
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sequential esterification-ring closing metathesis-Nozaki–Hiyama–Kishi strategy for constructing a natural product-like library of tetrahydrofuran-containing macrolides†
Daniel
Driedger‡
,
Darryl M.
Wilson‡
and
Robert
Britton
 *
*
Department of Chemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada. E-mail: rbritton@sfu.ca
First published on 6th March 2025
Abstract
Polyketide-like macrolides (pMLs) represent a privileged class of compounds with a high incidence of bioactivity, however their structural complexity challenges their synthesis and more general study. Here we report the synthesis of a library of tetrahydrofuran-containing pMLs underpinned by a robust and convergent build/couple/pair/couple synthetic approach. The library comprises 170 pMLs originating from 17 building blocks, 10 of which were synthesized using a proline-catalyzed α-chlorination aldol reaction. Northern and southern hemisphere building blocks were coupled using either an oxidation/Horner-Wadsworth-Emmons sequence or a saponification/Steglich esterification strategy. Coupled fragments were cyclized via ring closing metathesis to yield macrocycles 14–16 atoms in size, which we diversified using 3 eastern side chain vinyl iodides through a Nozaki–Hiyama–Kishi reaction.
Introduction
Macrocycles (rings ≥12 atoms in size) are increasingly recognized as valuable frameworks in drug discovery due to their ability to bind to low-druggability targets, including the disruption of protein–protein interactions (PPIs).1,2 The unique binding of macrolides to protein surfaces benefits from numerous functional groups strategically displayed over a large surface area and a conformational pre-organization provided by the macrocyclic skeleton.3 Despite often violating established drug-like physicochemical rules, macrocycles also display surprisingly effective pharmacokinetic properties and cellular uptake, which has been attributed in part to their ability to temporarily mask polar groups through conformational interconversions.3,4 Therefore, it is no surprise that >80 macrocyclic compounds have been approved as drugs, most of which are derived from natural products,1,5 and the discovery of new bioactive macrocycles remains an important field of research.2Among the several sub-categories of macrocyclic natural products are polyketide-like macrolides (pMLs), distinguished by a macrocyclic lactone often containing contiguous methyl and hydroxyl groups, and unsaturations consistent with natural products produced via polyketide synthases (Fig. 1). pMLs are abundant in nature, representing 6% of all microbial secondary metabolites, and over-represented among approved macrocyclic drugs.1,6,7 Aside from natural product isolation campaigns, the discovery of new pMLs is challenged by the demanding total synthesis campaigns required for their production8–10 as well as the scarcity of compound libraries that approach the complexity of such compounds.11,12
 | ||
| Fig. 1 A selection of polyketide-like macrolide natural products. Biselide A, fijianolide A, and phormidolide A are also classified as THF-containing macrolides. | ||
Recently, we reported a strategy termed the Targeted Sampling of Natural Product space (TSNaP)13 that was designed to identify and assess regions of chemical space bounded by tetrahydrofuran-containing macrolides (Fig. 2A).14 Using TSNaP, we computationally assembled a collection of virtual pMLs using pML-inspired building blocks. We then prioritized the virtual pMLs according to similarity scores with natural THF-containing macrolides (e.g., biselide A, fijianolide A, phormidolide A, Fig. 1) and synthesized a library of 170 tetrahydrofuran-containing pMLs based on their similarity scores. Finally, we screened this library in a panel of whole-cell biological assays, revealing hit rates exceeding those typically encountered in small molecule libraries. Hits included antibacterial pMLs 120c and 111c, as well as pMLs such as 115Ac which showed an activity profile similar to that of known apoptosis-inducing compounds (Fig. 2A). Considering the wide scope of the TSNaP study, our initial report placed an emphasis on the rationale behind the library design, our process for selecting target compounds, and the results from biological testing of the library. Here, we instead disclose the entire breadth of the library synthesis, which represented a considerable effort (>300 synthetic steps) made possible by a handful of powerful synthetic methods and a convergent approach. We also discuss the synthetic challenges encountered in assembling such a pML library, and highlight useful insights gained from this synthesis campaign.
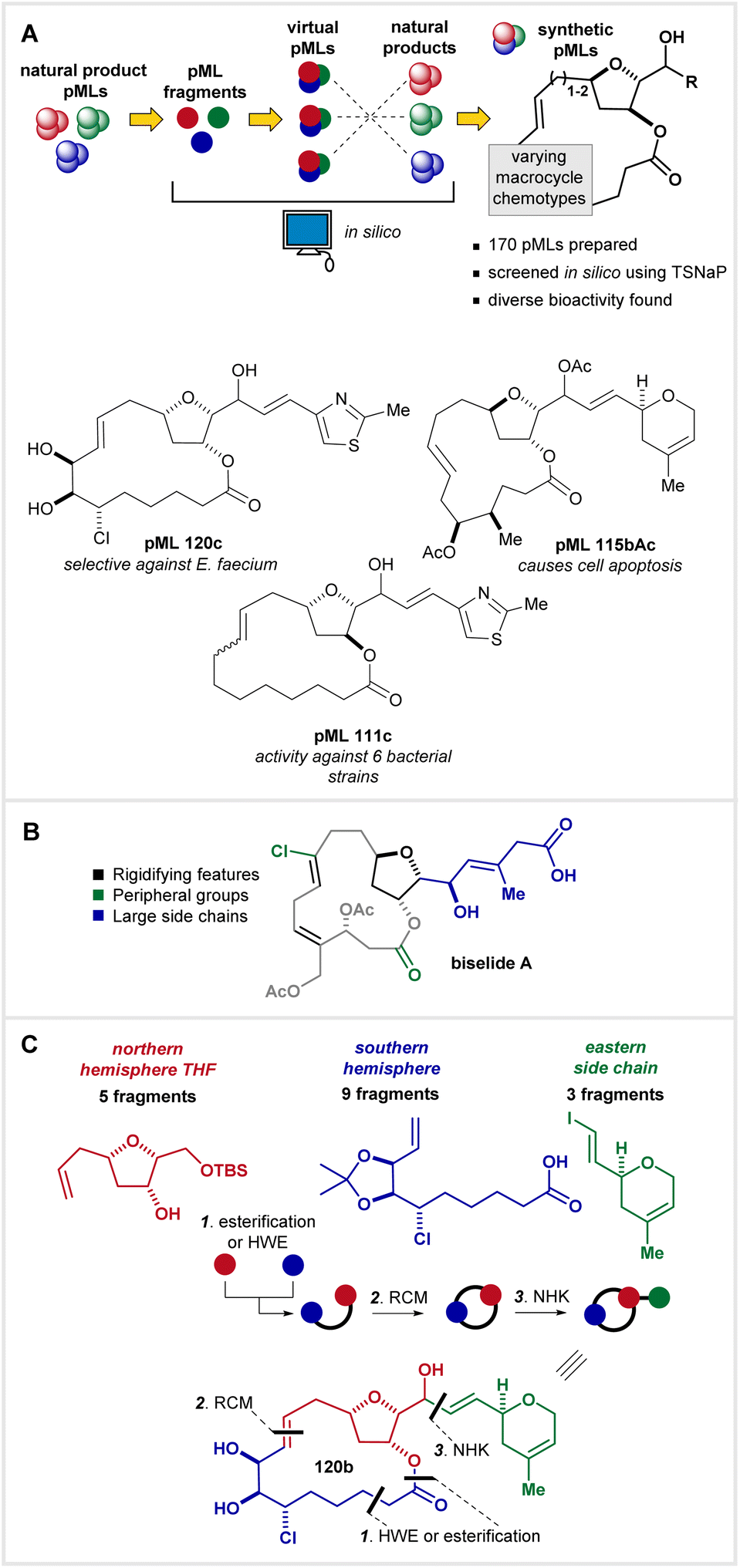 | ||
| Fig. 2 (A) Summary of the TSNaP approach and a sample of hit molecules from the biological testing. (B) Conserved features amongst bioactive macrocycles as highlighted in biselide A. (C) Our generalized strategy for the assembly of the pML library. | ||
Results and discussion
Synthetic strategy
