
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical photochemical difluorosulfoximination of alkenes and propellanes†
Simone
Baldon
 a,
Julien
Paut
ab,
Elsa
Anselmi
bc,
Guillaume
Dagousset
b,
Béatrice
Tuccio
e,
Giorgio
Pelosi
d,
Sara
Cuadros‡
*a,
Emmanuel
Magnier‡
*b and
Luca
Dell’Amico‡
*a
a,
Julien
Paut
ab,
Elsa
Anselmi
bc,
Guillaume
Dagousset
b,
Béatrice
Tuccio
e,
Giorgio
Pelosi
d,
Sara
Cuadros‡
*a,
Emmanuel
Magnier‡
*b and
Luca
Dell’Amico‡
*a
aDepartment of Chemical Sciences, University of Padova, Via Francesco Marzolo 1, 35131, Padova, Italy. E-mail: sara.cuadroshuertas@unipd.it; luca.dellamico@unipd.it
bInstitut Lavoisier de Versailles, Université Paris-Saclay, 45 avenue des Etats-Unis, 78035 Versailles, France. E-mail: emmanuel.magnier@uvsq.fr
cUniversité de Tours, Faculté des Sciences et Techniques, 37200 Tours, France
dDepartment of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17, 43124, Parma, Italy
eAix Marseille Univ., CNRS, ICR, 13013 Marseille, France
First published on 14th March 2025
Abstract
Herein, we report a metal-free divergent visible-light driven method for the synthesis of fluorinated sulfoximines. Both olefins and propellanes efficiently undergo difluorosulfoximination with yields up to 77% (65 examples). The process is general and robust and tolerates diverse functional groups, including esters, ethers, ketones, silyl groups, silyl ethers or boronic esters. The functionalization of diverse bioactive ingredients (8 examples) and various product manipulations demonstrate the synthetic usefulness of the developed synthetic platform. Finally, we rationalized the divergent reaction mechanism by performing Stern–Volmer quenching and EPR experiments that revealed the key activity of a difluoroalkyl sulfoximine radical.
Introduction
Sulfoximines have gained increasing popularity in pharmaceutical and agrochemical research as versatile isosteric replacement of sulfones, sulfonamides, carbonyl compounds, amines or alcohols, among others (Fig. 1a).1 These compounds possess interesting properties, such as high chemical stability, increased solubility in water2 and multiple hydrogen bond donor/acceptor sites. Further, the N-atom adds value to the structure by adding intrinsic chirality, with the possibility of exploring the 3D-chemical space with defined exit vectors. On the other hand, replacing hydrogen atoms with highly electronegative fluorine atoms is a common strategy to modulate properties, reactivity and conformations of molecules.3 In this regard, F-containing sulfoximines have found applications as key elements in bioactive compounds (Fig. 1a) or functional materials.4 However, general synthetic methods for the preparation of these classes of fluorinated compounds are still largely underdeveloped.4a In recent years, we undertook a research line for the development of effective methodologies to achieve the construction of F-containing sulfoximines 1 (Fig. 1b).5 Interestingly, these structures 1 have exhibited synthetic versatility in photoredox catalytic systems,6 with reactivity influenced by the