
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical ammonia oxidation with a homogeneous molecular redox mediator†
Tarisha
Gupta
 *,
Sanyam
,
Shivani
Saraswat
,
Anirban
Mondal
* and
Biswajit
Mondal
*
*,
Sanyam
,
Shivani
Saraswat
,
Anirban
Mondal
* and
Biswajit
Mondal
*
Department of Chemistry, IIT Gandhinagar, Palaj, Gujarat-382055, India. E-mail: mondal.biswajit@iitgn.ac.in
First published on 2nd July 2025
Abstract
Ammonia (NH3) is a promising carbon-free energy carrier due to its high energy density and hydrogen storage capacity. Its utilization in energy systems relies on the ammonia oxidation reaction (AOR), which is critical for direct ammonia fuel cells (DAFCs) and hydrogen production. Herein, we explore a robust and inexpensive ferrocene-based molecular electrochemical mediator, N-pyridylferrocenecarboxamide (Fcpy), for AOR. The Fcpy-mediated AOR exhibits the N2 faradaic efficiency (FE) of 94.7%, along with the concomitant production of H2 (FE = 87.3%). Mechanistic studies reveal the crucial role of H-bonding through the pyridyl moiety of Fcpy in facilitating N–H bond activation. Computational analysis further corroborates the observed reaction pathways, providing deeper insights. This work highlights the potential of molecular catalysts to advance ammonia oxidation and underscores their role in sustainable energy systems.
Introduction
The increasing global energy demand and the urgency to mitigate CO2 emissions from fossil fuel combustion necessitate the transition to sustainable, carbon-free energy sources.1 Among the promising candidates, ammonia (NH3) has garnered significant attention due to its high energy density, ease of liquefaction under moderate conditions, and established global production infrastructure.2–4 While ammonia is predominantly recognized for its applications in fertilizers and industrial chemicals, it is increasingly being explored as an alternative fuel and hydrogen carrier in the transition toward green energy solutions.5–9 It has a hydrogen storage capacity of 17.7 wt%, a high energy density of 3000 Wh kg−1, and a capacity for safe storage and transportation under moderate pressures.10,11 Compared to hydrogen, which faces challenges in storage and transportation, ammonia offers practical benefits due to its well-established infrastructure and the ability to detect leaks at very low concentrations. Furthermore, ammonia can be synthesized via emerging green electrochemical methods, significantly reducing greenhouse gas emissions associated with the traditional Haber–Bosch process.1,12 The utilization of ammonia in energy applications relies on its electrochemical oxidation through the ammonia oxidation reaction (AOR).13 AOR is a pivotal step for electricity generation in direct ammonia fuel cells (DAFCs) and hydrogen production via ammonia splitting (Scheme 1).14–16 However, the widespread application of ammonia-based energy systems is constrained by sluggish AOR kinetics, high overpotentials, and catalyst degradation.11,17,18 | ||
| Scheme 1 Electrochemical reactions at the anode and cathode during electrochemical AOR (eAOR). | ||
Molecular catalysts offer distinct advantages that facilitate detailed mechanistic insights into ammonia activation.1,19–21 Recent advances highlight the potential of transition-metal-based systems to address these challenges (Scheme 2).22–34 Despite these promising developments, AOR remains underexplored compared to other catalytic processes like water oxidation. Challenges include activating the strong N–H bonds in ammonia, competition from side reactions, and catalyst stability under operational conditions.35,36 A recent report demonstrated electrocatalytic ammonia oxidation via proton-coupled electron transfer (PCET) using pyridyl-functionalized ferrocene mediators that activate NH3 through hydrogen bonding to a pendant pyridine.37
 | ||
| Scheme 2 Selected molecular electrocatalysts reported for mediated AOR in chronological order. | ||
This article explores the application of our recently reported molecular redox mediator, N-pyridylferrocenecarboxamide (Fcpy),38 for eAOR. There is an emphasis on understanding the site-separated PCET pathways and addressing the current limitations. Despite the lower stability of ferrocene in the presence of a base, we aim to unlock the full potential of ammonia as a sustainable energy carrier and accelerate its integration into carbon-free energy cycles.
Result and discussion
The cyclic voltammetry (CV) studies were performed in common non-aqueous solvents, acetonitrile (MeCN), and dimethyl sulfoxide (DMSO)32 using a 0.1 M n-tetrabutylammonium tetrafluoroborate (TBAF) supporting electrolyte and a glassy carbon (GC) working electrode. A reversible redox feature is observed at 0.22 V vs. Fc+/0 for Fcpy owing to the Fe+/0 process of the ferrocene moiety.38 Saturation of NH3 was done by bubbling aqueous NH3 solution in 4 mL of electrolyte containing 100 mM TBAF for 20 minutes (for experimental setup, see Fig. S1†), resulting in an NH3 concentration of 0.5 M in MeCN and 1.0 M in DMSO as calculated using the NMR method (Fig. S2†). The addition of NH3 resulted in a substantial increase in the oxidation current, indicating electrocatalytic ammonia oxidation. In MeCN, the onset potential at the inflection of catalytic current from the background was measured as 20 mV vs. Fc+/0 (Fig. 1a), corresponding to an overpotential of ∼960 mV (for detailed discussion, refer Section S1.3 of ESI†).37 The onset potential at 10 μA was measured as 94 mV vs. Fc+/0, corresponding to an overpotential of 1.03 V. Similarly, Fcpy exhibits AOR in DMSO with an onset potential of 79 mV (Fig. 1b). Foot-of-the-wave analysis (FOWA) was used for the kinetic analysis and determination of the apparent rate constant (kobs) for the mediated AOR in both solvent systems (Fig. 1c and Section S1.4†).39 The mediator shows better performance in terms of kobs in DMSO (6.0 s−1) than in MeCN (0.3 s−1) in the current range of 15–25 μA. For better understanding, kobs was determined in an additional range of 9–15 μA at the foot of the catalytic wave (summarized in Table S1 and Fig. S3†), where it is also found that the rate is higher in DMSO, which may be attributed to the higher solubility of NH3 in DMSO as compared to MeCN.32,37 Using the peak-current method kobs for the mediated AOR in MeCN is calculated as 0.046 ± 0.002 s−1 (Fig. S4 and eqn (S5) in Section S1.5†). The apparent difference between rate constants from FOWA and the peak current method arises because FOWA reflects the intrinsic, ideal catalytic rate excluding side reactions, while the peak current method captures true experimental factors such as catalyst degradation and side reactions, leading to a lower apparent rate. MeCN is a widely reported solvent for most non-aqueous homogeneous systems, and the thermodynamic potential for ammonia oxidation is well-studied in this medium.40 Keeping these in consideration, all further analyses were performed using the MeCN solvent. | ||
| Fig. 1 Cyclic voltammograms of Fcpy-mediated vs. blank AOR: N2 purged (dotted) and NH3 purged (bold) in (a) MeCN and (b) DMSO; (c) corresponding FOWA for mediated AOR in MeCN (red) and DMSO (green). Experimental conditions: 1 mM Fcpy (mediator); 0.5 M NH3 in MeCN and 1.0 M NH3 in DMSO; 100 mM TBAF as supporting electrolyte in the respective solvent; glassy carbon, Pt wire, and Ag/AgCl were used as the working, counter, and reference electrodes, respectively; 298 K; the potential is adjusted with respect to Fc+/0. | ||
To investigate the kinetics of the AOR, the electrocatalytic current was studied as a function of the concentration of mediator, ammonia, and exogenous base (pyridine). In an ammonia-saturated solution, the electrocatalytic current increases linearly with the mediator (Fcpy) concentration (0.1–1.0 mM) (Fig. S5†), indicating that the reaction is first-order w.r.t. mediator. For the NH3 concentration dependence study, the aqueous ammonia solution was directly added to the acetonitrile to maintain the corresponding concentration. When the aqueous ammonia concentration was varied between 10–100 mM, the electrocatalytic current also increased linearly with [aq. NH3]1/2 (Fig. S6†), suggesting the process to be first-order w.r.t. the substrate, NH3. To rule out the role of water in aqueous ammonia solution during the AOR, a control experiment with the equivalent volume (taken during the aq. NH3 concentration dependence experiment) of water was added, which shows no dependence of water concentration on the reaction rate. This suggests that the electrocatalytic current is due to AOR and not because of OER (Fig. S7†). However, the electrocatalytic AOR current remains unchanged with increasing concentrations of exogenous base, pyridine (Fig. S8†), implying a PCET step. Overall, it was observed that the electrocatalytic AOR is first-order w.r.t. [Fcpy] and [aq. NH3], but zeroth-order w.r.t. [ext. py]. Thus, the rate law is established as rate = kcat[Fcpy][NH3].
Further, the reaction mixture of Fcpy and saturated NH3 was analyzed using 1H-NMR to get a mechanistic insight into ammonia interactions with the pyridine moiety of Fcpy. 1H-NMR spectra in CDCl3 revealed peak broadening and an up-field chemical shift of the py protons in the region 7–9 ppm, as shown in Fig. 2a. A similar set of experiments was repeated for the combination of pyridine and NH3, and that showed similar observations (Fig. 2b), confirming hydrogen bonding interactions between the pyridine and NH3. To exclude the possibility of any interaction with water (present in the aq. NH3 gas), a control experiment was performed by bubbling Ar from water for 1 h. There was no up-field chemical shift observed in this case as shown in Fig. S9.†
 | ||
| Fig. 2 1H-NMR spectra (500 MHz, 298 K, CDCl3): (a) Fcpy and (b) py solution upon bubbling aq. NH3 gas for 1 h. | ||
To assess the product formation during eAOR, controlled-potential electrolyses (CPE) for the mediated and non-mediated eAOR were conducted in MeCN using a 1 × 1 cm2 carbon cloth working electrode. The electrolysis was performed at 0.28 V (vs. Fc+/0) for 1 hour in a single-compartment cell with constant stirring. The chamber contents and headspace were fully purged with Argon to remove N2 and O2, followed by NH3 saturation. During bulk electrolysis, the current remained nearly constant around 0.2 mA (Fig. S10†) for mediated eAOR, while the charge consumption for Fcpy-mediated and non-mediated (blank) eAOR were 1.2C and 0.3C, respectively, under identical conditions (Fig. 3a). For the Fcpy-mediated eAOR electrolysis under otherwise similar conditions was also performed in a two-compartment cell (Fig. S11†). Before electrolysis, 0.2 mL sample from the headspace of the electrolysis cell was injected and analyzed using gas chromatography (GC), which indicates the presence of a slight amount of air. After 1 hour of electrolysis, the gaseous products were analyzed, revealing a H2/N2 ratio of 2.8 for the mediated AOR, with trace amounts of O2 observed (Fig. S12†). The % faradaic efficiencies (FE) were calculated for the anodic as well as cathodic reactions. For the 2e-process at cathode, the FE for H2 was found to be 87.3% (13.3% for blank AOR), whereas for the 6e-process at anode, FE of N2 was around 94.7% for the mediated AOR, as shown in Fig. 3b. However, please note that the N2 quantification is extremely difficult due to excess N2 in the background air (before electrolysis) and any unavoidable air leakage in the cell during electrolysis.7 Apart from gaseous products, the possibility of other products such as nitrite and nitrate41 dissolved in the electrolyte solution was also assessed. Using the modified Griess colorimetric method (for details, refer to Section S1.6b†).42 Only the nitrite was detected with FE of <1%, but no nitrate was formed.
 | ||
| Fig. 3 (a) Constant potential electrolysis at 0.28 V (vs. Fc+/0) for the same duration without (blue) and with (red) 1 mM Fcpy using 1 × 1 cm2 carbon-cloth as working electrode. Experimental conditions: 100 mM TBAF as supporting electrolyte in acetonitrile, 3-electrode setup in an undivided cell, 298 K; (b) summary of product quantification results after electrolysis with an average of three measurements. | ||
UV-Vis spectra of the electrolyte solution before and after electrolysis showed no significant change in peak positions (Fig. S13†). However, the intensity of the bands decreases (vide infra). Furthermore, the dynamic open-circuit potential (OCP) was measured and averaged for 5 minutes as −0.48 V, −0.81 V, and −0.92 V for Fcpy only, NH3 only, and the mixture of Fcpy + NH3, respectively (Fig. S14†). The non-mediated AOR requires more driving force, as evident from its less negative OCP value. The highly negative OCP value of NH3 in the presence of the electrochemical mediator (Fcpy) indicates the feasibility of mediated AOR in the presence of Fcpy.43 To check for the homogeneity of the catalytic system, multiple cycle CV was performed in the presence of the mediator, followed by a rinse test (Fig. S15†).44 After the Fcpy-mediated AOR CV, the glassy carbon electrode was rinsed with dry MeCN. There was a visible deposition on the electrode surface. The same electrode was then inserted into an electrochemical cell containing a fresh NH3/MeCN/0.1 M TBAF solution to check for the eAOR in the absence of soluble Fcpy. The rinse test showed only a current corresponding to the blank NH3 oxidation (similar to grey bold CV, corresponding to blank NH3 oxidation in Fig. 1a), confirming that the electrocatalysis is not due to the heterogeneous species formed during the CV cycles. To understand the nature of heterogeneous deposition, FE-SEM and EDS analyses of the rinsed carbon cloth electrode after mediated eAOR CPE experiments were further performed. The SEM images showed the presence of layers grown over the electrode surface, which is absent after blank eAOR (Fig. S16†). The elemental mapping and EDS data also confirm that the Fe is deposited on the rinsed carbon cloth electrode after Fcpy-mediated eAOR electrolysis (Fig. S17†).
To evaluate the role of the pyridinic nitrogen in facilitating H-bonding with NH3 and its influence on catalytic activity, the pyridine arm in the ligand framework was replaced with a phenyl group (in N-phenylferrocenecarboxamide, FcPh). This structural modification, which eliminates the potential for hydrogen bonding with NH3, led to a notable decrease in catalytic current, suggesting lower rate as shown in Fig. S18.† This highlights the importance of the pyridine N-site in the catalytic process. Additionally, to probe the effect of base strength, the pyridine moiety (pKa = 12.53) was replaced with 2-methylpyridine (pKa = 13.32) to afford N-(6-methylpyridin-2-yl)ferrocenecarboxamide (FcpyMe). A modest increase in catalytic activity was observed with this substitution, as shown in Fig. S19,† suggesting that increased basicity may enhance NH3 activation via H-bonding in the PCET mechanism.
Computational studies
All quantum mechanical calculations were performed using Gaussian09, with GaussView utilized for visualization.45 Density functional theory (DFT) calculations were conducted using the B3LYP functional and the 6-311++g(d,p) basis set. The conductor-like polarizable continuum model (CPCM) with acetonitrile as the solvent (ε = 35.09) was applied to account for solvent effects. At this theory level, the mediated reaction mechanism with ammonia was investigated. Frequency calculations were performed to determine the Gibbs free energy changes (ΔG) for each reaction step. The reaction mechanism was analyzed in four key steps, as shown in Fig. 4a (step 1 to step 4).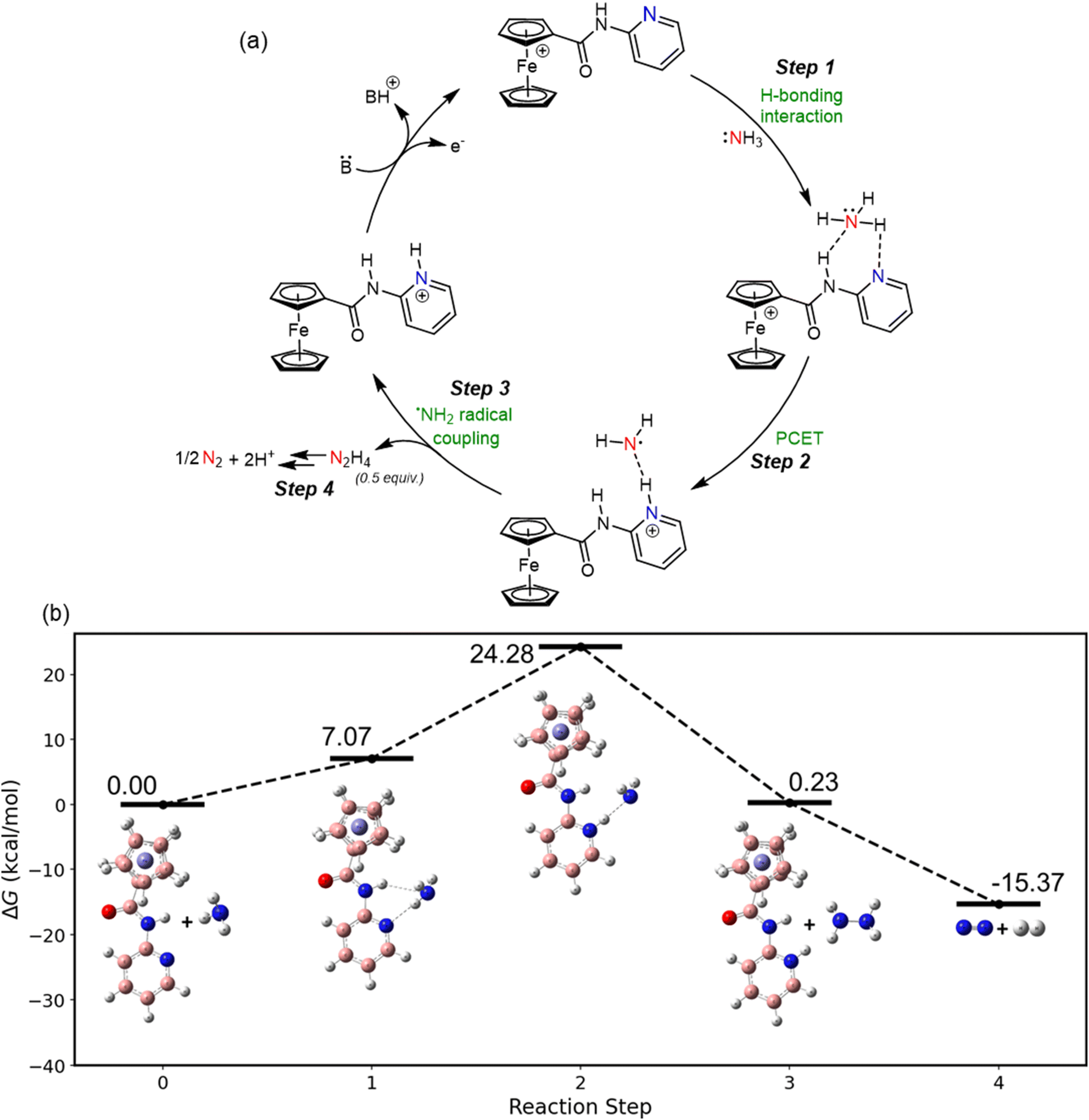 | ||
| Fig. 4 (a) Scheme for the proposed catalytic cycle for eAOR in MeCN using Fc+py (generated at anode) as a mediator; (b) free energy profile diagram illustrating the reaction pathway between Fcpy and ammonia in MeCN. | ||
Fig. 4b shows that the ferrocenium-pyridine ion (Fc+py) reacts with ammonia (step 1), with a calculated Gibbs free energy change of 7.07 kcal mol−1. This step is entropically unfavorable due to the loss of randomness of the gaseous ammonia molecule upon association with the Fc+py ion, resulting in a negative entropy change. The association occurs via hydrogen bond formation between the nitrogen atom of the Fc+py and the hydrogen atom of ammonia, as evident from Fig. 2. The hydrogen bond is strong, with a bond distance of 2.19 Å between the nitrogen atom of pyridine and the hydrogen atom of ammonia. Fig. S20† provides the other relevant distances. In the subsequent step (step 2), proton-coupled electron transfer (PCET) occurs, where a hydrogen atom from ammonia is transferred to the pyridine group of the Fc+py mediator. This step is thermodynamically unfavorable, with a Gibbs free energy change of 17.21 kcal mol−1. The third step involves the formation and release of hydrazine (N2H4), a reaction-driving step that is thermodynamically favorable, with a Gibbs free energy change of −24.05 kcal mol−1 (step 3). Hydrazine is commonly reported as a strong reductant and can itself get electrochemically oxidized at a very low potential (as shown in Fig. S21†).46–48 Soon after hydrazine forms as an intermediate during the eAOR, it gets oxidised into half a mole of nitrogen gas (N2) in the final step (step 4). This step is highly thermodynamically favorable due to gaseous molecule production, with a Gibbs free energy change of −15.60 kcal mol−1. We have also estimated the kinetic isotope effect (KIE) for the PCET step through computational analysis (details provided in Section S1.7 of the ESI†). The calculated KIE value of 1.2 indicates a measurable isotope effect. This presumably indicates PCET step as rate-limiting in the reaction mechanism.
Additionally, to analyze changes in the electron density of the Fcpy mediator during its conversion from its neutral (Fcpy) to cationic form (Fc+py), an electron density difference plot was generated. The plot is shown in Fig. 5. The plot reveals two distinct lobes: the red lobes (electron-deficient region indicating electron density loss) and the blue lobes (electron-dense region indicating electron density gain). This visualization demonstrates that the lone pair electron density of ammonia, shown in red in the neutral form, shifts towards the Fc+py mediator, represented by the red lobe of ammonia in the cationic form. Similarly, the electron density plot for both neutral and cationic forms is shown in Fig. S22,† and the electron density mapped electrostatic potential diagram is provided in Fig. S23.† The color of the intensity scales varies from red to blue, showing the potential change from negative to positive. The blue color is on the atoms with an electropositive character or electron deficiency, while the red is more on the species with more electron density or electronegative potential.
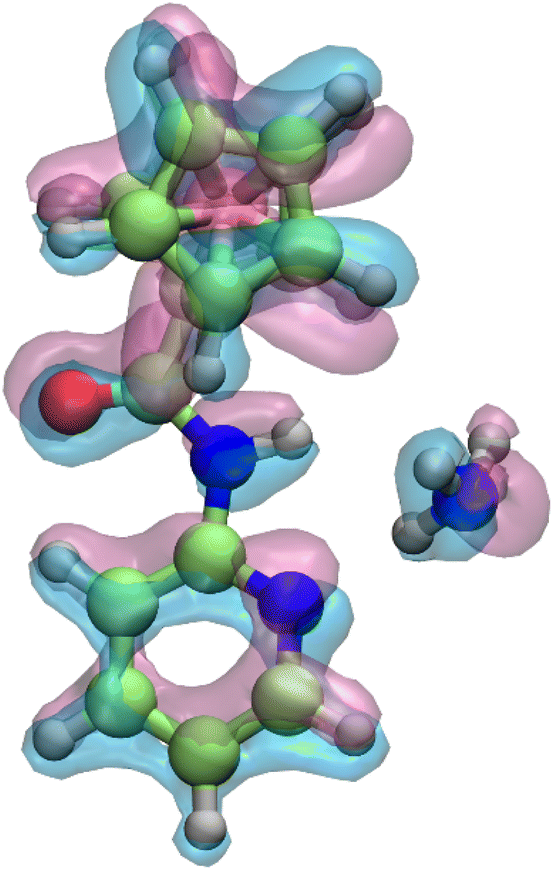 | ||
| Fig. 5 Electron density difference plot comparing the neutral (red) and cationic (blue) forms of the Fcpy with ammonia, with an isovalue of 0.025, showing the redistribution of electron density upon ionization. | ||
So far, all reported molecular catalysts for the AOR operate via a mechanism that involves the coordination of ammonia to the redox-active metal center, followed by PCET steps. This coordination is generally considered a prerequisite for facilitating the activation and subsequent oxidation of ammonia. However, in this study, we propose an alternative catalytic cycle in which ammonia oxidation proceeds without direct coordination to the redox-active cofactor. Instead, the oxidation mechanism is driven by ammonia association stabilized by weak interactions, followed by electron transfer and proton transfer steps. As discussed earlier, a recent report demonstrated electrocatalytic ammonia oxidation via PCET using pyridyl-functionalized ferrocenium mediators that activate NH3 through hydrogen bonding to a pendant pyridine.37 Our work employs a mechanistically analogous strategy, however, it exhibits a more positive redox potential, higher diffusion coefficient, and significantly enhanced catalytic rates in both MeCN and DMSO, while advantageous in terms of NH3 source, i.e., electrocatalytic performance under aqueous-compatible conditions (Table S2†). This departure from the conventional coordination-driven approach provides new insights into the mechanistic diversity of molecular eAOR catalysis and suggests that effective ammonia oxidation can be achieved through alternative non-coordination-based pathways.
Conclusions
This study underscores the potential of a molecular electrochemical mediator, N-pyridylferrocenecarboxamide (Fcpy), to carry out ammonia oxidation via site-separated PCET mechanism. By achieving impressive faradaic efficiencies for both nitrogen (N2) and hydrogen (H2) production, the Fcpy-mediated eAOR exemplifies the promise of leveraging PCET pathways to overcome critical challenges in ammonia utilization. While the stability of the catalyst presents an area for improvement, the mechanistic insights and computational validations outlined in this work offer valuable guidance for future catalyst design. The deviation from the conventional coordination-driven (ammonia to the redox-active metal center) approach provides new insights into the mechanistic diversity of molecular AOR catalysis and suggests that effective ammonia oxidation can be achieved through alternative non-coordination-based pathways. The proposed mechanism opens avenues for the design of catalysts that leverage non-traditional activation strategies. These findings highlight potential strategies to advance the development of more robust catalytic systems. Using aqueous ammonia as a substrate is a particularly noteworthy aspect of this work, offering a more practical and sustainable approach. As we continue to refine and optimize these systems, the integration of ammonia as a versatile energy carrier becomes an increasingly achievable goal, advancing the vision of a sustainable, carbon-neutral future.Data availability
The additional electrochemical, pre & post-electrolysis characterizations and computational details are available in the ESI.†Author contributions
B. M. and T. G. designed the research; T. G. and S. S. performed synthesis and electrochemistry; Sanyam and A. M. performed DFT; B. M., T. G. and S. S. analyzed the electrochemical data. Sanyam and A. M. analyzed computational data; B. M., T. G., and Sanyam wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely acknowledge IIT Gandhinagar internal grant for funding. T. G. acknowledges PMRF for the fellowship. Sanyam thanks CSIR for the fellowship. S. S. thanks SRIP-IIT Gandhinagar. Sanyam and A. M. thank IIT Gandhinagar for computational resources.Notes and references
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 17845–17858 CrossRef CAS PubMed.
- N. Kostopoulos, M. Robert and A. Sekretareva, ChemElectroChem, 2024, 11, 1–9 CrossRef.
- Y. Tian, Z. Mao, L. Wang and J. Liang, Small Struct., 2023, 4, 2200266 CrossRef CAS.
- H. Y. Liu, H. M. C. Lant, J. L. Troiano, G. Hu, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2022, 144, 8449–8453 CrossRef CAS PubMed.
- H. Liu, X. Xu, D. Guan and Z. Shao, Energy and Fuels, 2024, 38, 919–931 CrossRef CAS.
- A. M. Beiler, W. Li, A. Denisiuk, E. Palomares and A. Llobet, J. Energy Chem., 2024, 92, 292–295 CrossRef CAS.
- H. Roithmeyer, L. Sévery, T. Moehl, B. Spingler, O. Blacque, T. Fox, M. Iannuzzi and S. D. Tilley, J. Am. Chem. Soc., 2024, 146, 430–436 CrossRef CAS PubMed.
- M. F. Shabik, M. M. Hasan, K. A. Alamry, M. M. Rahman, Y. Nagao and M. A. Hasnat, J. Electroanal. Chem., 2021, 897, 115592 CrossRef CAS.
- A. M. Beiler, A. Denisiuk, J. Holub, F. J. Sánchez-Baygual, M. Gil-Sepulcre, M. Z. Ertem, D. Moonshiram, A. Piccioni and A. Llobet, ACS Energy Lett., 2023, 8, 172–178 CrossRef CAS PubMed.
- Z. Ni, J. Liu, Y. Wu, B. Liu, C. Zhao, Y. Deng, W. Hu and C. Zhong, Electrochim. Acta, 2015, 177, 30–35 CrossRef CAS.
- B. Zhou, N. Zhang, Y. Wu, W. Yang, Y. Lu, Y. Wang and S. Wang, J. Energy Chem., 2021, 60, 384–402 CrossRef CAS.
- H. Liu, H. M. C. Lant, C. Decavoli, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2025, 147, 1624–1630 CrossRef CAS PubMed.
- K. Yang, J. Liu and B. Yang, J. Catal., 2022, 405, 626–633 CrossRef CAS.
- Z. H. Lyu, J. Fu, T. Tang, J. Zhang and J. S. Hu, EnergyChem, 2023, 5, 100093 CrossRef CAS.
- H. S. Pillai, Y. Li, S. H. Wang, N. Omidvar, Q. Mu, L. E. K. Achenie, F. Abild-Pedersen, J. Yang, G. Wu and H. Xin, Nat. Commun., 2023, 14, 1–11 Search PubMed.
- F. Fang, Q. Cheng, M. Wang, Y. He, Y. Huan, S. Liu and T. Qian, ACS Nano, 2025, 19, 1260–1270 CrossRef CAS PubMed.
- S. Khan, S. S. Shah, M. A. R. Anjum, M. R. Khan and N. K. Janjua, Coatings, 2021, 11, 313 CrossRef CAS.
- C. Dai, X. Yuan, Y. Wang, X. Liu, C. Ju, L. Hu, S. He, R. Shi, Y. Liu, J. Zhang, Y. Zhu and J. Wang, Small, 2025, 2501582 CrossRef CAS PubMed.
- J. Li, F. Zhang, H. Xiong, Y. Cai and B. Zhang, Sci. China: Chem., 2024, 67, 3976–3993 CrossRef CAS.
- H. Y. Liu, H. M. C. Lant, C. C. Cody, J. Jelušić, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2023, 13, 4675–4682 CrossRef CAS.
- J. Xie, T. Yang, L. Hong, H. Li, B. Li, Z. Guo, Y. Liu and T. C. Lau, J. Am. Chem. Soc., 2025, 147, 14211 CrossRef CAS PubMed.
- M. D. Zott, P. Garrido-Barros and J. C. Peters, ACS Catal., 2019, 9, 10101–10108 CrossRef CAS.
- M. D. Zott and J. C. Peters, J. Am. Chem. Soc., 2021, 143, 7612–7616 CrossRef CAS PubMed.
- H. Toda, K. Kuroki, R. Kanega, S. Kuriyama, K. Nakajima, Y. Himeda, K. Sakata and Y. Nishibayashi, ChemPlusChem, 2021, 86, 1511–1516 CrossRef CAS PubMed.
- Y. Li, J. Y. Chen, Q. Miao, X. Yu, L. Feng, R. Z. Liao, S. Ye, C. H. Tung and W. Wang, J. Am. Chem. Soc., 2022, 144, 4365–4375 CrossRef CAS PubMed.
- M. D. Zott and J. C. Peters, ACS Catal., 2023, 13, 14052–14057 CrossRef CAS PubMed.
- F. Habibzadeh, S. L. Miller, T. W. Hamann and M. R. Smith, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2849–2853 CrossRef CAS PubMed.
- R. Borougeni, C. Greene, J. A. Bertke and T. H. Warren, ChemRxiv, 2019, 1–11 Search PubMed.
- S. Zhao, X. Zhang, G. Chen, T. Cheng, X.-L. Ding, S.-D. Zhong, S.-P. Yang, P. He and X.-Y. Yi, Inorg. Chem., 2024, 63, 23150–23157 CrossRef CAS PubMed.
- J. Holub, N. Vereshchuk, F. J. Sánchez-Baygual, M. Gil-Sepulcre, J. Benet-Buchholz and A. Llobet, Inorg. Chem., 2021, 60, 13929–13940 CrossRef CAS PubMed.
- M. J. Trenerry, C. M. Wallen, T. R. Brown, S. V. Park and J. F. Berry, Nat. Chem., 2021, 13, 1221–1227 CrossRef CAS PubMed.
- M. E. Ahmed, M. Raghibi Boroujeni, P. Ghosh, C. Greene, S. Kundu, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2022, 144, 21136–21145 CrossRef CAS PubMed.
- L. Liu, S. I. Johnson, A. M. Appel and R. M. Bullock, Angew. Chem., Int. Ed., 2024, 63, e202402635 CrossRef CAS PubMed.
- K. Nakajima, H. Toda, K. Sakata and Y. Nishibayashi, Nat. Chem., 2019, 11, 702–709 CrossRef CAS PubMed.
- J. H. Jang, S. Y. Park, D. H. Youn and Y. J. Jang, Catalysts, 2023, 13, 803 CrossRef CAS.
- D. Guo, F. Liu, T. Liu, Z. Ji, X. Fan, B. Sun, P. Zhang, F. Li and F. Li, ACS Catal., 2025, 8166–8173 CrossRef.
- M. E. Ahmed, R. J. Staples, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2025, 147, 6514–6522 CrossRef CAS PubMed.
- T. Gupta, N. Yati, N. Sanyam, A. Mondal and B. Mondal, ACS Catal., 2024, 14, 17690–17698 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- B. M. Lindley, A. M. Appel, K. Krogh-Jespersen, J. M. Mayer and A. J. M. Miller, ACS Energy Lett., 2016, 1, 698–704 CrossRef CAS.
- V. T. T. Phan, Q. P. Nguyen, B. Wang and I. J. Burgess, J. Am. Chem. Soc., 2025, 147, 10758 CrossRef CAS PubMed.
- H. Y. Liu, H. M. C. Lant, C. Decavoli, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2025, 147, 1624–1630 CrossRef CAS PubMed.
- B. Zhu, B. Dong, F. Wang, Q. Yang, Y. He, C. Zhang, P. Jin and L. Feng, Nat. Commun., 2023, 14, 1686 CrossRef CAS PubMed.
- Y.-H. Wang, Y.-J. Tseng, S. V. Kumbhar, H.-T. Chang, T. Yang, I.-C. Lu and Y.-H. Wang, Chem.–Eur. J., 2025, 31(10), e202403583 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheese-man, G. Scalmani, V. Barone, B. Mennucci and G. A. Peterssonet al., Gaussian 09, Revision E.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- F. Wang, J. B. Gerken, D. M. Bates, Y. J. Kim and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 12349–12356 CrossRef CAS PubMed.
- X. Cao, B. Wang and Q. Su, J. Electroanal. Chem., 1993, 361, 211–214 CrossRef CAS.
- G. Chen, P. He, C. Liu, X. F. Mo, J. J. Wei, Z. W. Chen, T. Cheng, L. Z. Fu and X. Y. Yi, Nat. Catal., 2023, 6, 949–958 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00730e |
| This journal is © The Royal Society of Chemistry 2025 |