Applying the TSNaP approach, the pML library members were designed to fulfill a number of the key criteria attributed to bioactive macrocycles by Whitty.3 These are exemplified in the natural product biselide A (Fig. 2B) and include: (i) rigidifying groups such as embedded rings and unsaturations, (ii) diverse heavy atom peripheral groups (e.g., OH, CH3, Cl), and (iii) one or more large substituents or side chains attached to the macrocycle. Each of these elements are accounted for amongst the 3 classes of building blocks used in the library synthesis, coupled together in various combinations using a build-couple-pair-couple (B/C/P/C) strategy.15,16 The northern hemisphere THF (5 distinct fragments, red, Fig. 2C), the southern hemisphere (9 distinct fragments, blue), and the eastern side chain (3 distinct fragments, green) comprise the assortment of library building blocks. After preparing all fragments (“build”), one of the five tetrahydrofuranols (differing in stereochemistry and chain length) was joined (“couple”) to a southern hemisphere enoic aldehyde or acid using either a Horner–Wadsworth–Emmons (HWE) reaction (for enoic aldehydes: 1st-generation approach) or a Steglich esterification (for enoic acids: 2nd-generation approach). Both approaches provided equivalent substrates for the subsequent ring closing metathesis (RCM) reaction to form a macrocycle (“pair”). After deprotection and oxidation of the northern tetrahydrofuranol, the material was divided into 3 portions, and a Nozaki–Hiyama–Kishi (NHK) reaction was used to join each of the 3 eastern side chain vinyl iodides with the macrocyclic aldehyde (“couple”). The synthesis of each individual library member was accomplished after the deprotection of any alcohols contained within the macrocycle, providing 3 library members per assembly sequence. This convergent and combinatorial B/C/P/C approach allowed us to generate a large library of pMLs with a reduced total step count and only a handful of building blocks (17 in total).Synthesis of northern hemisphere THF building blocks
With an aim of achieving a balance between similarity and dissimilarity from known THF-macrolide natural products and targeting ‘natural product-like’ chemical space, the stereochemistry of the tetrahydrofuranol series 1a–e (Fig. 3A) was chosen to overlap with that of the biselides,17 fijianolides (laulimalides),18,19 and phormidolide A (Fig. 1).20 With this challenge in mind, we relied on the proline-catalyzed α-chlorination aldol reaction (α-CAR) to create the northern THF fragments (Fig. 3B). This robust and scalable methodology was developed previously in our lab21,22 and has proven effective for the synthesis of many chloropolyols, which are excellent precursors to ribose analogues, carbohydrates and iminosugars.23–26 Notably, this process involves a dynamic kinetic resolution (DKR) and uses inexpensive D/L-proline and readily available achiral starting materials such as pent-4-enal (4) and 2,2-dimethyl-1,3-dioxan-5-one (6) to establish the 4 contiguous THF stereocentres in only 2 steps with high enantioselectivity and operational ease, enabling large scale parallel syntheses of early intermediates on multi-gram scale. Moreover, several of these early intermediates serve dual purpose in creating both northern and southern hemisphere building blocks. Each of the 8 possible stereoisomers (each resembling a 2-deoxy furanose, see 9a for numbering conventions) could be targeted by making binary decisions at 3 junctions: (i) the use of either L- or D-proline for the α-CAR (determines C1), (ii) either a 1,3-anti or 1,3-syn reduction of the resulting chlorohydrin (determines C4), and (iii) an optional Mitsunobu inversion of the exposed alcohol in 1a–c (determines C3). | ||
| Fig. 3 (A) Syntheses of northern hemisphere THF building blocks. (B) Scheme for the DKR proline-catalyzed α-CAR (α-chlorination aldol reaction). | ||
The tetrahydrofuranol synthesis is displayed in Fig. 3A, and begins with the aforementioned α-CAR between 2,2-dimethyl-1,3-dioxan-5-one (6) and either pent-4-enal (4) or hex-5-enal (5) to provide chlorohydrins 7a, ent-7a, and 7c in 44–46% yield (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 92% ee for 7a).21 Either an Evans–Saksena reduction27 or catecholborane reduction28 yielded diols 8a–c stereoselectively with yields of 56–76%. A microwave promoted cyclization involving the newly reduced alcohol and the secondary alkyl chloride delivered furan triols 9a–c in yields of 51–67%. The design of our library necessitated the deoxygenation of the C2 alcohol, to which the next 4 steps were dedicated, beginning with a selective protection using TIPDiSiCl2 (ref. 29) to yield alcohols 10a–c in good yield (73–83%). Triflation was followed by deoxygenation initially using nBu4NBH4/cyclohexene (12c, 44%) and after further optimization, NaBH4 (12a in 74%, 12b in 81% yield). The disiloxane protecting group was removed with either TBAF/THF at 0 °C (13c, 52%) or HCl/MeOH at 50 °C (13a in 92%, 13b in 96% yield) followed by selective re-protection using TBSCl/imidazole to furnish the first set of functional building blocks, 1a, 1b, and 1c in excellent yield (81–90%). C3 epimers 1d and 1e could be produced via a Mitsunobu inversion 2-step sequence using p-nitrobenzoic acid30 (76% and 86% yield over 2 steps, respectively). For our 1st-generation approach (vide infra), 1a–e were esterified with diethylphosphonoacetic acid (14) and DIC to complete the HWE-ready set of northern THF fragments 15a–e in excellent yield (85–98%). Overall, the 8–10 step sequence offered a stereochemically flexible and straightforward path toward ample quantities of 5 northern THF building blocks (>1 g in some cases).
:1 dr, 92% ee for 7a).21 Either an Evans–Saksena reduction27 or catecholborane reduction28 yielded diols 8a–c stereoselectively with yields of 56–76%. A microwave promoted cyclization involving the newly reduced alcohol and the secondary alkyl chloride delivered furan triols 9a–c in yields of 51–67%. The design of our library necessitated the deoxygenation of the C2 alcohol, to which the next 4 steps were dedicated, beginning with a selective protection using TIPDiSiCl2 (ref. 29) to yield alcohols 10a–c in good yield (73–83%). Triflation was followed by deoxygenation initially using nBu4NBH4/cyclohexene (12c, 44%) and after further optimization, NaBH4 (12a in 74%, 12b in 81% yield). The disiloxane protecting group was removed with either TBAF/THF at 0 °C (13c, 52%) or HCl/MeOH at 50 °C (13a in 92%, 13b in 96% yield) followed by selective re-protection using TBSCl/imidazole to furnish the first set of functional building blocks, 1a, 1b, and 1c in excellent yield (81–90%). C3 epimers 1d and 1e could be produced via a Mitsunobu inversion 2-step sequence using p-nitrobenzoic acid30 (76% and 86% yield over 2 steps, respectively). For our 1st-generation approach (vide infra), 1a–e were esterified with diethylphosphonoacetic acid (14) and DIC to complete the HWE-ready set of northern THF fragments 15a–e in excellent yield (85–98%). Overall, the 8–10 step sequence offered a stereochemically flexible and straightforward path toward ample quantities of 5 northern THF building blocks (>1 g in some cases).
Synthesis of southern hemisphere building blocks
Next, we turned our attention to the synthesis of the southern hemisphere fragments. Our intention for these fragments was to include a variety of pML peripheral groups (Me, OH, Cl) with differences in substitution pattern and stereochemistry, along with the following constraints required for incorporation into the envisioned macrocycles: (i) 6–10 carbons in length for optimal macrocycle ring size, (ii) a terminal alkene for RCM, and (iii) a terminal aldehyde or carboxylic acid for either an HWE reaction or Steglich esterification, respectively.We began by making use of the dual purpose THF-containing fragments 16a (13% from 8a) and 16b (18% from 8b) which were convenient acetonide-protected side-products of the microwave cyclization reactions from the northern THF fragment syntheses (Fig. 4A). Oxidation using Dess–Martin periodinane31 followed by a HWE reaction using Masamune–Roush conditions32 and triethylphosphonoacetate (17) afforded α,β-unsaturated esters 18a and 18b (68% and 72% over 2 steps). These two southern fragments were finalized via reduction using Stryker's reagent33,34 yielding esters 2a (32%) and 2b (55%), each only 3 additional steps from material in-hand.
 | ||
| Fig. 4 (A) Syntheses of THF-containing southern fragments 2a and 2b. (B) Syntheses of chloro-diol southern fragments 2c, 2d, and 2e. (C) Synthesis of southern fragment 2f. (D) Syntheses of southern fragments 2g and 2h. *Yield corresponds to the carboxylic acid product of an additional saponification step. | ||
Our next targets were chlorinated fragments based on manipulations of the keto-chlorohydrins 7a and 7c from the northern THF fragment synthesis. Treatment with InBr3 in anhydrous MeCN caused a migration of the 1,3-acetonide to the 1,2-acetonide delivering acyloins 19a and 19c (62% and 69% yields, respectively). Reduction of 19c with NaBH4 produced an inconsequential diastereomeric mixture of diols that was then cleaved with NaIO4 to give the intermediate aldehyde 20. The same HWE-Stryker's sequence from the synthesis of 2a and 2b yielded the completed enoic ester 2c in 25% over 4 steps from 19c (Fig. 4B). Unfortunately, it would later be found that this southern hemisphere fragment had a tendency to undergo trans-esterification after or during the deprotection of the final macrocyclic products (see 119a–c, Chart 1) due to the proximity of the alcohol to the ester (γ-lactone formation).
Not wanting to abandon the chloride-containing building blocks, we devised a synthesis of two related building blocks (2d and 2e) that exchanged the positions of the terminal alkene and ester, shifting the alcohols further away from the ester while retaining the chloride function (Fig. 4B). For the synthesis of 2e, DIBAL-H was used to reduce acetonide-migrated intermediate 19c in 63% yield. The mixture of diastereomeric diols (21c) was carried forward and silylated with TBSOTf/2,6-lutidine to give 22c in excellent yield (94%). Oxidative cleavage of the alkene function by means of catalytic K2OsO4·2H2O in the presence of NaIO4 proceeded smoothly to deliver aldehyde 23c in 78% yield. Next, olefination of aldehyde 23c with phosphonate 17 using an HWE reaction followed by hydrogenation (H2/PtO2) of the resultant α,β-unsaturated ester delivered saturated ester 24c in 93% yield over 2 steps. Finally, desilylation with HF/pyridine followed by a Garegg–Samuelsson olefination35 of the resultant diol cleanly afforded ester 2e (74% over 2 steps). Ester 2d was produced using an analogous route from acyloin 19a, and despite dramatically reduced yields (2% over 6 steps), enough material was available for incorporation into a set of 3 macrocycles.