N-substitution pattern (Fig. 1b). On one hand, N-tosyl and N-triflate sulfoximines (1, X= Ts or Tf, respectively) have been used as fluoroalkyl radical sources (pathway i).7 Here, a single electron transfer (SET) process from an excited photocatalyst to 1 triggers a C–S bond homolytic cleavage, while releasing fluorinated C-centered radicals (RF˙). These radicals have been subsequently engaged in radical additions with alkenes 3, ultimately forming fluoroalkyl products 4. In these strategies, the sulfoximine unit is not incorporated into the final target 4 but only serves as an auxiliary for the generation of reactive fluoroalkyl radicals. On the other hand, the group of Magnier utilized N-chloro substituted sulfoximines (1, X = Cl) in photoinduced SET reductions (pathway ii), promoting N–Cl homolytic bond scission while generating the corresponding N-sulfoximidoyl radical 2.8 The N-centered radical 2 has also been successfully engaged in radical addition processes with alkenes 3, offering a straightforward access to N-alkylated fluorinated products 5. Aiming at identifying novel reaction manifolds for 1, while addressing the increasing demands of fluorinated sulfoximines, we wonder whether an alternative scission pathway could be promoted to access difluoroalkyl sulfoximine radicals 6 (pathway iii), for which there are currently no available methods. Success in this endeavor will offer new avenues for the synthesis of CF2-sulfoximines via radical chemistry, while allowing the generation of novel F-containing building blocks.9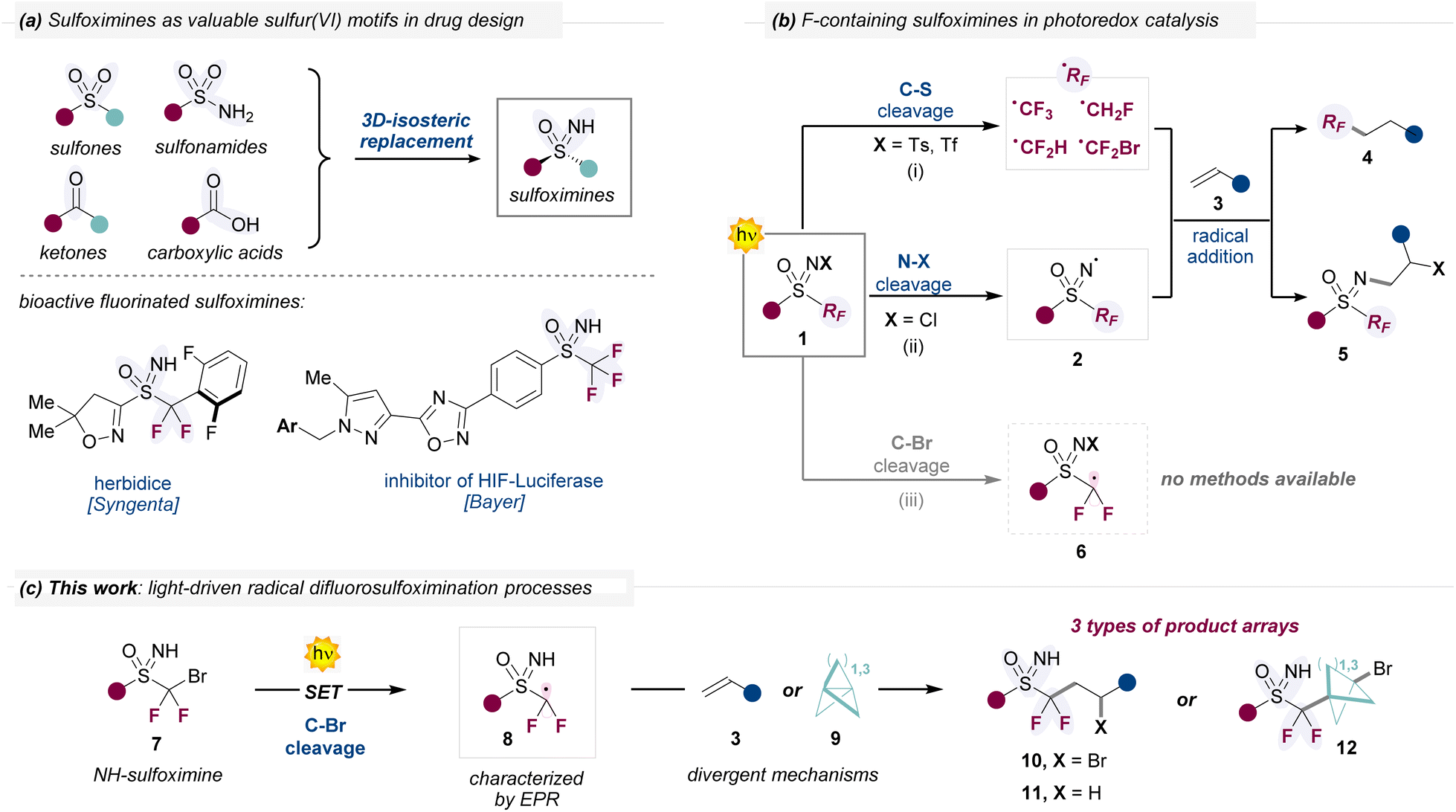 | ||
| Fig. 1 (a) Application of sulfoximines in drug design and examples of biologically active fluorinated sulfoximines. (b) Available reactivity pathways for fluorinated N-substituted sulfoximines 1, under photoredox catalyzed conditions. (c) This work: light-driven difluorosulfoximination of unactivated alkenes 3 and propellanes 9. PC: photocatalyst; SET: single electron transfer. | ||
Herein, we detail a mild organophotoredox strategy that gives access to unprecedented difluoroalkylsulfoximine radicals 8, while engaging them in divergent reaction manifolds (Fig. 1c). Our approach is based on the use of bromodifluoroalkyl NH-sulfoximines 7, which upon photoinduced SET, undergo preferentially C–Br homolytic cleavage to produce the target radicals 8. These intermediates successfully engage in different radical addition processes with unactivated alkenes 3 and propellanes 9, either via atom-transfer radical addition (ATRA) or hydrofunctionalization mechanisms.10 Using this divergent synthetic platform, we accessed a wide variety of difluoroalkyl sulfoximine products 10–12 (65 examples, up to 77% yield), including enantiomerically pure sulfoximines. On the other hand, the formation of the radical intermediate 8 was undoubtedly proven by means of spin trapping coupled with electron paramagnetic resonance (EPR) detection. Finally, product manipulations demonstrate the usefulness of the products, including the synthesis of novel classes of cyclic F-containing sulfoximines.
Results and discussion
We began our studies by evaluating the performance of different organophotoredox catalysts in the ATRA process between bromodifluoroalkyl sulfoximine 13 and 1-hexene 14 (Table 1). The first experiments were conducted at 20 °C, using acetonitrile as the solvent (0.5 M). Initially, we observed the formation of product 15 with the commercially available phenothiazine PC1 as well as with the dihydroacridine photocatalyst PC2 (entries 1 and 2). The NMR yield was raised up to 60% or 47%, by increasing the catalyst loading of either PC1 or PC2 from 5 to 15 mol% (entries 3 and 4). Finally, after an extensive PC screening (see ESI, Section C.1.5†), we identified the N-phenyl substituted dihydroacridine PC3 as the best PC, affording the product 15 in 60% NMR yield, after 10 h (entry 5). Alongside 15, we observed the formation of byproducts arising from the radical addition of ˙CF2Br radicals to 14 in variable NMR yields depending on the reaction conditions (see ESI Section C.3,† for further details). This suggests that, while C–Br bond scission predominates under these conditions, C–S bond scission is not entirely suppressed (vide infra).|
|
||||
|---|---|---|---|---|
| Entrya | PC (mol%) | λ (nm) | HAT donor |
15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 yieldb (%) :16 yieldb (%) |
| a Reactions performed at 0.1 mmol scale, using 1 equiv. of 13 and 3 equiv of 14 (see ESI, Section C). b The yield was determined by 1H-NMR analysis of the crude, using trichloroethylene as the internal standard. c Reaction time: 10 h. Isolated yields are in the parentheses. | ||||
| 1 | PC1 (5) | 400 | — | 33:— |
| 2 | PC2 (5) | 427 | — | 28:— |
| 3 | PC1 (15) | 400 | — | 60(42):— |
| 4 | PC2 (15) | 427 | — | 47:— |
| 5c | PC3 (15) | 427 | — | 60(52):— |
| 6 | None | 427 | — | No reaction |
| 7 | PC3 (15) | 427 | 17 | 5:51(47) |
| 8 | PC3 (15) | 427 | 18 | 12:40 |
| 9c | None | 427 | 17 | <5:55 |
| 10c | None | 400 | 17 | —:66(61) |
| 11 | PC3 (15) | Light off | 17 | No reaction |

|
||||
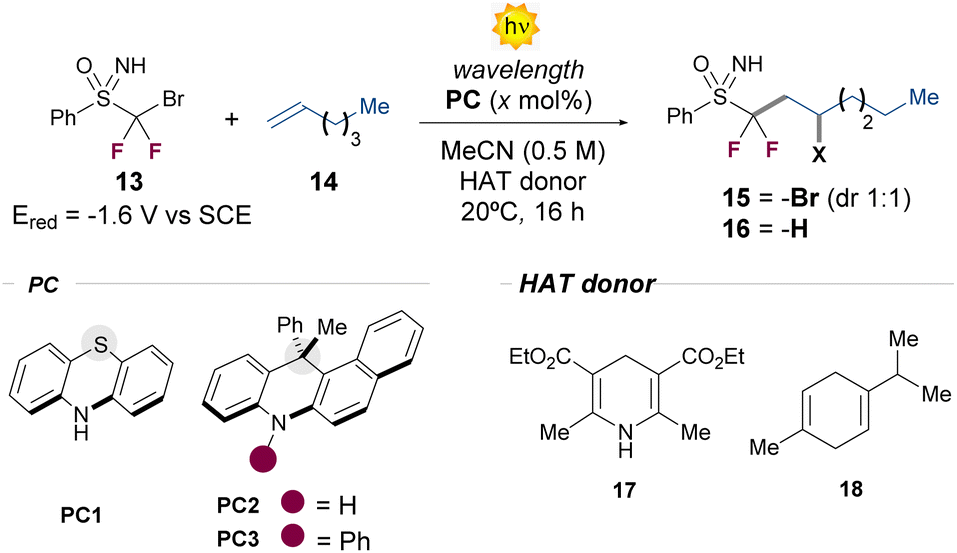
With this result in hand, we next investigated the possibility of going beyond the classical ATRA manifold in the presence of suitable hydrogen atom transfer (HAT) donors. To our delight, the addition of 1.5 equivalent of the Hantzsch ester 17 to the previously optimized conditions mainly afforded the product 16 in 47% isolated yield, with only traces of the ATRA product 15 (entry 7). Intriguingly, the process can be promoted without the use of a PC (entry 9), indicating the pivotal excited state reactivity of the Hantzsch ester 17 under the optimized reaction conditions.11 Taking into account the absorption profile of 17, we next tested other excitation wavelengths, identifying 400 nm as the most effective to obtain the hydrofunctionalization product 16 in higher yield (61% isolated yield, entry 10).
Generality of the process
Our next efforts were directed to assess the generality of the two divergent reaction manifolds (Table 2). A wide variety of unactivated terminal alkenes 3 bearing common functionalities in medicinal chemistry settings,12 including esters, ethers, silyl ethers, sulfones, boronates, or indole derivatives, were well tolerated in both reaction pathways, giving the corresponding products 19–34 in up to 68% isolated yield. As a general trend, we observed higher yields under the hydrofunctionalization conditions, in comparison with the ATRA reactivity.:1 (see ESI)
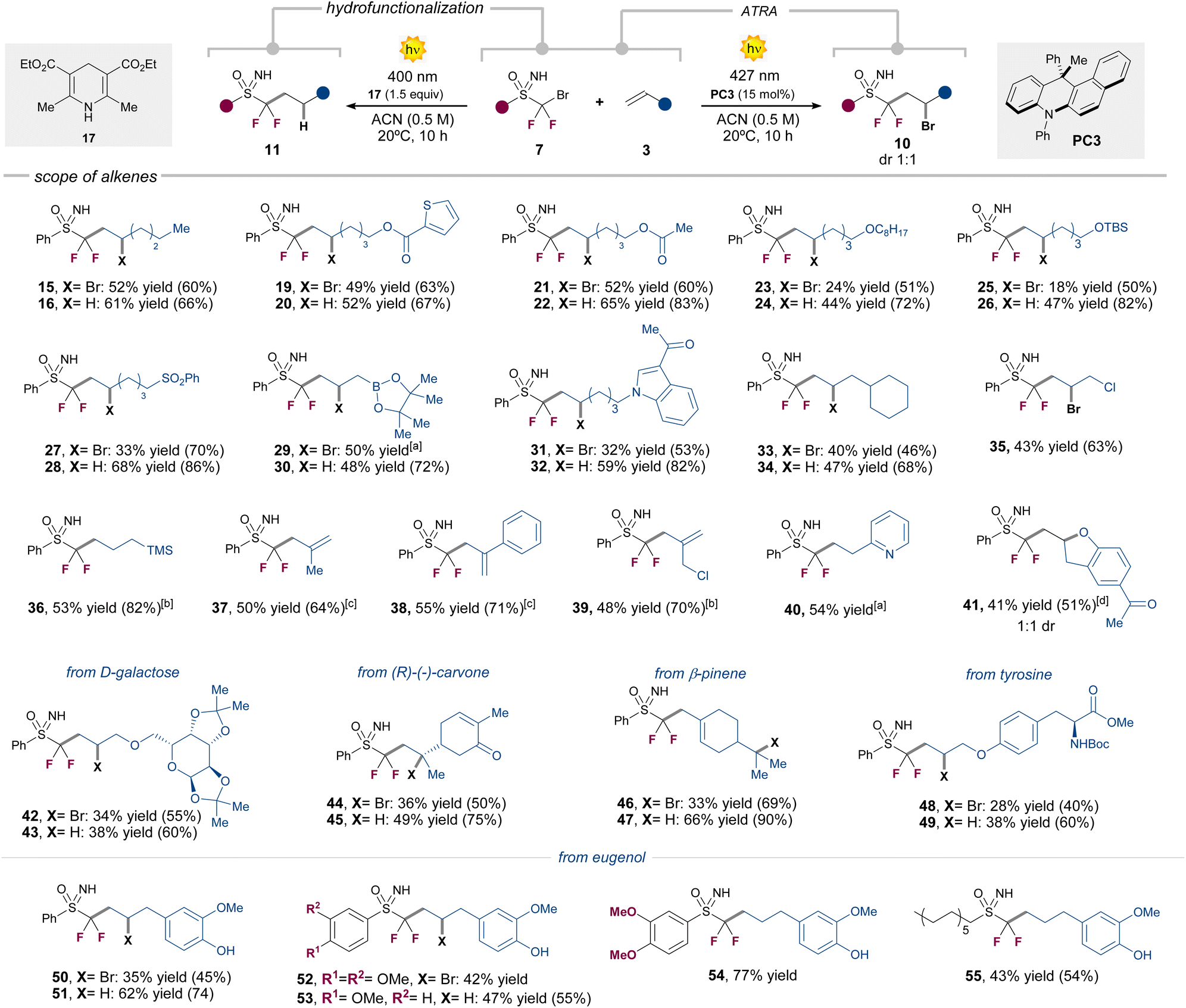
Allyl chloride proved to be reactive only in the ATRA pathway, delivering the highly versatile adduct 35 in 43% yield. On the other hand, allyl-TMS successfully afforded product 36 in 53% yield under the hydrofunctionalization conditions, while desilylated alkene products are formed under the ATRA reactivity (see ESI, Section E.1†). In a similar manner, product 37 was obtained from the use of the methyl-substituted allyl-TMS under the ATRA conditions (see ESI,† for other entries) Intriguingly, the phenyl-substituted allyl-TMS proved to be only reactive in the ATRA pathway, giving the corresponding desilylated product 38 in 55% yield. Alkenes bearing pyridine or –N(Boc)2 groups in terminal positions were only reactive in the hydrofunctionalization pathway, affording the products 40 and 91 (see ESI†) in 54% and 26% yield, respectively. Furthermore, inspired by previous reports,13 we also synthesized the functionalized benzofurane derivative 41, via sequential ATRA reaction followed by a nucleophilic substitution process, starting from 2-allylphenyl acetate (see ESI† for the synthesis of an indoline analogue). To our delight, the method proved to be amenable also to more complex biologically relevant substrates. We could use in both photochemical processes alkenes derived from D-galactose, carvone, β-pinene, tyrosine or eugenol (42–55, see ESI Section E.1† for other entries). Alkyl and mono-fluorinated N–Ts-protected sulfoximines were also competent substrates (55 and 98). We next evaluated the scope of sulfoximines 7 in the strain-release ATRA process with [1.1.1]-propellane 9 (Table 3).14 Here, likely thanks to the strain-release process, we observed higher yields, by only using 5 mol% of PC2.

Pleasingly, different aryl and alkyl substituted N–H or N–Ts sulfoximines were successfully engaged in this transformation, affording the ATRA products 56–70 in up to 71% yield. It is worth nothing that also [3.1.1]-propellanes can also be used as radical trapping agents, giving access to product 65–67 in 42–50% yield. These functionalized [3.1.1]-bicycloalkane units have been recently validated as bioisosteric replacements for meta-substituted arene rings, suggesting significant applications in drug design.15 Although this is not the main topic of this article, it should be pointed out that a major effort has been made to prepare novel sulfoximines. In the CF2Br series, five new sulfoximines were obtained by varying the aromatic ring on the sulfur atom (including a highly original pyridine ring). After reactions, the latter led to compounds 60 to 64. In addition, we engaged a N–Ts substituted fluorodibromo-sulfoximine in an unprecedented double sequential reduction/addition process of two [1.1.1]propellane units, with the formation of product 70 in 42% yield. Finally, original bromo fluoromethyl sulfoximine led to the synthesis of compounds 69 and 98 (the latter described in the ESI†). We next evaluated the possibility of performing a stereospecific version of the reaction by using chiral sulfoximine precursors, while assessing the configurational stability of the radical intermediate 8 (Fig. 2a).
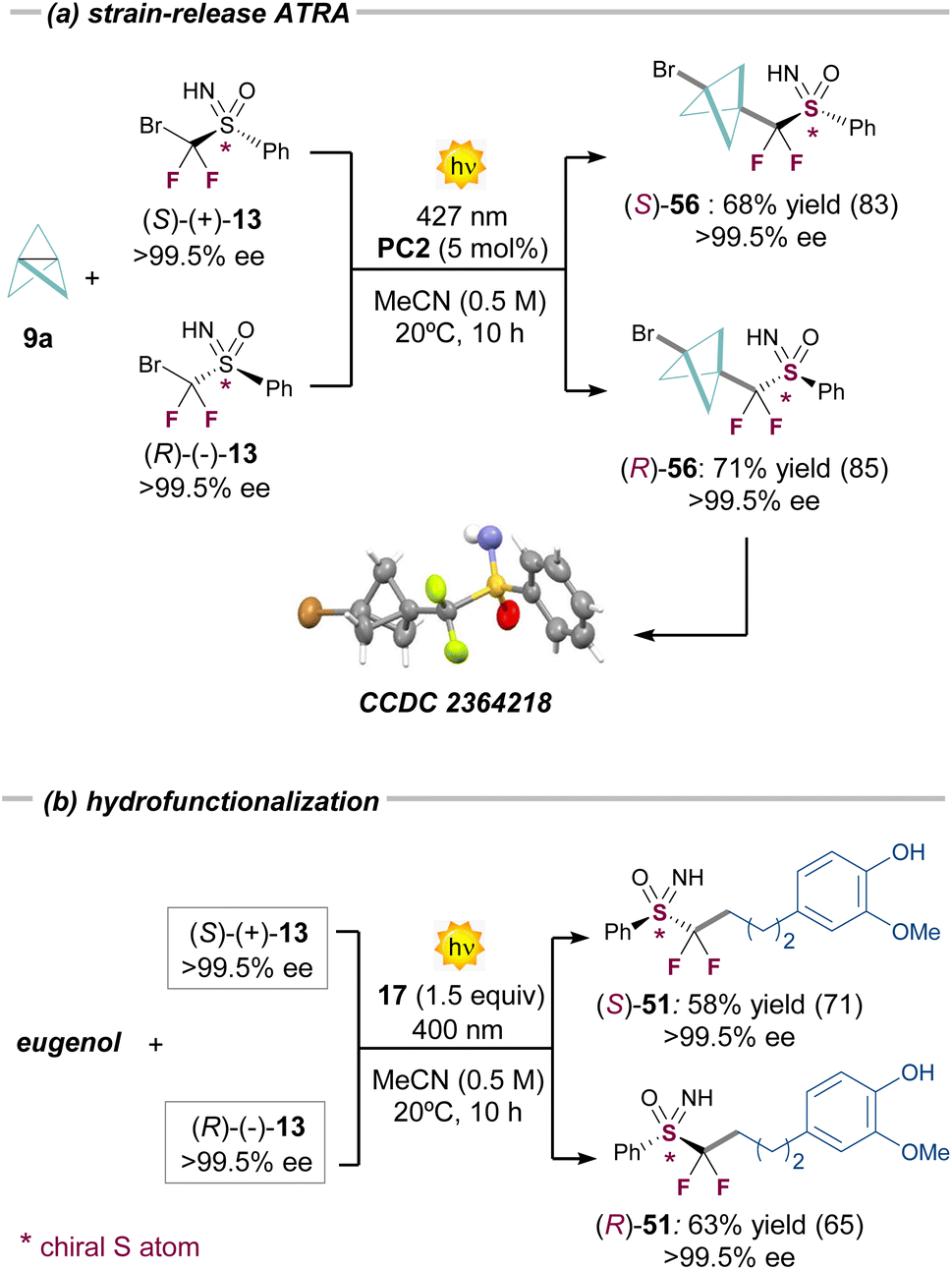 | ||
| Fig. 2 Reactions with enantiopure sulfoximines 13; (a) ATRA reaction, and (b) hydrofunctionalization. Yields refer to the isolated compounds. Values in the parentheses refer to 1H-NMR yields. | ||
To this end, we submitted both enantiomers of the bromodifluoroalkyl sulfoximine 13 to the different optimized reaction conditions developed in this study. Satisfactorily, the use of either the (S)- or (R)- enantiomers of 13 led to the formation of the enantiopure ATRA products (R)-56 and (S)-56, respectively (>99.5% ee). A similar result was observed in the hydrofunctionalization pathway using eugenol 3ab as the radical acceptor (Fig. 2b). In this case, the corresponding products (R)-51 and (S)-51 were also obtained without erosion of the chirality, corroborating that the chiral difluorosulfoximine radical 8 remains configurationally stable during both photochemical processes (see ESI Section G† for further examples).
Finally, to showcase the synthetic utility of the products, we carried out a series of product manipulations (Fig. 3). First, treatment of the ATRA product 21 with NaH (60% mineral oil) in DMF allowed the deprotonation of the –NH of the sulfoximine moiety, while triggering an intramolecular cyclization process to yield the 5-membered ring sulfoximine 71 in 25% yield. It is worth mentioning that there are no other methodologies to obtain these cyclic fluorinated sulfoximines, thus demonstrating the relevance of the obtained ATRA products to access other structures of higher complexity.
 | ||
| Fig. 3 Post functionalizations of some products. (a) Synthesis of F-containing cyclic sulfoximines. (b) Copper-mediated N-functionalization. Yields refer to the isolated compounds. | ||
Similarly, product 39 can also be treated with NaH (60% mineral oil) to induce intramolecular cyclization and yield the 6-membered ring sulfoximine 72 in 55% yield.
Furthermore, the hydrofunctionalized product 22 and the strain-released ATRA product 56 were successfully used in a copper catalyzed N-arylation process with phenyl iodide 73 and 3-iodopyridine 75,16 affording the N-phenyl substituted sulfoximine 74 and the N-pyridine substituted sulfoximine 76 in 87% and 50% yield, respectively.
Mechanistic investigations
We next focused on understanding the mechanisms accounting for the observed divergent reactivity. Under the ATRA conditions, PC3 or its EDA complex with the starting substrate (depending by the type of difluorosulfoximine used, see ESI, Section K.1† for further details) is the only absorbing species,9d whereas 17 is the main species absorbing at 400 nm in the hydrofunctionalization pathway (see ESI, Section K.1†). Stern–Volmer quenching studies showed that increasing amounts of the bromodifluoroalkyl sulfoximine 13 could effectively quench the emission of PC3, whereas no quenching was observed with the alkene 14 (Fig. 4a). Furthermore, the excited state redox potentials of both PC3 and 17 [Ered(PC3˙+/PC3*) = −2.30 V vs. SCE; Ered(17˙+/17*) = −2.28 V vs. SCE11a,17] are sufficiently negative to undergo a thermodynamically favoured SET reduction of the sulfoximine 13 [Ered(13/13˙−) = – 1.57 V vs. SCE]. | ||
| Fig. 4 (a) Stern–Volmer quenching studies. (b) EPR studies (detection key chiral radical). (c) Proposed mechanisms for the ATRA (left side) and hydrofunctionalization process (right side) developed in this study. | ||
We next carried out a spin trapping/EPR study to detect the key difluoroalkyl radical 8a, generated from the bromodifluoro sulfoximine 13 (Fig. 4b). A solution containing 13, PC3 and the spin trap 2-methyl-2-nitrosopropane (MNP) was prepared in acetonitrile. Upon irradiation under the standard reaction conditions, the signal reported in Fig. 4b was immediately observed. Its simulation (with a virtually perfect superimposition with the red dotted line) revealed the presence of a major radical species (over 90%) whose EPR spectrum showed twelve intense lines, due to hyperfine couplings with a nitrogen nucleus (aN = 11.6 G) and two fluorine nuclei at the α-position towards the nitrogen (aF1 = 22.1 G and aF2 = 14.2 G). On the basis of these hyperfine coupling constant (hfcc) values the spectrum recorded was assigned to the MNP adduct of a difluorinated carbon-centered radical and was perfectly compatible with the MNP-8a adduct (see Fig. 4b and ESI, Section J†). Because the CF2 group was linked to a chiral sulfur atom in 8a, the two fluorines in the spin adduct MNP-8a were not magnetically equivalent, as shown by the large difference between the two hfccs. Although this phenomenon is rather common in NMR, to the best of our knowledge this is the very first description of an EPR spectrum of a radical with two non-magnetically equivalent geminal fluorine nuclei.18 In addition, no EPR signal was detected before irradiation or in blank tests performed in the absence of either the substrate 13 or PC3.
Based on these experimental findings, we propose the divergent mechanism illustrated in Fig. 4c. On one hand, the ATRA pathway is initiated by a photoinduced SET process from the excited PC3* to sulfoximine 7, producing the difluorosulfoximine radical 8 and the radical cation of the photocatalyst (PC3˙+). The open-shell intermediate 8 participates in a radical addition process to alkene 3 (the same ATRA manifold with propellane 9 has been omitted for clarity), forming the intermediate 88. This radical (88) undergoes halogen atom transfer (XAT) with another molecule of sulfoximine 7, affording the ATRA product 10 along with the reactive radical 8. This chain propagation mechanism is supported by a relatively high quantum yield value of ϕ = 0.23 for 3 and 19.03 for 9. On the other hand, the hydrofunctionalization reaction is initiated by a photoinduced SET process from the excited Hantzsch ester 17* to 7, delivering the key radical 8 and the radical cation HEH˙+.17 In this scenario, the intermediate 77 formed after the radical addition undergoes a HAT process with another molecule of HEH to afford the desired product 11, alongside radical 78. Most likely, the rate of the HAT is faster than the XAT, thereby explaining the preferential formation of product 11 over 10. The very low quantum yield value registered for this process (ϕ = 0.02) suggests the activity of an in-cage mechanism. However, at the current stage we cannot exclude the operation of a very inefficient chain propagation mechanism, in which the radical 78 reduces 7, generating another reactive radical 8 and the Hantzsch pyridinium 79.19
Conclusions
In conclusion, we have developed a divergent synthetic platform for the efficient installation of valuable fluorinated sulfoximine moieties onto olefins and bicycloalkanes.20 Interestingly, by the rational selection of the reducing agent, either the excited PC3 or the Hantzsch ester 17, it is possible to channel the chemistry towards Br- or the H-containing fluorinated sulfoximine product, respectively. We have shown the generality of these processes with diverse classes of molecules, including olefins and propellanes bearing a large variety of functional groups (65 examples, up to 77% yield). Further, it was possible to transfer chiral sulfoximine radicals with complete stereoretention into the final products. Finally, we performed a series of product manipulations accessing structurally relevant synthetic targets such as cyclic 71 and 72. Mechanistic investigations revealed the key activity of the fluorinated radical 8 (characterized by EPR) that undergoes ATRA or hydrofunctionalization pathways.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. C., L. D. and E. M. conceived and directed the project. S. B., S. C., J. P., E. A., and G. D. performed the experiments and prepared the starting materials. S. B., S. C., J. P., and E. A. characterized the products and intermediates. B. T. performed the EPR analysis. G. P. performed the X-ray analysis. S. C., L. D. and E. M. wrote the manuscript with contributions from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MUR (Ministero dell’Università) PRIN2022PNRR23_01 (L. D.), and (European Research Council) ERC-Starting Grant 2021 SYNPHOCAT 101040025 (L. D.). The authors gratefully acknowledge the technical support units at the Department of Chemical Sciences (DiSC) of the University of Padova, in particular Dr Ilaria Fortunati, Samuel Pressi and Stefano Mercanzin. B. T. acknowledges the NIEHS for providing free public electron paramagnetic resonance software tools (Winsim simulation program and spin trap database). S. B. acknowledges UniPD for a doctoral fellowship. J. P. thanks the University of Paris-Saclay and the University of Padova for financial support via the grant BorsaDISC27 and Paris-Saclay grant as an international cotutelle collaboration. Dr Nicolas Vanthuyne (Aix Marseille Université, CNRS, UMR 7313- iSm2) is acknowledged for the preparative HPLC separation of the enantiomers of the sulfoximine 13.References
- (a) C. R Johnson, Comprehensive Organic Chemistry, 1st edn, 1979, vol. 3, p. 223 Search PubMed; (b) M. Reggelin and C. Zur, Synthesis, 2000, 1, 1 CrossRef; (c) S. G. Pyne, Sulfur Rep., 1999, 21, 281 CrossRef CAS; (d) M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225 CrossRef CAS PubMed; (e) U. Lücking, Angew. Chem., Int. Ed., 2013, 52, 9399 CrossRef PubMed; (f) Y. Han, K. Xing, J. Zhang, T. Tong, Y. Shi, H. Cao, H. Yu, Y. Zhang, D. Liu and L. Zhao, Eur. J. Med. Chem., 2021, 209, 112885 CrossRef CAS PubMed; (g) P. Mäder and L. Kattner, J. Med. Chem., 2020, 63, 14243 CrossRef PubMed; (h) U. Lücking, Org. Chem. Front., 2019, 6, 1319 RSC; (i) E. Boulard, V. Zibulski, L. Oertel, P. Lienau, M. Schäfer, U. Ganzer and U. Lücking, Chem. Eur J., 2020, 26, 4378 CrossRef CAS; (j) U. Lücking, Chem. Eur J., 2022, 28, e202201993 CrossRef PubMed . For selected recent examples, see: ; (k) S. Teng, Z. P. Schultz, C. Shan, L. Wojtas and J. M. Lopchuk, Nat. Chem., 2024, 16, 183 CrossRef CAS PubMed; (l) M. D. Glossbrenner, S. González-Granda, O. S. Nayal, E. A. Noten, C. M. Balintfy, D. A. Pratt and C. R. J. Stephenson, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-gw0xb; (m) Z. Zhong, B. J. W. Hocking, C. P. Brown, T.-K. Ma, A. J. P. White, D. J. Mann, A. Armstrong and J. A. Bull, Angew. Chem., Int. Ed., 2024, e202420028 Search PubMed; (n) N. S. Greenwood, Z. W. Boyer, J. A. Ellman and C. Gnamm, J. Med. Chem., 2025, 68, 4079 CrossRef CAS PubMed.
- E. Anselmi, B. Montigny, M. Lõkov, C. Kesküla, J. E. Soosaar, S. Tshepelevitsh, T. Billard, E. Magnier and I. Leito, Chem. Eur J., 2025, 31, e202402329 CrossRef CAS PubMed.
- Selected reviews: (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS; (d) V. Bizet and D. Cahard, Chem. Soc. Rev., 2014, 43, 135 RSC; (e) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem. Eur J., 2019, 25, 11797 CrossRef CAS PubMed; (f) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Chem. Eur J., 2021, 27, 17293 CrossRef CAS PubMed; (b) V. Bizet, R. Kowalczyk and C. Bolm, Chem. Soc. Rev., 2014, 43, 2426 RSC; (c) X. Shen and J. Hu, Eur. J. Org Chem., 2014, 4437 CAS , For selected examples of bioactive fluorinated sulfoximines, see: ; (d) Y. Zhu, R. B. Rogers and J. X. Huang, US Pat., US 20050228027, 2005 Search PubMed; (e) A. Plant, J. E. Boehmer and A. L. Peace, GB Pat., WO 2006037945, 2006 Search PubMed; (f) M. Haerter, H. Beck, P. Ellinghaus, K. Berhoerster, S. Greschat, K.-H. Thierauch and F. Suessmeier, German Pat., WO 2010054764, 2010 Search PubMed.
- (a) S. Chaabouni, J.-F. Lohier, A.-L. Barthelemy, T. Glachet, E. Anselmi, G. Dagousset, P. Diter, B. Pégot, E. Magnier and V. Reboul, Chem. Eur J., 2018, 64, 17006 CrossRef PubMed; (b) A.-L. Barthelemy, V. Certal, G. Dagousset, E. Anselmi, L. Bertin, L. Fabien, B. Salgues, P. Courtes, C. Poma, Y. El-Ahmad and E. Magnier, Org. Process Res. Dev., 2020, 24, 704 CrossRef CAS; (c) E. Magnier, Emerging Fluorinated Motifs: Synthesis, Properties, and Applications, 2020, vol. 2, 675 Search PubMed; (d) A.-L. Barthelemy and E. Magnier, C. R. Chim, 2018, 21, 711 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS; (c) M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed.
- (a) T. Koike, Chem. Rec., 2023, 23, e202300032 CrossRef CAS PubMed; (b) M. Briand, L. D. Thai, F. Bourdreux, N. Vanthuyne, X. Moreau, E. Magnier, E. Anselmi and G. Dagousset, Org. Lett., 2022, 24, 9375 CrossRef CAS PubMed; (c) T. Duhail, S. Messaoudi, G. Dagousset, J. Marrot, C. André-Barrès, E. Magnier and E. Anselmi, Adv. Synth. Catal., 2023, 365, 2392 CrossRef CAS.
- A. Prieto, P. Diter, M. Toffano, J. Hannedouche and E. Magnier, Adv. Synth. Catal., 2019, 361, 436 CAS.
- (a) N. Erdeljac, K. Bussmann, A. Schöler, F. K. Hansen and R. Gilmour, ACS Med. Chem. Lett., 2019, 10, 1336 CAS; (b) S. Meyer, J. Häfliger and R. Gilmour, Chem. Sci., 2021, 12, 10686 CAS; (c) Y. Kraemer, C. Ghiazza, A. N. Ragan, S. Ni, S. Lutz, E. K. Neumann, J. C. Fettinger, N. Nöthling, R. Goddard and J. Cornella, Angew. Chem., Int. Ed., 2022, 61, e202211892 CAS; (d) S. Cuadros, G. Goti, G. Barison, A. Raulli, T. Bortolato, G. Pelosi, P. Costa and L. Dell'Amico, Angew. Chem., Int. Ed., 2023, 62, e202303585 Search PubMed.
- While various difluoroalkylating agents, including sulfone derivatives such as BrCF2-SO2Ph, have been used for the incorporation of difluoroalkyl moieties into organic compounds, no radical ATRA or hydrofunctionalization processes have been developed with the corresponding difluoroalkylsulfoximines. For recent reviews on difluoroalkylations, see: (a) S. Barata-Vallejo and A. Postigo, Molecules, 2019, 24, 4483 CAS; (b) A. Lemos, C. Lemaire, A. Luxen and A. Synt, Adv. Synth. Catal., 2019, 361, 1500 CAS; (c) R. Jia, X. Wang and J. Hu, Tetrahedron Lett., 2021, 75, 153182 CAS; (d) J. Sheng, K.-J. Bian, Y.-M. Su, G.-X. Liao, R. Duan, C. Li, Z. Liu and X.-S. Wang, Org. Chem. Front., 2020, 7, 617 CAS.
- (a) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Org. Biomol. Chem., 2019, 17, 6936 CAS; (b) X. Huang, S. Luo, O. Burghaus, R. D. Webster, K. Harms and E. Meggers, Chem. Sci., 2017, 8, 7126 RSC; (c) H. Yan, Y. Liu, X. Feng and L. Shi, Org. Lett., 2023, 25, 8116 CrossRef CAS PubMed.
- P. Ertl, E. Altmann and J. M. McKenna, J. Med. Chem., 2020, 63, 8408 CrossRef CAS PubMed.
- V. Corti, J. Dosso, M. Prato and G. Filippini, J. Org. Chem., 2023, 88, 6008 CrossRef CAS PubMed.
- (a) K. B. Wiberg, S. T. Waddell and K. Laidig, Tetrahedron Lett., 1986, 27, 1553 CrossRef CAS; (b) P. Bellotti and F. Glorius, J. Am. Chem. Soc., 2023, 145, 20716 CrossRef CAS PubMed; (c) S. Cuadros, J. Paut, E. Anselmi, G. Dagousset, E. Magnier and L. Dell'Amico, Angew. Chem., Int. Ed., 2024, e202317333 CAS.
- (a) N. Frank, J. Nugent, B. R. Shire, H. D. Pickford, P. Rabe, A. J. Sterling, T. Zarganes-Tzitzikas, T. Grimes, A. L. Thompson, R. C. Smith, C. J. Chofield, P. E. Brennan, F. Duarte and E. A. Anderson, Nature, 2022, 611, 721 CAS; (b) T. Iida, J. Kanazawa, T. Matsunaga, K. Miyamoto, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2022, 144, 21848 CrossRef CAS PubMed.
- Y. Macé, B. Pégot, R. Guillot, C. Bournaud, M. Toffano, G. Vo-Thanh and E. Magnier, Tetrahedron, 2011, 67, 7575 CrossRef.
- G. S. Yedase, S. Venugopal, P. Arya and V. R. Yatham, Asian J. Org. Chem., 2022, 11, e202200478 CrossRef.
- D. R. Duling, J. Magn. Reson., Ser. B , 1994, 104(2), 105 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- S. Baldon, L. Dell'Amico and S. Cuadros, Eur. J. Org Chem., 2024, e202400604 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2364218. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01068c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |