
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA triply linked propellane-nanoring hybrid serving as a good host†
Yan
Chen‡
a,
Xingyu
Chen‡
b,
Lin
Li‡
 c,
Xiangping
Chen‡
a,
Jianlong
Xia
b and
Lei
Zhang
*a
c,
Xiangping
Chen‡
a,
Jianlong
Xia
b and
Lei
Zhang
*a
aBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: zhl@mail.buct.edu.cn
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Center of Smart Materials and Devices, Wuhan University of Technology, Wuhan 430070, P. R. China
cKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, College of Chemistry and Materials Science, Zhejiang Normal University, Jinhua 321004, P. R. China
First published on 11th March 2025
Abstract
A novel propellane-nanoring hybrid, TPPTI-[9]CMP, was synthesized by triply combining [9]cyclo-meta-phenylene ([9]CMP) with triperyleno[3,3,3]propellane triimides (TPPTI). This structure features two [9]CMP subunits, which necessarily fill the voids of TPPTI and promote assembly of the hybrid to form a porous superstructure held together by attractive dispersion between the [9]CMP subunits of neighboring molecules. In the structure, three large spatial cavities are formed, which allow efficient binding of up to three C60 within a single hybrid. Transient absorption spectroscopy revealed that TPPTI-[9]CMP and C60 interact with each other to form a stable complex and produce long-lived triplet states. Notably, the hybrid can adsorb ethane (C2H6) with very excellent selectivity over ethylene (C2H4), leading to a highly selective C2H6/C2H4 separation.
Introduction
Triptycene is the smallest three-dimensional (3D) rigid, paddlewheel-shaped polyaromatic system and an important component of low-density materials with applications in sensors, molecular machines, and supramolecular chemistry, due to the large internal molecular free volume (IMFV) conferred by its shape-persistent geometry (Fig. 1).1–7 In particular, the π-extended triptycenes, wherein larger ring systems are fused to the triptycene core, are conceptually interesting and practically useful, as this extension significantly increases IMFV above the bridgehead sites and between the blades.8–19 In contrast, an alternative paddlewheel-shaped molecule, trinaphtho[3.3.3]propellane (TNP) (Fig. 1), though scarcely studied, has a larger π-surface and higher rigidity than triptycene, which facilitates the formation of ordered organic solids with great porosity and large surface areas.20–23 Interest in π-extended TNPs has intensified, and recent studies have investigated their electronic and assembly properties,24–28 diradicaloid and triradicaloid character,29,30 gas separation,31 and use as emitters in organic light-emitting diodes.32,33 In 2019, we reported a new propellane, triperyleno[3,3,3]propellane triimides (TPPTI) (Fig. 1), which is composed of three perylene monoimide units fused on a [3,3,3]propellane.34,35 This propellane exhibits promising characteristics, including high solubility, large π-surface, and strong electron-accepting capability. Notably, the versatile chemistry possibilities associated with TPPTI offer great opportunities for the development of structurally and chemically distinctive 3D compounds. However, the large flat π-blades of TPPTI are prone to promoting self-association by π-stacking, thereby lowering solubility and disfavoring the formation of homogeneous mixtures or cocrystals with nonplanar partners. In addition, it is reasonable to assume that the large free volume of TPPTI, especially around its bridgeheads, partially frustrates and inhibits packing.36,37 | ||
| Fig. 1 Chemical structures of triptycene, trinaphtho[3.3.3]propellane, and triperyleno[3,3,3]propellane triimides with internal molecular free volume elements. | ||
To turn TPPTI into effective hosts, an effective way is to make derivatives devised to block π-stacking, increase solubility, and facilitate cocrystallization. Here we report a new [n]cyclo-meta-phenylene ([n]CMP)-bearing propellane hybrid, TPPTI-[9]CMP, wherein the [9]cyclo-meta-phenylene ([9]CMP) subunits are triply linked to the TPPTI framework. It is assumed that the spatial orientation of the consecutively linked cyclophenylenes, accompanied by the conformational changes of [n]CMP, would substantially enhance excellent flexibility and porosity, which, in turn, would result in unique self-assembly or new properties.38–40 The molecular geometry of the 3D hybrid provides three nanometer-sized cavities in the structure, as one can imagine that the [9]CMP subunits partially occupy the voids near the bridgeheads, promoting crystallization in a controlled orientation. This combination of a nanoring and propellane could thus open up new possibilities for supramolecular self-assembly that blend the flexibility and porosity expected for [n]CMP with enhanced binding affinities known for π-extended propellanes.