To supplement the collection of southern fragments originating from the α-CAR chemistry, we also generated 3 enoic alcohols using alternative methods for synthesizing polyketide-like motifs. The simplest of these is PMB alcohol 2f containing 2 adjacent stereocentres (Fig. 4C). The synthesis follows a route reported by Krische and co-workers,36 beginning with a diastereoselective allylation of the silyl-protected alcohol 25b, which proceeded in 58% yield. TBDPS deprotection of the resultant alcohol 27 (94%) followed by a PMB protection (80%) and reductive opening using DIBAL-H (89%) furnished primary alcohol 2f in 39% over 4 steps.
The more complex fragments 2g and 2h, containing 3 and 4 stereocentres, respectively, required more onerous syntheses using an Evans-aldol strategy inspired by and overlapping with past syntheses (Fig. 4D).36–40 Synthesis of 2g began with the oxidation of mono-TBDPS protected propanediol 25a (1 step from 1,3-propanediol) under Swern conditions.41 The resultant crude aldehyde was immediately subjected to an Evans-aldol reaction with the boron enolate produced from enolization of 31 with Bu2BOTf/Et3N,37,42 affording syn aldol adduct 32a in 73% yield over 2 steps. Next, 32a was reduced with NaBH4 in MeOH/H2O (81%) followed by exposure to dimethyl acetal 29 in the presence of CSA to furnish acetal 34a in 84% yield. Selective opening of the acetal with DIBAL-H gave PMB protected alcohol 35a in 84% yield. Swern oxidation of the primary alcohol function followed by allylation of the resultant aldehyde with allyl magnesium bromide furnished 36a as the major diastereomer (64% overall yield, 2 steps, d.r. = 7:2). Subsequent exposure of 36a to TBSOTf/2,6-lutidine delivered protected polyol 37a. Finally, selective deprotection of the primary OTBDPS group was achieved with NH4F in warm (60 °C) MeOH43 to deliver the southern hemisphere building block 2g in 75% yield. The entire sequence was repeated to produce 2h in similar fashion and yield using the Roche ester-derived methyl congener 25b as a starting point (Fig. 4D).
The last of the 9 fragments for the southern hemisphere of the macrocycle library was commercially available 9-decenoic acid (2i), which was used in macrocycles that we envisioned would act as a control or baseline to highlight the importance of the peripheral groups as it pertains to biological activity.
Coupling of northern and southern fragments
With all northern hemisphere THF and southern hemisphere fragments completed, we began the process of macrocycle assembly by combining fragments in unique combinations. Our 1st-generation coupling strategy involved an HWE reaction between one of phosphonates 15a–e and enoic aldehydes derived from alcohols 2e, 2f, 2g, and 16a. As exemplified by the coupling of alcohol 2g with phosphonate 15a (Fig. 5A), the alcohol first undergoes oxidation via Dess–Martin periodinane, followed by an HWE reaction with 15a under Masamune–Roush conditions.32 All reactions proceed fairly smoothly with yields of 56–91%. Later it was discovered that the additional degree of unsaturation due to the newly formed α,β-unsaturated ester was detrimental to the success of the downstream ring closing metathesis (vide infra), therefore, the olefin was selectively reduced using Stryker's reagent, providing the coupled products 39, 41, 43, 45, and 47 in moderately good yield (65–87%). | ||
| Fig. 5 (A) Oxidation of northern hemisphere THFs and HWE coupling with southern hemisphere fragments. (B) Saponification of northern hemisphere THFs and Steglich esterification coupling with southern hemisphere fragments. | ||
The 1st-generation HWE approach was originally devised for facile use of keto-chlorohydrin products (such as 7a–c) as southern hemisphere fragments and coupling partners. However, once it became clear that the keto-chlorohydrins were not compatible the RCM and that a reduction of the newly formed double bond was necessary, a 2nd-generation coupling strategy was devised. In the revised approach, we performed an HWE/reduction sequence on southern fragment aldehydes in advance, using triethyl phosphonoacetate (17) (see the synthesis of 2a–2e, Fig. 4), and after reduction then saponification, we obtained carboxylic acids which could be esterified with northern THF alcohols 1a–e. Overall, this bypassed the inclusion of the Stryker's reduction in the linear sequence as well as the phosphonate coupling step in the northern THF fragment synthesis, meanwhile providing the same molecular structures as would be produced via the 1st-generation approach.
The 2nd-generation strategy is exemplified by the coupling of enoic ester 2d and THF alcohol 1e (Fig. 5B). Ester 2d underwent a saponification using LiOH in a mixture of THF and water, and the crude material was subjected to a Steglich esterification with alcohol 1e using DIC as the coupling reagent to form ester 48 (83% over 2 steps). Using this method, esters 49–54 were also synthesized in excellent yields (86–98%) proving the 2nd-generation modification to be advantageous. All products obtained via the 1st and 2nd-generation coupling strategies could be used directly in the pivotal RCM reaction that would follow.
Preliminary ring closing metathesis (RCM) experiments
To mitigate the risk associated with creating highly strained macrocycles, we designed an experiment to assess the viability of different southern hemisphere building blocks in RCM reactions with the goal of identifying both the ring-size and tolerated functionalities. This experiment began with preparing a handful of diene RCM precursors using the 1st-generation HWE coupling strategy (Fig. 6). THF phosphonates 15a and 15c were coupled to easily prepared aldehydes or ketones (pent-4-enal 4, chlorohydrin 7a, and aldehydes 55 and 56) providing potential ring sizes of 12–16 atoms. Substrates were then optionally reduced (via Stryker's reagent) or deprotected (via TBAF) or neither, in order to study the effects of unsaturation and protecting groups on ring-closure. RCM results were analysed using high-resolution mass spectrometry (HRMS), which gave a qualitative picture of the reaction outcome including the formation or absence of product [M + H]+ and the formation of the undesired homodimeric side-product [2M + H]+. | ||
| Fig. 6 RCM experiments to assess the formation of 12–15-membered rings, analyzed by HRMS. | ||
Our first RCM experiment aimed to form the highly strained 12-membered macrocycle 57, which unsurprisingly led to exclusive formation of macrocyclic homodimers. Similarly, attempted closure of the slightly larger 13-membered macrocycle series 58–60 was largely unsuccessful regardless of whether the alcohol function was protected (58vs.59) and the lactone was unsaturated or saturated (58vs.60). Attempted RCM to form the 14-membered macrocycle 61 led to exclusive formation of homodimeric macrocyclic products, however the successful RCM of the saturated congener 62 indicated that a reduction of the enoate function may promote ring closing in these systems. Motivated by this encouraging result, we reasoned that further alleviation of ring strain was required to enable facile formation of the desired monomeric macrocycles.
To ease ring strain, we attempted the formation of 14-membered macrocycle series 63–65 incorporating a less conformationally constrained southern hemisphere fragment. Disappointingly, the formation of 14-membered macrocycle 63 was not observed, however formation of desilylated congener 64 was detected, albeit in relatively small amounts and accompanied by several other unidentified products. Gratifyingly, saturated analogue 65 exclusively formed the desired RCM product, further indicating the importance of removing the C2–C3 olefin function. Finally, we evaluated formation of a heavily protected 15-membered macrocycle 66, which resulted in formation of both the monomeric and homodimeric RCM products.
These results led to a few key conclusions: (i) the homodimer can be disfavoured by constructing macrocycles with a minimum ring size of 14 atoms, (ii) various peripheral functionalities are well tolerated, however, α,β-unsaturated esters should be avoided as they impart additional ring strain and/or cause competing undesired metathesis events, and (iii) limiting the use of bulky protecting groups is beneficial for the desired RCM processes. These insights allowed us to better predict which southern hemisphere fragments would be compatible in the key RCM reaction and therefore which building blocks should be prioritized for synthesis.
Synthesis of eastern fragment building blocks
The final set of building blocks to be synthesized was the eastern side chain fragment, each a vinyl iodide designed for an NHK coupling and modelled after side chains found in bioactive natural products. As such, we prioritized methyl amide 3a that resembles the side chain found in haterumalides44 and biselides,17,45 dihydropyran 3b that mimicked the side chain in the fijianolides18,19 and thiazole 3c found in the epothilones (Fig. 1, 3a–c found in Fig. 7).46 | ||
| Fig. 7 Syntheses of the vinyl iodide-containing eastern fragments 3a–c. | ||
Our efforts towards the syntheses of the side chains began with the methyl amide 3a as outlined in Fig. 7. Here, but-2-ynol (67) was subjected to a known carboalumination–iodination to deliver vinyl iodide 6847 in good yield (80%), and the alcohol functionality was subsequently oxidized with chromic acid to furnish carboxylic acid 69 in 78% yield. Finally, treatment of acid 69 with DIC/HOBt generated the active ester in situ, which was then coupled with methylamine to furnish methyl amide 3a in 54% yield.
The dihydropyran 3b and thiazole 3c side chains were prepared according to known sequences previously reported by Paterson (Fig. 7).48,49 Synthesis of dihydropyran 3b initiated from a hetero-Diels–Alder (HDA) reaction50,51 between TBS protected glycolaldehyde 70 and known diene 72 in the presence of Jacobsen's organochromium HDA catalyst 71 and proceeded smoothly to deliver 73 in 66% yield (>95% ee). Reduction of the methyl ether with Et3SiH/BF3·OEt2 and subsequent desilylation under acidic conditions (HCl/MeOH) gave primary alcohol 74 in 79% over both steps. Next, sequential Dess-Martin oxidation, homologation with the Ohira–Bestmann reagent (generated in situ from 75 and 76), and hydrozirconation–iodination delivered vinyl iodide 3b in 30% yield over 3 steps.
Via a related sequence, thiazole 3c was prepared from commercially available 77 through homologation to furnish the desired alkyne 78 in 74% yield. Finally, hydrozirconation–iodination of alkyne 78 delivered vinyl iodide 3c in 70% yield and concluded our efforts on the syntheses of the eastern side chains.
RCM and NHK coupling for diversification
With all fragments prepared, we turned to the critical juncture in the library synthesis, the RCM and NHK diversification sequence. An example sequence is illustrated in Fig. 8, starting from the diene 53. A ring closing metathesis using Grubbs 2nd-generation catalyst heated to 60 °C in toluene afforded the macrocyclic product 79 in 78% yield with a d.r. of 4:1 E/Z. TBS deprotection with HF/pyridine produced 80 (73%) with its revealed exocyclic alcohol to be oxidized by Dess–Martin periodinane. In general, these macrocyclic aldehydes proved to be highly unstable, and the crude material was used immediately for the accompanying NHK reaction in all cases. Additionally, it was found that the use of freshly prepared Dess–Martin periodinane31 aided in reducing the amount of hydrated aldehyde, which proved to be a major source of material loss. After oxidation of 80, the resultant aldehyde was divided into 3 equal portions and coupled to each of the 3 eastern side-chain vinyl iodides via an NHK reaction.52 Coupled products 81a–c were isolated in moderate yield (49–59%) with a range of diastereomeric ratios (6:1 to 1:1). The 11 additional examples of the RCM/NHK sequence are summarized in Fig. 8, for a total of 12 sets of macrocyclic products (36 total products), and rings ranging from 14–16 atoms in size. In most cases, the RCM strongly favored the E macrocycle, with the exception of a few cases in which the Z isomer was formed in sufficient quantity to be used for the NHK coupling (93a–c and 99c, Fig. 8). In general, the yields for the NHK were modest and in some cases low (<30%) with diastereomeric ratios covering the full range (1:1 to >19:1). In most cases, these diastereomers were inseparable by chromatography and were left as such. Additionally, we made the intentional decision to leave the stereochemistry of the diastereomeric alcohol unassigned, as determination of the configuration would require derivatization which would be complicated by limits in product supply at this point.
 | ||
| Fig. 8 RCM, TBS deprotection, oxidation, and NHK diversification of all macrocyclic precursors. aThiazole 84c made by subjecting to 82 to (1) DDQ, pH 7 buffer, CH2Cl2, rt and (2) TBSOTf, 2,6-lutidine, CH2Cl2, rt before undergoing 1° TBS deprotection and oxidation/NHK conditions as above. bStewart-Grubbs catalyst. cd.r. not determined. | ||
However, available literature on the NHK reaction suggests a modest, but clear, preference for the formation of the Felkin–Anh or polar Felkin–Anh products in acyclic52–55 systems and some cyclic56 systems. Notably, NHK couplings with tetrahydrofurfurals57,58 including macrocyclic tetrahydrofurfurals8,52,59 have been shown to proceed with a modest preference for the polar Felkin–Anh products. Thus, it is likely that where diastereoselectivity was observed, the major isomer is the polar Felkin–Anh product. Interestingly, when the stereocentres at positions C2 and C3 in the THF were syn-configured, the diastereoselectivity of the NHK was increased relative to comparable compounds in which these stereocentres were anti-configured (e.g., compare d.r. for 105a–c and 108a–c). This enhanced selectivity is consistent with, and may arise from, a 4-coordinate56,60 polar Felkin–Anh transition state where an oxygen atom of the syn-configured tetrahydrofurfural is capable of coordinating to the Cr(III) metal centre (Fig. 8). In contrast, a similar stabilizing interaction is not possible when the stereocentres at C2 and C3 are anti-configured, which may explain the poorer stereoselectivity in these cases. We note that this hypothesis about coordination in the syn-configured macrocyclic tetrahydrofurfurals remains speculative.
Final library member deprotections and acetylation
The final stage in the library synthesis was the deprotection of peripheral macrocyclic alcohols. Chart 1 pictures the entire macrocycle library, with any number of 3 deprotection conditions applied to each macrocycle. DDQ and pH 7 buffer in CH2Cl2 was used to remove the PMB group (conditions W), 70% HF/pyridine in THF was used to remove the TBS group (conditions X), and HCl in MeOH was used to remove the acetonide group from vicinal diols (conditions Y). The macrocyclic peripheral groups, determined by the southern hemisphere fragment, fall into 3 general categories: class I with stereogenic methyls (115–118), class II containing the chlorodiol (119–122), and class III consisting of an embedded tetrahydrofurandiol (123–125), plus the set of hydrocarbon macrocycles 111a–c (class IV). Deprotections proceeded smoothly in most cases, however there were a few instances of undesired side-reactions. In the cases of 118c and 119a–c a translactonization occurred during the final deprotection to form a 5- or 6-membered lactone. Another anomaly in the library was the serendipitous oxidation of the allylic alcohol in substrate 84e during oxidative deprotection of the PMB protecting group, providing 115e as an additional library member (its alcohol congener was eventually made using a TBS-protected macrocycle, 115c). Altogether, we were able to prepare 85 different macrocycles (including diastereomers). This includes the preparation of an analogue lacking a side-chain for each family of compounds, completing sets of 4 analogues per unique macrocycle. Additionally, many compounds represented a mixture of two NHK diastereomers which were both included in the total count.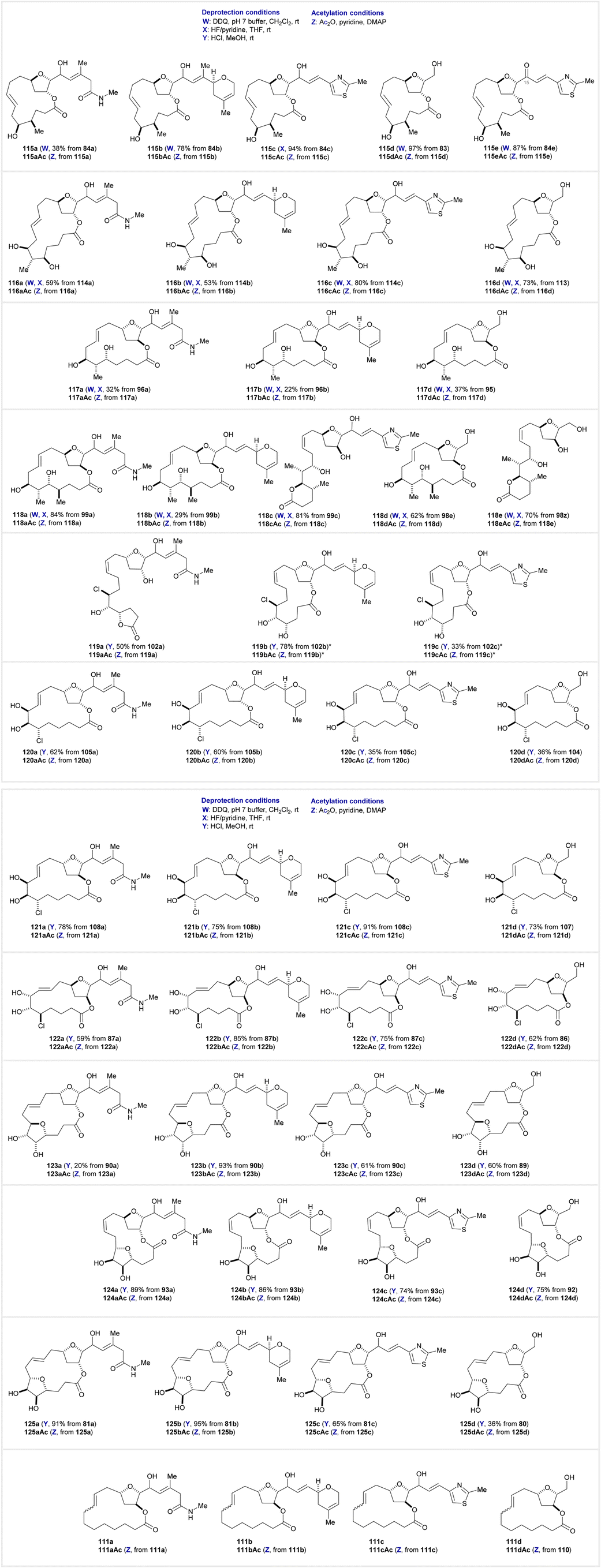 | ||
| Chart 1 Contd. | ||
To explore the effects of acetylation on bioactivity via cell permeability,61,62 we also peracetylated a small portion (∼1 mg of each compound (conditions Z)), to form 115–125Ac and 111Ac, doubling the count of library members to 170. As previously mentioned, the biological activity of this library was evaluated and discussed in detail in a recent publication.13
Conclusion
In summary, we synthesized a library of 170 pMLs varying in complexity, stereochemistry (6–9 stereocentres), ring size (14–16 atoms), peripheral groups (Me, OH, Cl, embedded THF), and large side-chains (amide, dihydropyran, and thiazole-containing). The synthetic campaign was enabled by numerous powerful synthetic methods, including the α-CAR reaction which was used to make 10 of the 14 chiral building blocks used in the library. Additionally, a compatibility screen of RCM reactions was used to guide library selection, followed by a challenging late-stage NHK reaction to diversify final products. We hope that the wide scope of reactions can act as a valuable resource for researchers, as well as provide a template for similar library syntheses to be accomplished on different families of molecules.Data availability
The experimental procedures, characterization data and 1H and 13C NMR spectroscopic data generated in this study are provided in the ESI.†Author contributions
D. D., D. M. W. and R. B. conceptualized the synthetic work, both D. D. and D. M. W. synthesized and characterized all compounds. Writing was done by D. D. and D. M. W. and supervision, funding acquisition, revision, and editing was done by R. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Natural Sciences and Engineering Research Council (NSERC) of Canada was received by R. B. (Discovery Grant: 2019-06368), D. M. W. (CGS-M & PGS-D) and D. J. D. (CGS-M & CGS-D).Notes and references
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- D. G. Jimenez, V. Poongavanam and J. Kihlberg, J. Med. Chem., 2023, 66, 5377–5396 CrossRef.
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco, D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef CAS.
- A. Whitty, M. Zhong, L. Viarengo, D. Beglov, D. R. Hall and S. Vajda, Drug Discovery Today, 2016, 21, 712–717 CrossRef CAS PubMed.
- L. A. Viarengo-Baker, L. E. Brown, A. A. Rzepiela and A. Whitty, Chem. Sci., 2021, 12, 4309–4328 RSC.
- P. Milton, J. I. D. Hamley, M. Walker and M. G. Basáñez, Expert Rev. Anti-Infect. Ther., 2020, 18, 1067–1081 CrossRef CAS.
- M. J. Yu, W. Zheng and B. M. Seletsky, Nat. Prod. Rep., 2013, 30, 1158–1164 RSC.
- V. R. Challa, D. Kwon, M. Taron, H. Fan, B. Kang, D. Wilson, F. P. J. Haeckl, S. Keerthisinghe, R. G. Linington and R. Britton, Chem. Sci., 2021, 12, 5534–5543 Search PubMed.
- A. K. Ghosh and Y. Wang, J. Am. Chem. Soc., 2000, 122, 11027–11028 CrossRef CAS PubMed.
- S. A. May and P. A. Grieco, Chem. Commun., 1998, 1597–1598 RSC.
- Y. K. Kim, M. A. Arai, T. Arai, J. O. Lamenzo, E. F. Dean, N. Patterson, P. A. Clemons and S. L. Schreiber, J. Am. Chem. Soc., 2004, 126, 14740–14745 CrossRef CAS.
- I. B. Seiple, Z. Zhang, P. Jakubec, A. Langlois-Mercier, P. M. Wright, D. T. Hog, K. Yabu, S. R. Allu, T. Fukuzaki, P. N. Carlsen, Y. Kitamura, X. Zhou, M. L. Condakes, F. T. Szczypiński, W. D. Green and A. G. Myers, Nature, 2016, 533, 338–345 CrossRef CAS PubMed.
- D. M. Wilson, D. J. Driedger, D. Y. Liu, S. Keerthisinghe, A. Hermann, C. Bieniossek, R. G. Linington and R. A. Britton, Nat. Commun., 2024, 15, 2534 Search PubMed.
- A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Álvarez, Chem. Rev., 2013, 113, 4567–4610 Search PubMed.
- T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48–56 Search PubMed.
- K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288–10317 CrossRef CAS PubMed.
- T. Teruya, K. Suenaga, S. Maruyama, M. Kurotaki and H. Kigoshi, Tetrahedron, 2005, 61, 6561–6567 CrossRef CAS.
- D. Corley, R. Herb, R. Moore and P. Scheur, J. Org. Chem., 1988, 53, 3644–3646 CrossRef CAS.
- E. Quiñoá, Y. Kakou and C. Phillip, J. Org. Chem., 1988, 53, 3642–3644 Search PubMed.
- R. T. Williamson, A. Boulanger, A. Vulpanovici, M. A. Roberts and W. H. Gerwick, J. Org. Chem., 2002, 67, 7927–7936 CrossRef CAS.
- M. Bergeron-Brlek, T. Teoh and R. Britton, Org. Lett., 2013, 15, 3554–3557 CrossRef CAS PubMed.
- M. Meanwell, G. Fehr, W. Ren, B. Adluri, V. Rose, J. Lehmann, S. M. Silverman, R. Rowshanpour, C. Adamson, M. Bergeron-Brlek, H. Foy, V. R. Challa, L. C. Campeau, T. Dudding and R. Britton, Commun. Chem., 2021, 4, 1–9 CrossRef.
- A. Kaghad, D. Panagopoulos, G. Caballero-García, H. Zhai and R. Britton, Nat. Commun., 2023, 14, 1–8 Search PubMed.
- M. Bergeron-Brlek, M. Meanwell and R. Britton, Nat. Commun., 2015, 6, 1–6 Search PubMed.
- R. A. Ashmus, Y. Wang, M. González-Cuesta, D. T. King, B. Tiet, P. A. Gilormini, J. M. García Fernández, C. Ortiz Mellet, R. Britton and D. J. Vocadlo, Org. Biomol. Chem., 2021, 19, 8057–8062 RSC.
- M. Meanwell, M. Sutherland and R. Britton, Can. J. Chem., 2017, 96, 144–147 CrossRef.
- D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560–3578 CrossRef CAS.
- D. A. Evans and A. H. Hoveyda, J. Org. Chem., 1990, 55, 5190–5192 CrossRef CAS.
- W. T. Markiewicz, Tetrahedron Lett., 1980, 21, 4523–4524 CrossRef CAS.
- S. F. Martin and J. A. Dodge, Tetrahedron Lett., 1991, 32, 3017–3020 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- M. A. Blanchette, W. Choy, J. T. Davis, A. P. Essenfeld, S. Masamune, W. R. Roush and T. Sakai, Tetrahedron Lett., 1984, 25, 2183–2186 CrossRef CAS.
- W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291–293 CrossRef CAS.
- D. W. Lee and J. Yun, Tetrahedron Lett., 2005, 46, 2037–2039 CrossRef CAS.
- J. Garegg and B. Samuelsson, Synthesis, 1976, 6, 469–470 Search PubMed.
- D. C. Schmitt, A. M. R. Dechert-Schmitt and M. J. Krische, Org. Lett., 2012, 14, 6302–6305 CrossRef CAS PubMed.
- A. B. Smith, K. P. Minbiole, P. R. Verhoest and M. Schelhaas, J. Am. Chem. Soc., 2001, 123, 10942–10953 CrossRef CAS PubMed.
- T. K. Chakraborty, S. Ghosh and S. Dutta, Tetrahedron Lett., 2001, 42, 5085–5088 CrossRef CAS.
- T. K. Chakraborty and A. K. Chattopadhyay, J. Org. Chem., 2008, 73, 3578–3581 CrossRef CAS PubMed.
- A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962–5963 CrossRef CAS PubMed.
- A. J. Mancuso, S. L. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480–2482 CrossRef CAS.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- W. Zhang and M. J. Robins, Tetrahedron Lett., 1992, 33, 1177–1180 CrossRef CAS.
- N. Takada, H. Sato, K. Suenaga, H. Arimoto, K. Yamada, K. Ueda and D. Uemura, Tetrahedron Lett., 1999, 40, 6309–6312 CrossRef CAS.
- H. Kigoshi and I. Hayakawa, Chem. Rec., 2007, 7, 254–264 CrossRef CAS.
- K. Gerth, N. Bedorf, G. Höfle, H. Irschik and H. Reichenbach, J. Antibiot., 1996, 49, 560–563 CrossRef CAS.
- C. L. Rand, D. E. Van Horn, M. W. Moore and E. Negishi, J. Org. Chem., 1981, 46, 4093–4096 CrossRef CAS.
- I. Paterson, H. Bergmann, D. Menche and A. Berkessel, Org. Lett., 2004, 6, 1293–1295 CrossRef CAS.
- I. Paterson, D. Menche, A. E. Håkansson, A. Longstaff, D. Wong, I. Barasoain, R. M. Buey and J. F. Díaz, Bioorg. Med. Chem. Lett., 2005, 15, 2243–2247 CrossRef CAS.
- A. G. Dossetter, T. F. Jamison and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 2398–2400 CrossRef CAS.
- K. Gademann, D. E. Chavez and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 114, 3185–3187 CrossRef.
- A. Gil, F. Albericio and M. Álvarez, Chem. Rev., 2017, 117, 8420–8446 CrossRef CAS PubMed.
- M. D. Lewis and Y. Kishi, Tetrahedron Lett., 1982, 23, 2343–2346 CrossRef CAS.
- J. Mulzer, T. Schulze, A. Strecker and W. Denzer, J. Org. Chem., 1988, 53, 4098–4103 CrossRef CAS.
- J. Mulzer, L. Kattner, A. R. Strecker, C. Schröder, J. Buschmann, C. Lehmann and P. Luger, J. Am. Chem. Soc., 1991, 113, 4218–4229 CrossRef CAS.
- J. Pospíšil, C. Müller and A. Fürstner, Chem.–Eur. J., 2009, 15, 5956–5968 CrossRef PubMed.
- J. Lamariano-Merketegi, A. Lorente, A. Gil, F. Albericio and M. Álvarez, Eur. J. Org. Chem., 2015, 235–241 CrossRef CAS.
- A. Vinaykumar, B. Surender and B. V. Rao, RSC Adv., 2023, 13, 22824–22830 RSC.
- I. Hayakawa, M. Ueda, M. Yamaura, Y. Ikeda, Y. Suzuki, K. Yoshizato and H. Kigoshi, Org. Lett., 2008, 10, 1859–1862 CrossRef CAS PubMed.
- D. W. C. Macmillan and L. E. Overman, J. Am. Chem. Soc., 1995, 117, 10391–10392 CrossRef CAS.
- M. Martínez-Martínez, C. Coscolín, G. Santiago, J. Chow, P. J. Stogios, R. Bargiela, C. Gertler, J. Navarro-Fernández, A. Bollinger, S. Thies, C. Méndez-García, A. Popovic, G. Brown, T. N. Chernikova, A. García-Moyano, G. E. K. Bjerga, P. Pérez-García, T. Hai, M. V. Del Pozo, R. Stokke, I. H. Steen, H. Cui, X. Xu, B. P. Nocek, M. Alcaide, M. Distaso, V. Mesa, A. I. Peláez, J. Sánchez, P. C. F. Buchholz, J. Pleiss, A. Fernández-Guerra, F. O. Glöckner, O. V. Golyshina, M. M. Yakimov, A. Savchenko, K. E. Jaeger, A. F. Yakunin, W. R. Streit, P. N. Golyshin, V. Guallar and M. Ferrer, ACS Chem. Biol., 2018, 13, 225–234 CrossRef PubMed.
- J. Morstein, A. Capecchi, K. Hinnah, B. Park, J. Petit-Jacques, R. C. Van Lehn, J. L. Reymond and D. Trauner, J. Am. Chem. Soc., 2022, 144, 18532–18544 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00591d |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |