 33, 2210098 Search PubMed.
- X. B. Liao, C. L. Pan, H. X. Yan, Y. Zhu, Y. S. Pan and C. J. Yin, Chem. Eng. J., 2022, 440, 135930 CrossRef CAS.
- K. Kim, M. Kim, H. Lee, D. W. Chung and J. Kim, Small, 2024, 20, e2402341 Search PubMed.
- S. Chong, L. Yuan, Q. Zhou, Y. Wang, S. Qiao, T. Li, M. Ma, B. Yuan and Z. Liu, Small, 2023, 19, 2303985 CrossRef CAS PubMed.
- Y. Wu, Q. Zong, Y. Zhuang, Q. Wang, C. Liu, Q. Zhang, D. Tao, J. Zhang, J. Wang and G. Cao, Chem. Eng. J., 2024, 491, 152064 CrossRef CAS.
- D. Bin, W. Huo, Y. Yuan, J. Huang, Y. Liu, Y. Zhang, F. Dong, Y. Wang and Y. Xia, Chem, 2020, 6, 968–984 CAS.
- X. Tan, F. Zhang, D. Chen, P. Wang, Y. Liu, C. Meng and Y. Zhang, Chem. Eng. J., 2024, 489, 151119 CrossRef CAS.
- X. Tan, F. Zhang, D. Chen, J. n. Gong, J. Sun, C. Meng and Y. Zhang, J. Colloid Interface Sci., 2024, 669, 2–13 Search PubMed.
- K. Zhu, W. Jiang, Z. Wang, W. Li, W. Xie, H. Yang and W. Yang, Angew Chem. Int. Ed. Engl., 2023, 62, e202213368 Search PubMed.
- H. Geng, M. Cheng, B. Wang, Y. Yang, Y. Zhang and C. C. Li, Adv. Funct. Mater., 2019, 30, 1907684 CrossRef.
- L. Chen, H. Nie, S. Zhou, G. Cao and A. Pan, Sci. China Mater., 2023, 66, 3453–3460 CrossRef CAS.
- Y. Hou, P. Ma, F. Long, M. Liu, Y. Zheng, L. Sun, J. Shi, K. Niu, J. Su, Y. Ma and Y. Gao, ACS Nano, 2024, 18, 27358–27371 CrossRef CAS PubMed.
- S. Hu, X. Huang, L. Zhang, G. Li, S. Chen, J. Zhang and X. Liu, Adv. Funct. Mater., 2023, 33, 2214161 Search PubMed.
- Y. Song, Q. Pan, H. Lv, D. Yang, Z. Qin, M. Y. Zhang, X. Sun and X. X. Liu, Angew. Chem., Int. Ed., 2021, 60, 5718–5722 Search PubMed.
- X. Mu, Y. Song, Z. Qin, J. Meng, Z. Wang and X.-X. Liu, Chem. Eng. J., 2023, 453, 139575 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, crystallography data and additional figures. See DOI: https://doi.org/10.1039/d5sc01210d |
| This journal is © The Royal Society of Chemistry 2025 |