Results and discussion
A two-step synthesis of TPPTI-[9]CMP (Scheme 1a) started from hexabrominated TPPTI 1, which was prepared using the synthetic method developed by our group.34 A six-fold Suzuki coupling of 1 with m-terphenyl boronic ester 2 provided 3 in 80% yield, which was converted to the desired hybrid TPPTI-[9]CMP in 50% yield by a six-fold intramolecular nickel-mediated Yamamoto-type homocoupling reaction. This 3D hybrid is much more soluble than TPPTI, and its 1H NMR spectrum revealed eight nonequivalent aromatic protons, indicating the highly dynamic symmetry of the structures in solution. Additionally, the model compound [9]CMP was prepared by nickel-mediated cyclotrimerization of m-terphenyl 4, and its molecular structure was unambiguously confirmed by X-ray crystallography (Scheme 1b). | ||
| Scheme 1 (a) Synthetic steps to TPPTI-[9]CMP; (b) synthetic step to [9]CMP. Crystal structure and crystal packing of [9]CMP are shown. | ||
Single crystals of TPPTI-[9]CMP suitable for X-ray diffraction analysis were formed from slow diffusion of methanol into chloroform solution. The X-ray structure confirmed the formation of the hybrid, which adopts a quasi-D3h symmetric conformation (Fig. 2a). The three perylene monoimide units in the TPPTI subunit show a slight degree of twisting, presumably due to enhanced packing forces. In the structure of TPPTI-[9]CMP, the geometry of the [9]CMP subunit is different from that of parent [9]CMP, as measured by the C–C bond lengths and the dihedral angles between neighboring m-phenylene units. Interestingly, the [9]CMP subunit has a decrease in strain compared to parent [9]CMP (Fig. S11†). These observations confirmed that the configuration of [9]CMP is inherently deformable, which can vary in response to changes in substitution, packing, or other factors. As one structural feature, the three perylene monoimide units and two [9]CMP units are responsible for creating three cavities in the structure, which can efficiently obstruct π-stacking, giving the hybrid significant porosity and solubility in organic solvents even though its molecular structure is large. Thus, TPPTI-[9]CMP is assumed to be a good partner to cocrystallize with C60. Indeed, fluorescence quenching titration of TPPTI-[9]CMP with C60 in toluene showed fluorescence quenching (Fig. S5†). The titration data were fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding mode, yielding a binding constant of 7.7 × 104 M−1, with no other fitting models observed. In contrast, fluorescence titration of TPPTI with C60 showed no complexation-induced quenching process (Fig. S7†). Furthermore, slow diffusion of methanol into the solution with equimolar amounts of TPPTI-[9]CMP and C60 in toluene formed the crystals suitable for single-crystal X-ray diffraction, which revealed a 3:1 host–guest complex (C60)3@TPPTI-[9]CMP (Fig. 2c). In the complex, each C60 molecule is located within one cavity, and the centroid–centroid distances between the central benzene rings in the perylene monoimide units, with C60 guests range from 6.33 to 6.97 Å. In addition, each C60 is effectively interacting with two m-phenylene units by C–H⋯π and C⋯C contacts. In comparison to the free host, the binding of C60 results in slight changes in the [9]CMP subunit, in which the two m-phenylene units slightly rotate in order to tune the cavity size to accommodate the C60 guest in an approximate fashion. Independent gradient model (IGM) analysis further confirmed that the strength of the binding comes from intermolecular interactions between C60 and perylene monoimide and [9]CMP units (Fig. 2d). These results suggested that the presence of the [9]CMP subunit appears to provide more binding sites than pristine TPPTI, leading to stronger binding affinity towards C60. As another structural feature of TPPTI-[9]CMP and (C60)3@TPPTI-[9]CMP, the t-butyl groups in [9]CMP subunits engage in multiple dispersion interactions to align the molecules in a columnar fashion in the direction of the propellane axis (Fig. 2b, e, and 2f), resulting in 3D porous organic frameworks in the solid state.41,42
:1 binding mode, yielding a binding constant of 7.7 × 104 M−1, with no other fitting models observed. In contrast, fluorescence titration of TPPTI with C60 showed no complexation-induced quenching process (Fig. S7†). Furthermore, slow diffusion of methanol into the solution with equimolar amounts of TPPTI-[9]CMP and C60 in toluene formed the crystals suitable for single-crystal X-ray diffraction, which revealed a 3:1 host–guest complex (C60)3@TPPTI-[9]CMP (Fig. 2c). In the complex, each C60 molecule is located within one cavity, and the centroid–centroid distances between the central benzene rings in the perylene monoimide units, with C60 guests range from 6.33 to 6.97 Å. In addition, each C60 is effectively interacting with two m-phenylene units by C–H⋯π and C⋯C contacts. In comparison to the free host, the binding of C60 results in slight changes in the [9]CMP subunit, in which the two m-phenylene units slightly rotate in order to tune the cavity size to accommodate the C60 guest in an approximate fashion. Independent gradient model (IGM) analysis further confirmed that the strength of the binding comes from intermolecular interactions between C60 and perylene monoimide and [9]CMP units (Fig. 2d). These results suggested that the presence of the [9]CMP subunit appears to provide more binding sites than pristine TPPTI, leading to stronger binding affinity towards C60. As another structural feature of TPPTI-[9]CMP and (C60)3@TPPTI-[9]CMP, the t-butyl groups in [9]CMP subunits engage in multiple dispersion interactions to align the molecules in a columnar fashion in the direction of the propellane axis (Fig. 2b, e, and 2f), resulting in 3D porous organic frameworks in the solid state.41,42
 | ||
| Fig. 2 (a) Crystal structure of TPPTI-[9]CMP. (b) View of the packing in TPPTI-[9]CMP, as seen down the propellane axis. (c) Crystal structure of complex (C60)3@TPPTI-[9]CMP. (d) Optimized superstructure and the intermolecular binding iso-surface of (C60)3@TPPTI-[9]CMP. (e and f) Crystal packing of TPPTI-[9]CMP and (C60)3@TPPTI-[9]CMP (the substituted groups and the solvent molecules are omitted for clarity). | ||
The electronic structures of [9]CMP and TPPTI-[9]CMP were probed via density functional theory (DFT) calculations at the B3LYP/6-31G(d) level of theory. The calculated lowest unoccupied molecular orbital (LUMO) of [9]CMP is distributed over the whole ring, while the highest occupied molecular orbital (HOMO) is partially distributed on the ring (Fig. 3a). For TPPTI-[9]CMP, the HOMO is delocalized across all three perylene monoimide units and the HOMO-1 and HOMO-2 are energetically degenerate, which are delocalized on only two units. The LUMO and LUMO+1 are degenerate and delocalized over two of the three units (Fig. 3b and S9†). The HOMO-3 and HOMO-4 are degenerate and partially delocalized over the [9]CMP subunits; however, no significant π-electron orbital mixing between the TPPTI and [9]CMP subunits is observed (Fig S10†). TPPTI-[9]CMP and TPPTI exhibit a very similar absorption profile in the wavelength range between approximately 480 to 640 nm, although slightly red-shifted (∼13 nm), as the main transitions are localized on the TPPTI subunit (Fig. 3c). Additionally, TPPTI-[9]CMP possesses one major absorption at 266 nm, which correlates with the [9]CMP subunit, corresponding to the HOMO-2 and HOMO-3 to LUMO+3 and LUMO+4 transitions (Table S2†). As expected, both TPPTI-[9]CMP and TPPTI have similar fluorescence profiles, fluorescence quantum yields (87% vs. 85%), and lifetimes (4.4 ns vs. 3.8 ns) (Fig. 3c and S12†). In comparison, note that [9]CMP has a poorer fluorescence quantum yield of 10% and a shorter lifetime of 1.0 ns. This suggests that the TPPTI subunit is dominating the excited state processes for the hybrid.
 | ||
| Fig. 3 (a and b) Calculated molecular orbitals of [9]CMP and TPPTI-[9]CMP. (c) UV-vis spectra (solid line) of [9]CMP, TPPTI, and TPPTI-[9]CMP and emission spectra (dashed line) of TPPTI, and TPPTI-[9]CMP. (d) Cyclic voltammogram and differential pulse voltammogram of TPPTI-[9]CMP. | ||
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) studies were performed in dichloromethane, and the redox potentials were determined relative to ferrocene/ferrocenium (Fc/Fc+). Compared to perylene monoimide (PMI) exhibiting two reversible one-electron reduction waves and a reversible one-electron oxidation wave,34TPPTI-[9]CMP undergoes a six-electron reversible reduction process, which involves a reversible one-electron wave at −1.44 V, corresponding to the formation of a singly reduced PMI radical anion, two reversible one-electron waves at −1.53 V, corresponding to the formation of two singly reduced PMI radical anions, three reversible one-electron waves at −1.99, −2.21, and −2.41 V, corresponding to the formation of three singly reduced PMI dianions, and a reversible one-electron wave at −2.18 V, corresponding to the formation of a singly reduced PMI dianion. In addition, the CV of TPPTI-[9]CMP shows three one-electron oxidation waves at 0.84, 0.98, and 1.15 V, corresponding to the formation of three singly oxidized PMI radical cations.
Furthermore, the excited-state evolution process for TPPTI-[9]CMP and (C60)3@TPPTI-[9]CMP was investigated by femtosecond and nanosecond transient absorption spectroscopy (fs- and ns-TA). Upon excitation at 520 nm, TPPTI-[9]CMP in chloroform initially shows a broad excited state absorption (ESA) band from 600 to 750 nm and ground-state bleaching (GSB) bands at 528 and 568 nm (Fig. 4a–c). Within the ultrafast timescale (∼1 ps), the ESA band is split into two bands at 620 nm and 680 nm and the overall spectral shape does not change throughout the test window. Specifically, the intensity and dynamics of the sub-peak at ∼610 nm are controlled by solvent polarity (Fig. S13†). We attributed the ESA band at ∼610 nm to the CT state character of TPPTI-[9]CMP, which originates from the vibrational relaxation process of the photoinduced excited states. We further clarify the assignment for the CT state with normalized comparison of decay profiles and global analysis for fs-TA in toluene and chloroform (Fig. S14 and S15†). In addition, the spectral shapes in ns-TA are in accordance with those in fs-TA, which are assigned to the singlet state with CT character with a lifetime of 3–4 ns (Fig. S17 and S18†).
 | ||
| Fig. 4 2D contour maps, transient absorption spectra and decay profiles of TPPTI-[9]CMP (a–c) and (C60)3@TPPTI-[9]CMP (d–f) in dilute chloroform solution with excitation at 520 nm and 530 nm, respectively. | ||
In the case of (C60)3@TPPTI-[9]CMP, the broad ESA band from 600 nm to 750 nm and GSB band at 570 nm initially occur upon excitation at 530 nm (Fig. 4d–f). Subsequently, both ESA and GSB signals decay rapidly within the instrument resolution time, and ESA converts into two bands with CT character, indicating a fast CS process. Then a new ESA band at 500 nm appears, and the GSB band red-shifts to 580 nm. The growth and decay processes of new species are out of the test window. The lifetime of this long-lived state for (C60)3@TPPTI-[9]CMP can reach up to 300–400 μs (Fig. S19 and S20†), which is significantly prolonged relative to TPPTI-[9]CMP. The sensitization experiment revealed that the spectra of the sensitized triplet state for TPPTI-[9]CMP is in good coincidence with that of long-lived species, indicating that the triplet state is mainly located on TPPTI-[9]CMP (Fig. S21†).
On the basis of the above discussion, we inferred that TPPTI-[9]CMP might relax into the charge transfer state upon excitation (Fig. S22†), which decays to the ground state through a radiative pathway, leading to near-quantitative fluorescence quantum yield. In contrast, the CS process might be accelerated in (C60)3@TPPTI-[9]CMP, which subsequently undergoes a charge recombination process to produce triplet states with the mechanism of radical-pair intersystem crossing (RP-ISC) (Fig. S22†).43,44 The long-lived triplets are proved to be located on TPPTI-[9]CMP and decay with a lifetime of hundreds of microseconds. The charge separation and RP-ISC process compete with the fluorescence process, resulting in severe fluorescence quenching in (C60)3@TPPTI-[9]CMP.
The intrinsic porosity and large π-framework in TPPTI-[9]CMP motivate us to further investigate its potential for ethane (C2H6)/ethylene (C2H4) separation.45,46 The packing of the hybrids in the solid state is determined by intermolecular C–H⋯π and C–H⋯O interactions, resulting in a 3D porous network with honeycomb-shaped channels. The porous network contains void spaces, with pore sizes of 11.7 Å × 5.9 Å (Fig. S23†). The nitrogen sorption isotherm measured at 77 K revealed that TPPTI-[9]CMP has a Brunauer–Emmett–Teller (BET) surface area of 165 m2 g−1 (Fig. S24†). The single-component adsorption experiments were performed on C2H6 and C2H4 at different temperatures (273, 283, and 298 K) using activated TPPTI-[9]CMP (Fig. S25†), which revealed that the adsorption of C2H6 consistently exceeds that of C2H4 across the entire pressure range at different temperatures. At 298 K and 1 bar, the adsorbed amount of C2H6 for TPPTI-[9]CMP reached approximately 32.4 cm3 g−1, which is significantly higher than that of C2H4 (16.5 cm3 g−1) (Fig. 5a). The isosteric heats of adsorption (Qst) for C2H6 were calculated to be between 17.2 and 21.4 kJ mol−1 using the virial equation (Fig. 5c and Table S5†), which are higher than those of C2H4 (11.6 to 16.3 kJ mol−1), indicating a stronger gas bonding affinity for C2H6.
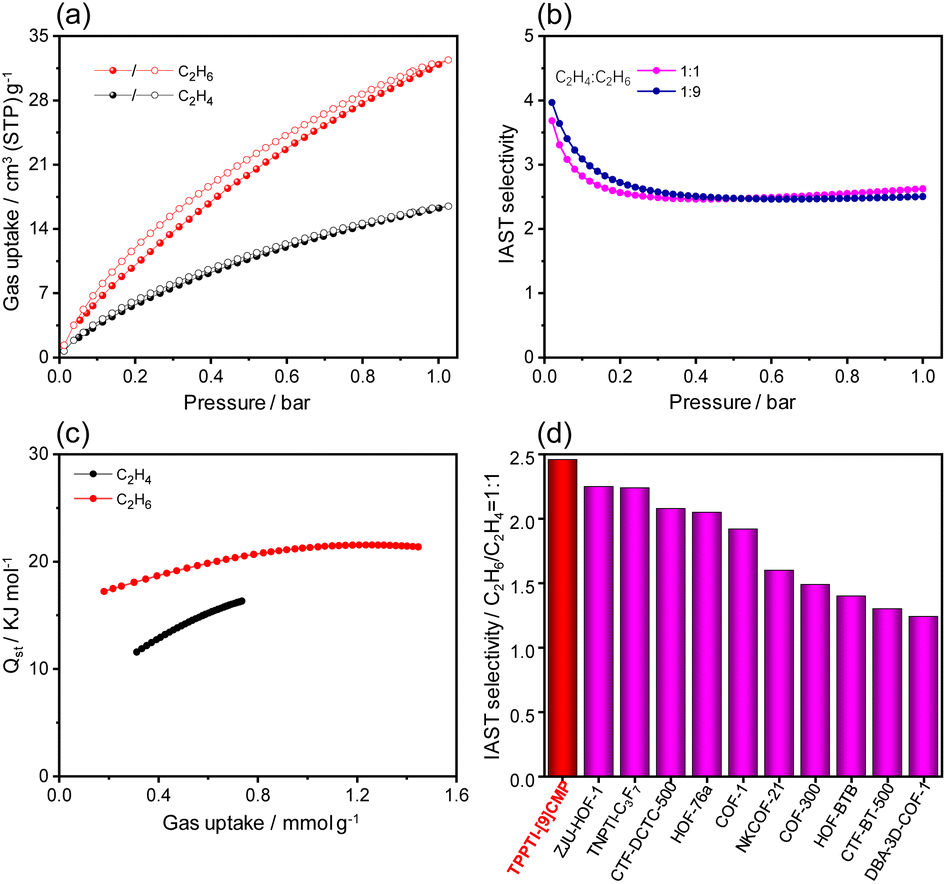 | ||
| Fig. 5 (a) Single component adsorption isotherms of C2H6 and C2H4 at 298 K. (b) Qst curves for TPPTI-[9]CMP. (c) IAST selectivity of C2H6/C2H4 (1:1, v/v, without gas carrier) mixtures at 298 K. (d) Comparison of IAST selectivities of the C2H6/C2H4 (1:1, v/v) mixture for TPPTI-[9]CMP with previously reported pure organic C2H6-selective adsorbents at 298 K and 1 bar. | ||
These results are further supported by dispersion-corrected density-functional theory calculations (DFT-D), which predicted the static binding energies for C2H6 and C2H4 to be approximately 32.7 and 25.4 kJ mol−1, respectively. The increased binding energy can primarily be attributed to the enhanced steric complementarity between the C2H6 molecule and the cavity of TPPTI-[9]CMP when compared to C2H4.47 This is likely due to more efficient C–H⋯π interactions and hydrogen bonding for C2H6 than for C2H4 (Fig. S28†). Our calculations also indicated that the C2H6 molecules preferentially locate in the cavity and the C–H⋯N hydrogen bonding might play a dominant role in C2H6-selectivity (Fig. S28†). In addition, the selectivity of TPPTI-[9]CMP towards C2H6 and C2H4 is estimated to be 2.46 by the ideal solution theory (IAST, Fig. 5b). This value surpasses those reported for other high-performance C2H6-selective organic adsorbents at 298 K and 1 bar (Fig. 5d and Table S4†).48 A breakthrough experiment was finally conducted in a fixed-bed column using a binary gas mixture of C2H6/C2H4 (50/50, v/v) at 298 K (Fig. S26†). C2H4 first breaks through the packed column at 40 seconds, while C2H6 begins to break through after 258 seconds. During this interval, polymer-grade purity (>99.95%) C2H4 can be collected. Overall, these results highlight the potential of the hybrid for efficient gas separation.
Conclusions
In conclusion, we have designed and synthesized a nanoring-propellane hybrid, in which two [9]CMP units are triply linked to the TPPTI framework. A comparison between TPPTI and the hybrid reveals that the presence of the [9]CMP unit plays an important role in the crystallization and assembly of the hybrid. Of particular interest is that crystallizing this hybrid produces a superstructure with significant porosities for guest inclusion. We believe that the development of this unique hybrid with the nanoring unit and propellane framework will allow its potential usage for binding a variety of guest species, fullerene extraction, and gas separation, overcoming some of the crystallinity and assembly limitations that have previously hindered the development of this field.Data availability
Detailed synthesis and characterization of the related compounds, crystal data for [9]CMP, TPPTI-[9]CMP, and (C60)3@TPPTI-[9]CMP, along with 1H NMR, 13C NMR, high-resolution mass spectra (HRMS) of the related compounds, CV, UV, and PL measurements, PL titration, transient absorption measurements, gas adsorption tests, and theoretical calculation details, are available in the ESI.†Author contributions
L. Z and J. Xia conceived and designed the project. Y. C and X. C synthesized and characterized the compounds. X. C performed transient absorption measurements. L. L performed gas adsorption measurements. All authors discussed the results and contributed to the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2022YFC2106100) and the National Natural Science Foundation of China (NSFC) (22175013).Notes and references
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649 CrossRef CAS.
- Y. Jiang and C.-F. Chen, Eur. J. Org Chem., 2011, 32, 6377 CrossRef.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181 CrossRef CAS PubMed.
- J.-R. Mistry, S. Montanaro and I. A. Wright, Mater. Adv., 2023, 4, 787 RSC.
- Y. Han, Z. Meng, Y.-X. Ma and C.-F. Chen, Acc. Chem. Res., 2014, 47, 2026 CrossRef CAS PubMed.
- J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301 RSC.
- C.-F. Chen and Y. Han, Acc. Chem. Res., 2018, 51, 2093 CrossRef CAS PubMed.
- Y. Duan, G. Zhang, X. Liu, F. Shi, T. Wang, H. Yan, H. Xu and L. Zhang, J. Org. Chem., 2022, 87, 8841 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Chem. Eur J., 2015, 21, 17308 CrossRef CAS PubMed.
- S. Montanaro, D. G. Congrave, M. K. Etherington and I. A. Wright, J. Mater. Chem. C, 2019, 7, 12886 RSC.
- D. Reinhard, F. Rominger and M. Mastalerz, J. Org. Chem., 2015, 80, 9342 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Org. Lett., 2014, 16, 704 CrossRef CAS PubMed.
- K. Baumgärtner, M. Hoffmann, F. Rominger, S. M. Elbert, A. Dreuw and M. Mastalerz, J. Org. Chem., 2020, 85, 15256 CrossRef PubMed.
- Z. Liu, W. Song, S. Yang, C. Yuan, Z. Liu, H.-L. Zhang and X. Shao, Chem.–Eur. J., 2022, 28, e202200306 CrossRef CAS PubMed.
- S. R. Peurifoy, E. Castro, F. Liu, X. Y. Zhu, F. Ng, S. Jockusch, M. L. Steigerwald, L. Echegoyen, C. Nuckolls and T. J. Sisto, J. Am. Chem. Soc., 2018, 140, 9341 CrossRef CAS PubMed.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774 CrossRef CAS PubMed.
- D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. Song, M. Lackinger, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450 CrossRef CAS PubMed.
- M. K. Amin, C. Ye, S. Pang, Y. Liu, D. Taylor, G. S. Nichol and N. B. McKeown, Chem. Sci., 2024, 15, 14968 RSC.
- G. Dyker, J. Körning, P. G. Jones and P. Bubenitschek, Angew Chem. Int. Ed. Engl., 1993, 32, 1733 CrossRef.
- G. Dyker, J. Körning, F. Nerenz, P. Siemsen, S. Sostmann and A. Wiegand, Pure Appl. Chem., 1996, 68, 323 CrossRef CAS.
- G. Dyker, T. Kerl, J. Körning, P. Bubenitschek and P. G. Jones, Tetrahedron, 2000, 56, 8665 CrossRef CAS.
- T. Kubo, S. Miyazaki, T. Kodama, M. Aoba, Y. Hirao and H. Kurata, Chem. Commun., 2015, 51, 3801 RSC.
- T. Kodama, S. Miyazaki and T. Kubo, ChemPlusChem, 2019, 84, 599 CrossRef CAS PubMed.
- K. Kato, S. Tanaka, N. Seto, K. Wada, M. Gon, S. Fa, S. Ohtani, K. Tanaka and T. Ogoshi, Chem. Commun., 2023, 59, 7080 RSC.
- T. Kodama, Y. Hirao, T. Nishiuchi and T. Kubo, ChemPlusChem, 2017, 82, 1006 CrossRef CAS PubMed.
- S. Kawai, O. Krejčí, T. Nishiuchi, K. Sahara, T. Kodama, R. Pawlak, E. Meyer, T. Kubo and A. S. Foster, Sci. Adv., 2020, 6, eaay8913 Search PubMed.
- K. Kato, Y. Uchida, T. Kaneda, T. Tachibana, S. Ohtani and T. Ogoshi, Chem. Asian J., 2024, 19, e202400080 Search PubMed.
- T. Kodama, M. Aoba, Y. Hirao, S. M. Rivero, J. Casado and T. Kubo, Angew. Chem., Int. Ed., 2022, 61, e202200688 Search PubMed.
- T. Kodama, Y. Hirao and T. Kubo, Precis. Chem., 2023, 1, 183 CrossRef CAS.
- Y. Chen, Y. Zhao, Y. Zhao, X. Chen, X. Liu, L. Li, D. Cao, S. Wang and L. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202401706 CrossRef CAS PubMed.
- L. Hua, Y. Liu, B. Liu, Z. Zhao, L. Zhang, S. Yan and Z. Ren, Nat. Commun., 2022, 13, 7828 CrossRef CAS PubMed.
- Z. Zhao, S. Yan and Z. Ren, Acc. Chem. Res., 2023, 56, 1942 CrossRef CAS PubMed.
- L. Lv, J. Robert, C. Xiao, Z. Jia, W. Jiang, G. Zhang, C. Risko and L. Zhang, Chem. Sci., 2019, 10, 4951 RSC.
- L. Lv, W. Sun, Z. Jia, G. Zhang, F. Wang, Z. Tan and L. Zhang, Mater. Chem. Front., 2020, 4, 3539 RSC.
- L. J. Abbott, N. B. McKeown and C. M. Colina, J. Mater. Chem. A, 2013, 1, 11950 Search PubMed.
- C. L. Hilton, C. R. Jamison, H. K. Zane and B. T. King, J. Org. Chem., 2009, 74, 405 CrossRef CAS PubMed.
- C. Zhou, R. Li, Y. Chen, X. Liu, H. Zhao, X.-Q. Yang, Z. Ren, D. Wang, Z. Wang and L. Zhang, CCS Chem., 2024, 6, 2427 Search PubMed.
- K. Ikemoto, R. Kobayashi, S. Sato and H. Isobe, Angew. Chem., Int. Ed., 2017, 56, 6511 Search PubMed.
- K. Ikemoto and H. Isobe, Bull. Chem. Soc. Jpn., 2021, 94, 281 CrossRef CAS.
- B. Kohl, M. V. Bohnwagner, F. Rominger, H. Wadepohl, A. Dreuw and M. Mastalerz, Chem. Eur J., 2016, 22, 646 CrossRef CAS PubMed.
- L. Ueberricke and M. Mastalerz, Chem. Rec., 2021, 21, 558 Search PubMed.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122 RSC.
- X. Wang, Y. Song, G. Pan, W. Han, B. Wang, L. Cui, H. Ma, Z. An, Z. Xie, B. Xu and W. Tian, Chem. Sci., 2020, 11, 10921 Search PubMed.
- D. S. Sholl and R. Lively, Nature, 2016, 532, 435 CrossRef PubMed.
- L. Li, R.-B. Lin, R. P. Krishna, H. Li, S. Xiang, H. Wu, J. Li, W. Zhou and B. Chen, Science, 2018, 362, 443 Search PubMed.
- K. Su, W. Wang, S. Du, C. Ji and D. Yuan, Nat. Commun., 2021, 12, 3703 Search PubMed.
- X. Zhang, J.-X. Wang, L. Li, J. Pei, R. Krishna, H. Wu, W. Zhou, G. Qian, B. Chen and B. Li, Angew. Chem., Int. Ed., 2021, 60, 10304 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2416459 (for [9]CMP), 2416457 (TPPTI-[9]CMP), and 2416458 (for (C60)3@TPPTI-[9]CMP). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00713e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |