
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-dimensional conjugated metal–organic frameworks for electrochemical energy conversion and storage
Xiao
Li
a,
Xi
Su
b,
Tan
Su
*b,
Long
Chen
 *b and
Zhongmin
Su
*ab
*b and
Zhongmin
Su
*ab
aSchool of Chemical and Environmental Engineering, Jilin Provincial Science and Technology Innovation Centre of Optical Materials and Chemistry, Jilin Provincial International Joint Research Center of Photo-functional Materials and Chemistry, Changchun University of Science and Technology, Changchun 130012, China. E-mail: zmsu@nenu.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, 130012, China. E-mail: sutan_jlu@jlu.edu.cn; longchen@jlu.edu.cn
First published on 27th February 2025
Abstract
Effective electrocatalysts and electrodes are the core components of energy conversion and storage systems for sustainable carbon and nitrogen cycles to achieve a carbon-neutral economy. Two-dimensional conjugated metal–organic frameworks (2D c-MOFs) have emerged as multifunctional materials for electrochemical applications benefiting from their similarity to graphene with remarkable conductivity, abundant active sites, devisable components, and well-defined crystalline structures. In this review, the structural design strategies to establish active components with a maximum degree through redox-active ligand assembly in 2D c-MOFs are briefly summarized. Next, recent representative examples of 2D c-MOFs applied in electrocatalysis (hydrogen/oxygen evolution and oxygen/carbon dioxide/nitrogen reduction) and energy storage systems (supercapacitors and batteries) are introduced. The synergistic effect of multiple components in 2D c-MOFs is particularly emphasized for enhanced performance in electrochemical energy conversion and storage systems. Finally, an outlook and challenges are proposed for realizing more active components, elucidating the reaction mechanism involving the derived structures, and achieving low-cost economy in practical applications.
1. Introduction
With the ever-increasing energy consumption and concomitant serious environmental problems associated with fossil fuels, high value-added chemical production and energy storage relying on renewable electrochemical devices have become an overwhelming trend for carbon-neutral economies.1,2 Owing to their advantages of eco-friendliness, simple manipulation, and flexible configuration, electrochemical energy conversion and storage systems led to advanced progress in recent decades, including water-splitting electrolyzers, CO2/N2-reduction electrolyzers, fuel cells, supercapacitors, metal–air batteries, and rechargeable batteries.3–5 Electrocatalysts and electrode materials are the core components of electrochemical devices and should possess the intrinsic features of well-defined structures with abundant active sites, efficient charge/mass transfer routes, and long-term chemical and structural stability to ensure excellent availability.6,7 To date, the practical applications of most electrochemical materials are limited by their unsatisfactory energy conversion efficiency resulting from their high energy barriers and sluggish reaction kinetics.8,9 Precious metal-based materials are the most efficient electrocatalysts and electrode materials, but their high cost, scarcity, and poor stability restrict their wide application in electrochemical devices.10–12 There is an urgent need to develop highly effective, low-cost materials with clear structure–property relationships to address the above challenges and stimulate the rational design and regulation of electrochemical devices.Metal–organic frameworks (MOFs) represent a class of typical crystalline materials with highly ordered porosity assembly by coordination bonds between metal ions/clusters and ligands and have various applications in gas sensing and storage, photo/electrocatalysis, and batteries, among others.13–15 Owing to their insulating properties, traditional 3D MOFs are commonly used as precursors in electrochemical energy systems through pyrolysis, in which the corresponding products of carbon or metal oxides inherit the merits of large surface areas and abundant active sites, resulting in improved electrochemical performance.16 However, uncertain fine structures greatly impede the investigation of structure–property relationships.17 Two-dimensional conjugated MOFs (2D c-MOFs) are typically constructed using π-conjugated ligands equipped with ortho-substituted functional groups and transition metal ions through a square-planar coordination configuration.18 2D c-MOFs have demonstrated prominent applications, such as electronics, gas sensing, catalysis, magnets, batteries, etc.19–23 Benefitting from the graphene-like structures, 2D c-MOFs exhibit two types of electron transfer pathways: strong in-plane conjugation and weak interaction of out-of-plane π–π stacking, which cooperatively result in decent intrinsic electrical conductivities.24 Moreover, together with the abundant and accessible active sites exposed in the planes and channels, effective mass transport, and device feasibility, 2D c-MOFs can be utilized as multifunctional materials for electrochemical energy storage and transformation via integration into electrolyzers and batteries.25
Currently, the investigation of 2D c-MOFs is mainly devoted to the synthesis of new types of structures based on molecular design and the search for new unexploited applications. For electrochemical applications, 2D c-MOFs have been used only for a few decades. Challenges remain regarding insufficient performance and unambiguous structural–property correlations. Several recent excellent reviews on MOFs and carbon-rich conjugated frameworks have discussed ligand design, synthetic methods, conductive mechanisms for electrode materials, and electrocatalysts.26–29 In this article, we highlight and sum up recent progress in 2D c-MOFs for applications in electrochemical energy conversion and storage systems, focusing on the synergistic effect of redox π-conjugated ligands and metal ions in charge transfer and mass transport processes. First, the design of redox ligands is introduced. Subsequently, representative 2D c-MOFs used in electrocatalytic water splitting, oxygen/carbon/nitrogen reduction for energy conversion, supercapacitors, and metal-ion batteries for energy storage are introduced. This review offers insights into the synergistic effects of ligands and metal ions to establish the maximum number of active sites, thereby supporting the emerging improvement of 2D c-MOFs in electrochemical energy applications.
2. Ligand design strategies, active site engineering, and tailor-made functions
Conjugated ligands with π-electron delocalization and redox properties are considered to be electrochemically active components that play vital roles in energy storage and conversion processes, including charge/mass transfer, key intermediate adsorption, product formation, and desorption.30–32 The ligands used to construct 2D c-MOFs are usually equipped with ortho-substituted functional groups, such as –OH, –NH2, –SH, and –SeH. Transition metal ions such as Co2+, Cu2+, Mn2+, Sn2+ and Zn2+, in 2D c-MOFs display square-planar coordinated modes through d-π conjugation, driving the growth in the second dimension for two-dimensional conjugated structures, always alongside 1D channels in the third dimension through π–π stacking interactions. Against the background of current progress, designing and synthesizing more novel ligands cooperating with more redox-active sites, such as various unsaturated bonds and heteroatoms, is greatly significant in the development of the synthesis chemistry of 2D c-MOFs.The topological structures and channel types of 2D c-MOFs can be predicted based on the symmetry of the organic ligands, which offers promising opportunities for designing frameworks with special functions. Typically, trigonal organic ligands based on benzene and polyacene aromatic cores of with C3 or C6 symmetry are inclined to form hexagonal lattices and channels.33 Tetragonal and rhombic lattices are often constructed using tetrasubstituted organic ligands with D2 symmetry containing porphyrin, phthalocyanine, and dibenzochrysen.34 Kagome lattices are often obtained using ligands with D6h symmetry, such as perthiolated coronene (PTC).35 Two stacking modes are most common in 2D c-MOFs, including the A–A stacking (face-to-face) mode and the slipped parallel mode, both of which feature one-dimensional ordered open channels. To date, with the exception of the first 2D c-MOF of Co-HHTP with clear single-crystal structures,36 most 2D c-MOFs are obtained in the form of polycrystalline powders, which cannot be tested by the single-crystal X-ray diffraction (SXRD) technique resulting from the rapid kinetics in the formation process and strong in-plane bonds of metal and heteroatoms.37 Considering the difficulty of controlling the coordination rate of metal ions and planar ligands, nonplanar organic linkers are promising candidates to synthesize planar 2D c-MOFs, first proposed by our group in 2020.34 A series of nonplanar ligands with various cores has been employed to construct planar 2D c-MOFs with high conductivity and enhanced crystallinity.38 The good solubility of nonplanar ligands ensures the easy synthesis and purification of high-quality single crystals. Flexible and decoratable structures can fulfill unprecedented topologies and custom-made functions through rational design and synthetic methods. For example, Cu-DCB-MOFs are synthesized utilizing a versatile “Rotor-Stator” strategy with highly crystalline structures, endowing zinc-ion hybrid supercapacitors with exceptional high performance.39
Multiple types of redox activities influenced by the metal ions and ligands in 2D c-MOFs play crucial roles in charge/mass transfer and metal ion storage for electrochemical energy conversion and storage applications. The square planar coordinated MX4 (M = Cu2+, Ni2+, Co2+, X = N, O, S, Se) is noted for its multiple chemical states and variations in electrochemistry, which can coexist in the same frameworks, such as Cu2+/Cu+ in Cu(HTQ).31 In contrast, unsaturated bonds, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN, exhibit redox activity via single/double-bond conversion. Recent representative examples of the redox ligands are summarized in Fig. 1. For example, HHTP in Zn-HHTP exhibits reversible oxidation and reduction through the CO to C–O· radical or C–O bond of semiquinone, which is responsible for the successive PF6− and Li+ storage in sodium-ion batteries.37 The unsaturated CO bonds also endow HHTQ-based 2D c-MOFs with multifunctional applications in the CO2RR, NO2 sensing, and lithium-ion batteries.41–43 For Zn3(HHTQ)2, a redox synergy strategy involving transition metal ions and ligands was used to balance the reactivity for NO2 sensing with remarkable cycling stability. Unsaturated CN bonds were confirmed to be the predominant active sites through coordination or redox interactions in the form of –N+– and conductivity variations. Cu3(HHTQ)2 displays excellent performance to reduce CO2 to methanol, with a low overpotential for high faradaic efficiency. The nitrogen-rich core of tircycloquinazadine (TQ) facilitates the capture of CO2 with a Gibbs free energy ΔG of −0.24 eV, showing good CO2 adsorption ability.42 For lithium storage, TQ was verified as a redox active component which reacted with Li+ for the first time, synergistically working with CuO4 in Cu-HHTQ, resulting in ultrahigh capacity and superior cyclability for lithium-ion batteries.43 In general, ligands with redox activity can establish the largest utilization degree of multicomponent 2D c-MOFs by synergistic effects, further helping to elucidate the structure–property relationships at the molecular and atomic levels, which can be beneficial for promising electrochemical materials through rational design and skillful synthesis of 2D c-MOFs.
O and CN, exhibit redox activity via single/double-bond conversion. Recent representative examples of the redox ligands are summarized in Fig. 1. For example, HHTP in Zn-HHTP exhibits reversible oxidation and reduction through the CO to C–O· radical or C–O bond of semiquinone, which is responsible for the successive PF6− and Li+ storage in sodium-ion batteries.37 The unsaturated CO bonds also endow HHTQ-based 2D c-MOFs with multifunctional applications in the CO2RR, NO2 sensing, and lithium-ion batteries.41–43 For Zn3(HHTQ)2, a redox synergy strategy involving transition metal ions and ligands was used to balance the reactivity for NO2 sensing with remarkable cycling stability. Unsaturated CN bonds were confirmed to be the predominant active sites through coordination or redox interactions in the form of –N+– and conductivity variations. Cu3(HHTQ)2 displays excellent performance to reduce CO2 to methanol, with a low overpotential for high faradaic efficiency. The nitrogen-rich core of tircycloquinazadine (TQ) facilitates the capture of CO2 with a Gibbs free energy ΔG of −0.24 eV, showing good CO2 adsorption ability.42 For lithium storage, TQ was verified as a redox active component which reacted with Li+ for the first time, synergistically working with CuO4 in Cu-HHTQ, resulting in ultrahigh capacity and superior cyclability for lithium-ion batteries.43 In general, ligands with redox activity can establish the largest utilization degree of multicomponent 2D c-MOFs by synergistic effects, further helping to elucidate the structure–property relationships at the molecular and atomic levels, which can be beneficial for promising electrochemical materials through rational design and skillful synthesis of 2D c-MOFs.
 | ||
| Fig. 1 Typical redox ligands reported for 2D c-MOFs. | ||
3. Applications in electrochemical energy conversion systems
In the field of electrochemical energy conversion, there are several main reactions, including the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), carbon dioxide reduction reaction (CO2RR), and nitrogen reduction reaction (NRR). The HER and OER can be integrated into a water electrolyzer for water splitting, and the OER and ORR can be combined for zinc–air batteries.44,45 For electrocatalytic reactions, the sluggish kinetics, high activation energy barriers, and large overpotentials heavily hinder the practical application of transition-metal-based electrocatalysts. Developing highly efficient non-precious metal electrocatalytic materials with clear structural–activity relationships is a major challenge.2D c-MOFs exhibit well-designed crystal structures and single-atom-like catalyst behavior, which can act as an ideal model for fundamental mechanistic investigations of structure–performance relationships at the atomic level.45 The conjugated planar structures contribute to intrinsic electrical conductivity, abundant active sites, and clear pathways for electron and mass transfer, positioning 2D c-MOFs as promising electrocatalysts. Furthermore, the flexible variation of components with ligands induces specific topologies that can effectively modulate the electronic structures of active sites, which is crucial for enhancing electrocatalytic performance.46,47 The effect of ligands on 2D c-MOFs has attracted increasing research attention.
Electrocatalytic activity is determined by electron delocalization and conductivity, both of which tightly correspond to the architecture of the ligands. Furthermore, the functional groups in the organic ligands usually act as synergistic catalytic sites.
3.1 ORR electrocatalysts
The ORR is a crucial half-reaction of fuel batteries and metal–air batteries. M–N–C are the most considered catalytically active sites for the ORR. In 2016, Ni3(HTTP)2 has been applied in the ORR for the first time, showing that the NiN4 units act as active sites.48 Following PcCuO8-Co on carbon nanotubes (CNT) in 2019, CuO4 motifs were confirmed as ORR active sites owing to the favorable optimization of the electronic structure, resulting in moderate interaction with the key intermediates of *OOH.49 Furthermore, the in-plane sites of conjugated ligands have also been proposed as ORR active sites originating from their large limiting potential and strong oxygen binding.50–52 In 2023, a di-copper-based 2D c-MOF of CuPc–CuO4 films combined with a Ni-nitrilotriacetic acid (Ni-NTA)-functionalized graphene electrode was investigated for the ORR using electrochemical resonance Raman spectroscopy.53 The reduction transitions of CuII → CuI and CuPcx increased π-conjugation, enabling catalysis. The CuII/CuI states of CuO4 and CuPc are the active sites for the ORR.Coordination atoms can adjust the electronic structures of the metal sites. In Fig. 2a, Dai and colleagues used density functional theory (DFT) to investigate the regulation effect of diverse organic ligands on the ORR activity in 2D c-MOFs.54 Co–2O based on HHTP displays superior ORR activity via the 4e− reduction route with a relatively low overpotential of 0.23 V (Fig. 2b and c). The Co sites in Co–2O possess a high-spin state, regulated by the synergistic effect of triphenylene and hydroxyl groups, which promotes interaction with the key intermediates of *OH for enhanced ORR performance. Moreover, all Co-MOFs displayed excellent electrochemical and thermodynamic stabilities. Jaramillo et al. introduced dual MN4 (M = Co, Ni, Cu) sites in a 2D phthalocyanine MOF as ORR electrocatalysts, in which one metal ion as M2 coordinates with an amine node and the other metal ion is bonded to the phthalocyanine center (M1) (Fig. 2d).55 The Co sites dominate the catalytic activity and control the layer distance. The low degree of saturated M–N bonds at the M2 sites promotes the strong adsorption of OH, displaying more dominance in catalytic activity, while the M1 sites greatly influence the selectivity. In Fig. 2e, the bimetallic combination of Ni–Co is predicted to obtain the highest ORR activity with 0.27 V as the minimum overpotential based on the optimized OOH*–OH* value. This work highlights the tailored function of electrocatalysis by tuning the strain of the metal sites and coordination environments.
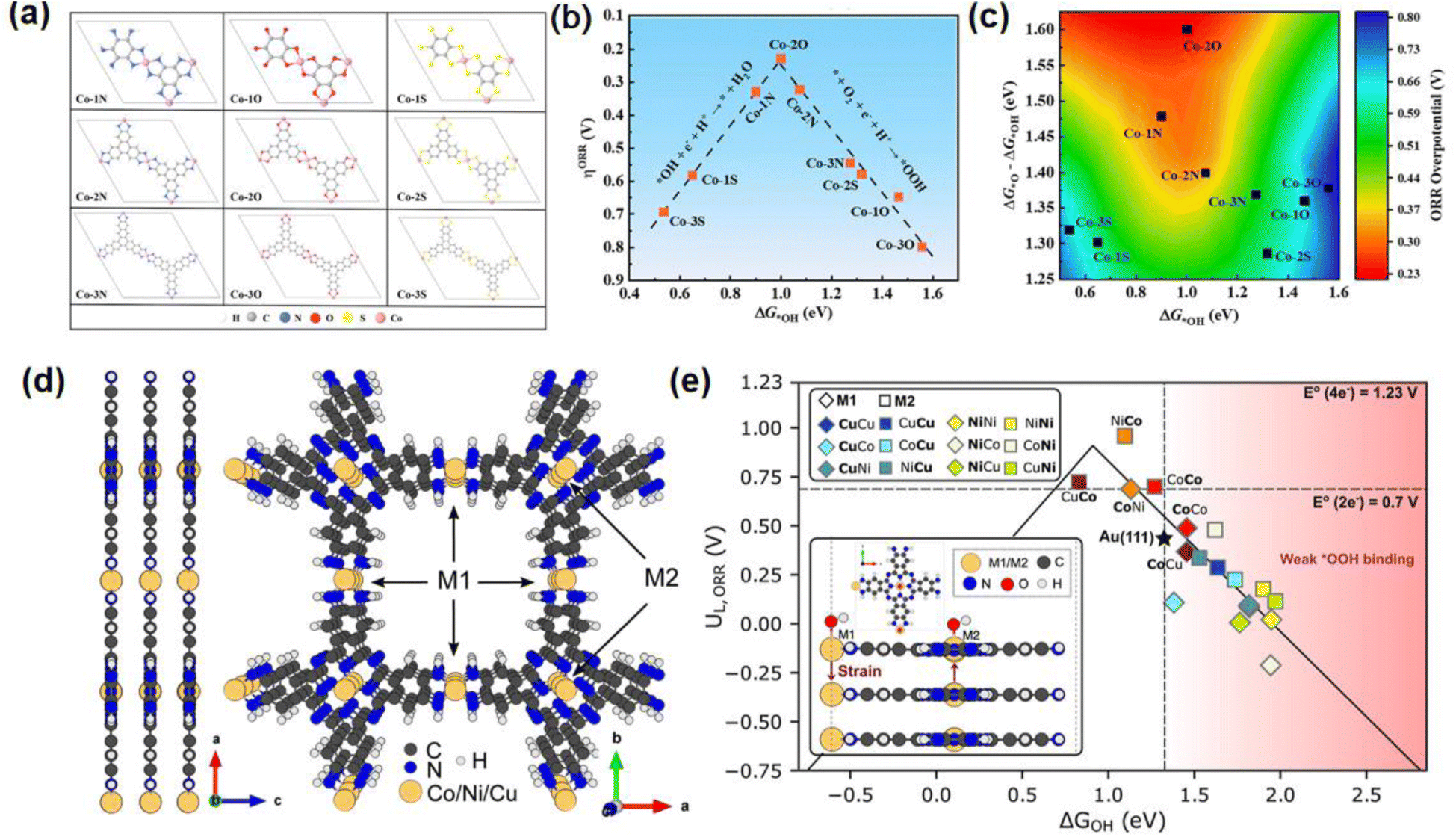 | ||
| Fig. 2 (a) Scheme of different organic ligands in 2D Co-MOFs. (b and c) Volcano plots of DG*OH versus overpotentials. (d) MOF structural model from side and top views. (e) Volcano plot of MOFs (each site) toward a four-electron ORR for flat layers. (a)–(c) were reproduced with permission from ref. 54. Copyright 2023 American Chemical Society. (d) and (e) were reproduced with permission from ref. 55. Copyright 2024 American Chemical Society. | ||
3.2 OER electrocatalysts
The OER is a key half reaction in electrocatalytic water splitting and metal–air batteries. The four-electron transfer process results in slow kinetics and high activation energy barriers of the OER; thus, effective and low-cost electrocatalysts are urgently needed. The CoN4 sites were verified to be efficient OER-active sites. The large d-π conjugation required for a fast electron transfer capability makes 2D c-MOFs highly promising OER electrocatalysts.As one of the smallest conjugated ligands, hexaaminobenzene (HAB) possesses dense redox-active sites that enable charge transfer between metal nodes and the plane of the ligand. The ultrathin 2D nanosheets of Co-HAB-NSs with a thickness about 4.5 nm, displayed a low overpotential of 310 mV at 10 mA cm−2 in KOH with good durability.56 The nitrogen richness of HAB endows Co-HAB-NSs with highly dense active sites for electrocatalysis. Through DFT calculations, the overpotentials of the carbon atoms from the benzene rings were lower than those of the Co sites, implying a higher catalytic activity. This work indicates that dense active sites from redox ligands can greatly improve the catalytic performance through strong π-d conjugation with 1D channels to accelerate charge transfer.
The larger conjugated ligands of HITP compared to the HAB-constructed 2D c-MOF of Co3(HITP)2 (Fig. 3a) presented a higher electrical conductivity of 1150 S m−1 than that of graphene.57Co3(HITP)2 displayed a remarkable OER performance with an overpotential of 254 mV to reach 10 mA cm−2 (Fig. 3b). Both the calculated density of states (DOS) and electronic band structure of Co3(HITP)2 are metallic, resulting from the p–d conjugation of pz orbitals from N atoms and the dyz and dxz orbitals from Co (Fig. 3c–e). The high content (23.44 wt%) of the active sites of the CoN4 sites ensured excellent OER performance. The structural flexibility through ligand control of 2D c-MOFs offers more opportunities for the OER through electronic structure modulation.
 | ||
| Fig. 3 (a) The structure of Co3(HITP)2. (b) LSV curves of Co3(HITP)2 and controlled samples. (c–e) Total and partial DOS of Co3(HITP)2. (f) Structure schematics of TMNxO4−x-TOT systems. (g) 2D volcano plot of Gibbs free energy difference between *O and *OH (DGO*–DGOH*) versus the ηOER and ηORR. (a)–(e) were reproduced with permission from ref. 57. Copyright 2020 Elsevier. (f) and (g) were adapted with permission from ref. 61. Copyright 2023 American Chemical Society. | ||
Co-HHTP also shows OER activity with an overpotential of 245 mV at 10 mA cm−2, exceeding most reported Co-MOFs.58 DFT calculations demonstrated the moderate d-band centers and favorable electrical conductivity of Co-HHTP, promoting the adsorption/desorption capability of oxygen-containing intermediates. In situ Raman spectroscopy confirmed the formation of CoOOH as the real active sites in the OER process.
Bifunctional OER/ORR catalysts based on 2D c-MOFs, such as NixCo3−x(HITP)2 (ref. 59) and (Mn/Fe)3(HIB)2,60 have been developed as bimetallic frameworks. Jing et al. proposed a simple descriptor as “spin momentum” of the metal center as the universal principle to assess the OER/ORR activity by machine learning activity (Fig. 3f).61 2D conjugated organic linkers provide different coordination environments as the core and functional groups work together. The hybridization of d–p interactions involves p orbitals from the O/N/C of ligands and d orbitals from metal ions, which redistribute the electronic states of the metal centers. Taking TMNxO4−x-TOTs as models, CoN4-TOT exhibits bifunctional OER/ORR activity with ηOER = 0.28 V and ηORR = 0.26 V in Fig. 3g. This work can guide the screening of 2D c-MOFs with various organic ligands and reveal the intrinsic correlation between coordination properties and catalytic performance.
The conjugation of ligands can largely decide the electrocatalytic process by enhancing charge-transfer kinetics. Using DFT calculations, Dai et al. reported a sequence of 2D c-MOFs (TM-HHTC) for the OER/ORR performance investigation.62 Co-HHTC exhibited superior OER/ORR activity with an overpotential of 0.21 V. Co metal sites and Co–O bonds show suitable d-band center values and medium charge accumulation, which are key factors for the OER/ORR activity. Moreover, the interaction between the 2s orbital of oxygen and the 4pz orbital of Co can regulate catalytic activity.
3.3 HER electrocatalysts
Electrocatalytic water splitting is considered as the most efficient and simple method for hydrogen production. In 2D c-MOFs, Co–S bonds are usually recognized as active sites for the HER, such as Co3(BHT)2 and Co3(HTTP)2.63 Unsaturated NiN2 sites also play a decisive role in the HER.64 Sarkar reported TMPc-Cu–O MOFs for water splitting, in which TM = Fe, Co, and Ni were coordinated with the core of Pc and Cu in the skeleton. CoPc–Cu–O displays smallest overpotentials of 0.11/0.65 V for the HER/OER, in which the C atoms from Pc units and Cu sites are confirmed as the active sites for the HER and OER, respectively. Cu3HHAE2 comprises CuO4 units and sp-carbons from the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond arylene–ethynylene macrocycle ligand (Fig. 4a and b).65 Due to the incorporation of electron deficient properties of acetylene units, the acidity of the functional group (–OH) increases, resulting in reversibility of the metal–ligand bonds. As shown in Fig. 4c, Cu3HHAE2 was used as a photoelectrode for hydrogen generation, as the first case of 2D c-MOFs, achieving approximately 260 μA cm−2 at 0 V versus a reversible hydrogen electrode (RHE). The CC bonds with low ΔGH* value of 0.17 eV serve as active sites for the HER (Fig. 4d).
C bond arylene–ethynylene macrocycle ligand (Fig. 4a and b).65 Due to the incorporation of electron deficient properties of acetylene units, the acidity of the functional group (–OH) increases, resulting in reversibility of the metal–ligand bonds. As shown in Fig. 4c, Cu3HHAE2 was used as a photoelectrode for hydrogen generation, as the first case of 2D c-MOFs, achieving approximately 260 μA cm−2 at 0 V versus a reversible hydrogen electrode (RHE). The CC bonds with low ΔGH* value of 0.17 eV serve as active sites for the HER (Fig. 4d).
 | ||
| Fig. 4 (a) The structure of Cu3HHAE2. (b) Views parallel to the ab plane. (c) Photocurrent–time plots for Cu3HHAE2 and HHAE cathodes at 0 V vs. RHE. On: illumination on; off: illumination off. (d) Free-energy diagram for H2- evolution via a single-site reaction pathway: 1, 2, 3, 4, 5, 6 denote different active sites of Cu3HHAE2. Reproduced with permission from ref. 65. Copyright 2022 Wiley. | ||
3.4 CO2RR electrocatalysts
The CO2 reduction reaction usually proceeds via various reaction routes with electron numbers ranging from 2 to 18, harvesting various products such as CO, CH3OH, HCOOH, CH4, C2H5OH, C2H4 and so on.66 The selectivity of the CO2RR depends on the type of metal ion, in which high-value-added carbon products are often generated by Cu species. The CO2RR involves three main steps: (1) CO2 molecules are adsorbed and activated on catalytic sites; (2) proton migration and electron transfer induce breakage of C–O bonds and formation of C–H bonds; and (3) release of the corresponding products and diffusion into the electrolyte. Successive and complex reaction steps result in huge challenges, such as high energy barriers, slow reaction kinetics, difficult separation of raw materials and products, and inferior selectivity and stability. 2D c-MOFs with planar structures with highly dense and exposed active sites and tailored structures can solve the above challenges by integrating multiple active components from redox ligands or dual metal sites.Feng and coworkers designed a bimetallic 2D c-MOF of PcCu–O8-Zn as an effective electrocatalyst through the CO2RR to produce CO.67 The selectivity of CO was 88% with a turnover frequency (TOF) number of 0.39 s−1 and long-time stability (>10 h). Moreover, syngas can be modulated from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 to 1:4H2/CO by changing working potentials and the metal centers. Operando spectroelectrochemistry and DFT calculations indicated the synergistic catalytic mechanism of the two metal units, in which ZnO4 can act as the catalytic site for the CO2RR, while CuN4 promotes the adsorption and protonation of CO2. Ligands with various heteroatoms and cores play decisive roles in the CO2RR. The coordination environment of the metal nodes can also participate in the catalytic process of the CO2RR.
:7 to 1:4H2/CO by changing working potentials and the metal centers. Operando spectroelectrochemistry and DFT calculations indicated the synergistic catalytic mechanism of the two metal units, in which ZnO4 can act as the catalytic site for the CO2RR, while CuN4 promotes the adsorption and protonation of CO2. Ligands with various heteroatoms and cores play decisive roles in the CO2RR. The coordination environment of the metal nodes can also participate in the catalytic process of the CO2RR.
2D c-MOFs based on benzene cores can exhibit excellent electrical conductivity and highly dense metal sites, which are attractive for the CO2RR. The use of rationally modified ligands can be investigated as an efficient method to improve the CO2RR of the objective products. In 2021, our group used a nitrogen-rich tricycloquinazoline (TQ)-based catechol ligand (HHTQ) to construct 2D c-MOFs: M3(HHTQ)2 (M = Cu, Ni) under solvothermal conditions (Fig. 5a).42 Alongside the graphene-like porous sheets and single-atom catalyst characteristics, Cu3(HHTQ)2 exhibited excellent CO2RR activity, in which the faradaic efficiency (FE) of the CH3OH product reached 53.6% at a low overpotential of 0.4 V (Fig. 5b–d). The introduction of the TQ moiety helped capture and catalyze CO2 synergistically with CuO4 for the CO2RR (Fig. 5e). Wang et al. reported a nitrogen-rich ligand-based 2D c-MOF (HATNA-Cu-MOF), as shown in Fig. 5f.68 The exposed N sites displayed high affinity for CO2. As shown in Fig. 5g–j, the HATNA-Cu-MOF exhibited excellent performance for the conversion of CO2 to CH4. The FE of CH4 was 78% at 8.2 mA cm−2 with good durability for 12 h at −1.5 V.
 | ||
| Fig. 5 (a) Synthesis of M3(HHTQ)2 (M = Cu, Ni), (b) LSV curves for Cu3(HHTQ)2 in Ar (grey line) or CO2 (orange line) saturated 0.1 M KHCO3 electrolyte with a scan rate of 5 mV s−1. (c) CH3OH FEs at different potentials. (d) The selectivity for each product (CH3OH and H2). (e) Free-energy profiles of CO2 on MO4 units. (f) Synthetic process of the HATNA-Cu-MOF. (g) LSV curves of the HATNA-Cu-MOF in Ar- and CO2-saturated 0.1 M KHCO3 electrolytes. (h) Potential dependent faradaic efficiencies of different reduction products. (i) Chronoamperometry and FE(CH4) of the HATNA-Cu-MOF at 1.5 V. (j) Comparison of CH4 faradaic efficiencies between the HATNA-Cu-MOF, HATNA-6OH and the model compound. (a)–(e) were adapted with permission from ref. 42. Copyright 2021 Wiley. (f)–(j) were reproduced with permission from ref. 68. Copyright 2021 Wiley. | ||
Heterodimetal Sn⋯Cu dual sites were integrated into CuSn-HAB to improve asymmetric C–C coupling in the CO2RR.69 CuSn-HAB depicts superior CO2 conversion activity for ethanol production, which shows an FE of 56(2)% with at 68 mA cm−2 at −0.57 V (vs. RHE), and no significant degradation of current density when operated for continuous 35 h. The SnN2O2 units showed enhanced affinity for oxygen, which facilitated the production of the key intermediate *OCH2. Compared with Cu⋯Cu sites, which tend to produce ethylene, Cu⋯Sn sites exhibit more thermodynamically favorable interactions between *CO and *OCH2 through asymmetric C–C coupling, yielding the ethanol product.
3.5 NRR electrocatalysts
The NRR for converting N2 to NH3 in 2D c-MOFs is still rare and in its infancy. Because of the relatively high energy (944 kJ mol−1 of NN bonds), N2 activation is the most challenging step for the NRR. Co3(HTTP)2 nanoparticles show an NH3 production rate of 22.14 μg h−1 mg−1 cat with an FE of 3.4% at −0.4 V vs. RHE.70 Jing et al. employed DFT calculations to elucidate the influence of heteroatoms in MN4 patterns on NRR activation.71 Mo3(C6S6)2 demonstrated the best activity with a minimal of 0.37 V, followed by the pathway of enzymatic nitrogenase. Babarao and coworkers investigated the NRR activity of the MN4 units in 2D porphyrin frameworks as single-atom catalysts (Fig. 6a).72 DFT calculations indicated that the bonding state of the M–N bonds and the antibonding state of the N–N bonds are crucial and contribute equally to the NN bond activation (Fig. 6b). As shown in Fig. 6c, the Ti-TCPP MOF exhibited exclusive NRR selectivity with a potential of −0.35 V in aqueous solutions. In the final step of protonation, Ti-TCPP exhibits a low kinetic energy barrier (1.43 eV, Fig. 6d), making Ti-TCPP a promising candidate electrocatalyst for the NRR. Xu et al. explored the 2D c-MOFs of TM3C18N6X12 as NRR electrocatalysts using DFT calculations.73 Coordinated atoms significantly influence the NRR performance of the 2D c-MOF based electrocatalysts. Os3C18S12N6 shows a lowest overpotential of 0.28 V through the distal pathway. The σ-acceptance-π* donation influences the adsorption and activation processes of N2. Two descriptors were proposed to assess the catalytic activity during the N2 adsorption process: the electronegativity of the metal atoms before and after the adsorption of N2 (ΔQTMXTM), and the valence electron transfer product.
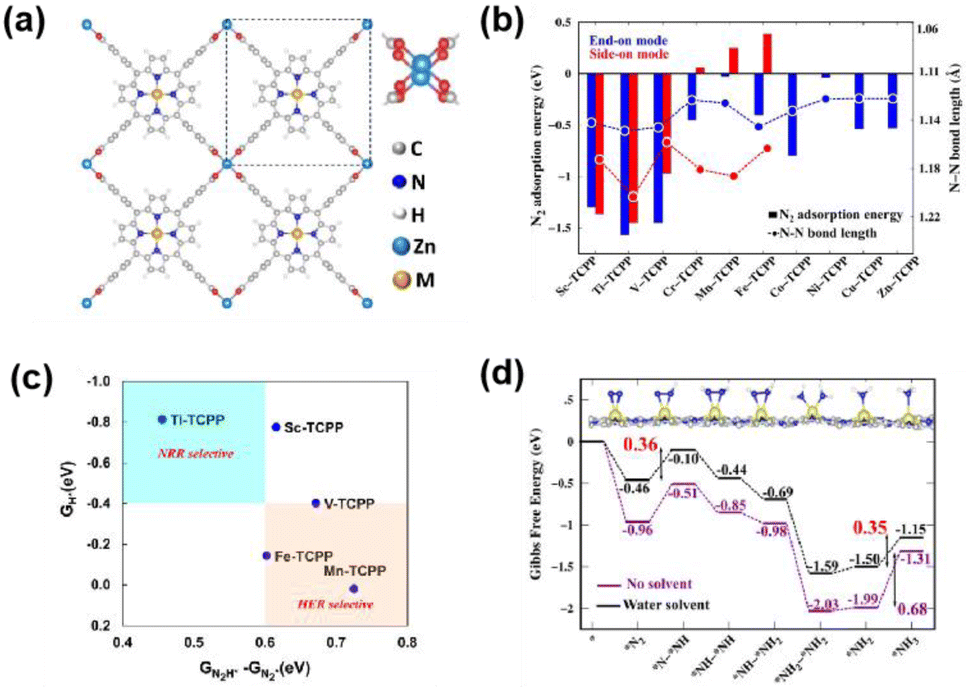 | ||
| Fig. 6 (a) The optimized structure of M-TCPP 2D-MOFs with the paddlewheel Zn2(CO2)4 linker. (b) N2 adsorption energies and N–N bond lengths of N2 adsorbed in perpendicular and parallel modes in M-TCPP MOFs. (c) NRR selectivity plots of the M-TCPP MOFs with lower limiting potentials. (d) Free energy diagram of the enzymatic NRR on Ti-TCPP. Reproduced with permission from ref. 72. Copyright 2024 American Chemical Society. | ||
4. Applications in electrochemical energy storage systems
Electrochemical energy storage systems mainly include supercapacitors and metal-ion batteries, which play significant roles in our daily lives and human society, and have attracted wide attention in the past few decades. Supercapacitors rely on charge storage via a surface-controlled process, delivering a ultralong cycle life, high power density, and free maintenance, but a low energy density. Metal-ion batteries with faradaic redox reactions by bulk diffusion-controlled processes generally focus on higher rate performance and energy density with long-term cycle stability and safety. Electrode materials play decisive roles in both supercapacitors and metal-ion batteries and can directly determine their performance. Large conductivity and high redox activity are two common features of highly active electrode materials. The higher conductivity can induce the faster charge transfer among active sites and substrates, which is beneficial for the energy storage process in supercapacitors and metal-ion batteries. For this purpose, 2D c-MOFs with high intrinsic electrical conductivity (up to 2500 S cm−1)74 and tunable redox sites (metal nodes and redox ligands) are integrated into the frameworks to facilitate fast electron transport and reversible migration of metal ions. Moreover, the regular channels and large surface area of 2D c-MOFs can provide clear pathways and space for ion transport and storage. 2D c-MOFs have attracted growing research interest in electrochemical energy storage fields and have shown excellent rate capability and splendid activity.4.1 Supercapacitors
The active materials in supercapacitors depend on different charge-storage capacitances, including physical ion adsorbents that induce electrical double-layer (EDL) capacitor materials and surface faradaic redox reactions, resulting in pseudocapacitor materials. In 2D c-MOFs, both metal nodes and redox organic linkers can exhibit capacity, often with different charge storage mechanisms, of which the synergistic contribution produces unprecedented supercapacitor performance.In 2017, Ni3HITP2 with a high conductivity exceeding 5000 S m−1,75 as the first case of 2D c-MOFs, exhibits gravimetric capacitance of 111 F g−1 and areal capacitance of 18 μF cm−2 at 0.05 A g−1, predominately by the double-layer capacitance mechanism. In 2018, Bao and colleagues reported Cu/Ni-HAB 2D c-MOFs with redox activity as submillimeter-thick electrochemical supercapacitors.76 Bulk Ni-HAB and Cu-HAB showed large capacitances of 420 and 215 F g−1, respectively, in which HAB reversible redox reactions contributed to the primary pseudocapacitors.
In 2020, we reported redox ligand DBC-based conjugated MOFs of Cu-DBC under solvothermal conditions, which showed a high electrical conductivity (≈1.0 S m−1) with typical semiconducting behavior (Fig. 7a).34 The charge storage mechanism of supercapacitors for Cu-DBC exhibited both pseudocapacitance and double-layer capacitance owing to the synergistic effect of the DBC ligands and copper bis(dihydroxy) nodes, resulting in a gravimetric capacitance of 479 F g−1, a large areal capacitance of 879 mF cm−2, and a volumetric capacitance of 22 F cm−3 (Fig. 7b). Recently, we synthesized a 2D c-MOF of Cu-TBC using a hexahydroxytribenzocoronene ligand (6OH-TBC) as the electrode material for supercapacitors (Fig. 7c).77 Large conjugated aromatic ligands can enhance the conductivity and stability of 2D c-MOFs. Featuring a decent electrical conductivity of ∼0.068 S m−1, Cu-TBC shows dominant capacitance at −0.4–0.6 V (vs. Ag/AgCl) with a gravimetric capacitance of 474.8 F g−1 at 0.2 A g−1 (Fig. 7d) in 0.1 M H2SO4.In Fig. 7e, the charge storage mechanisms were also influenced by the electric double layer and pseudocapacitance.
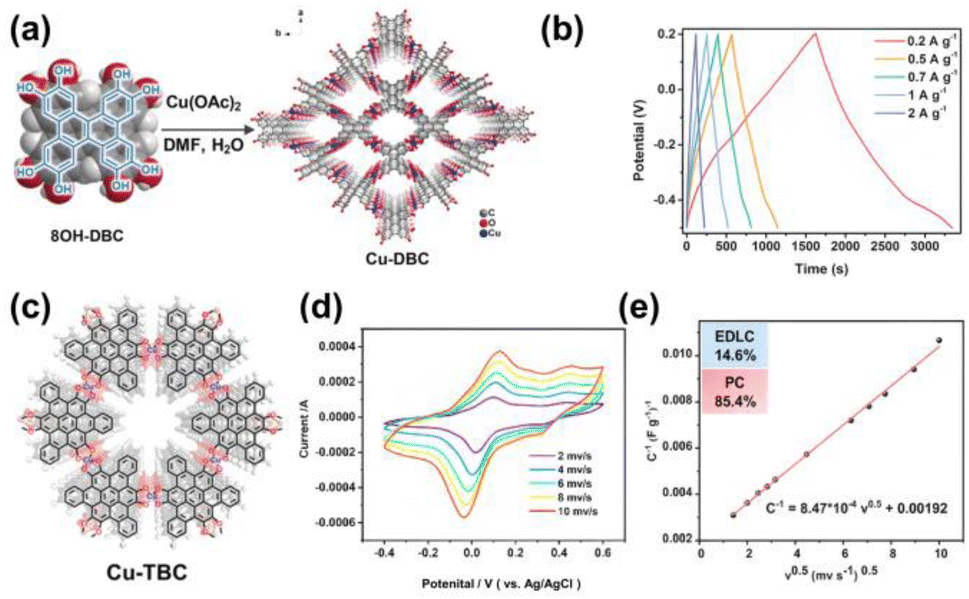 | ||
| Fig. 7 (a) Synthesis of Cu-DBC. (b) GCD profiles collected at different current densities. (c) Structure of Cu-TBC. (d) CV curves of Cu-TBC at different scan rates. (e) Trasatti plot of the reciprocal of the gravimetric capacitance (C g−1) against the square root of the scan rate. (a) and (b) Were reproduced with permission from ref. 34. Copyright 2020 Wiley. (c)–(e) Were reproduced with permission from ref. 77. Copyright 2023 Royal Society of Chemistry. | ||
The Feng group developed NH4+ hybrid supercapacitors (HSC) using a 2D c-MOF of Cu-HHB to immobilize iodine (Cu-HHB/I2) as the cathode material (Fig. 8a).78 Cu-HHB/I2 features an excellent areal capacitance of 111.7 mF cm−2 at 0.4 mA cm−2, shown in Fig. 8b. Cu–O4 nodes in Cu-HHB can act as both binding sites for iodine and redox-active sites. As shown in Fig. 8c, the synergy between CuO4 and I2via redox reactions contributes to the high pseudocapacitance. In another example, as displayed in Fig. 8d, Bonaccorso and coworkers developed porous Cu-HHB through a surfactant-assisted method as negative electrodes with activated carbon as positive electrodes for hybrid supercapacitors.79 The Cu-HHB based HSC exhibits high cell specific capacity (22.1 mA h−1 g−1 at 0.1 A g−1) and specific energy (15.55 W h kg−1) with 77% cycling stability. Combined charge storage mechanisms, including EDLC- and battery-type faradaic behavior, have been proposed as a result of the redox reaction of Cu2+ to Cu+/Cu0 from CuO4 units and catechols to benzoquinones of the HHB ligands. Recently, Feng et al. synthesized a 2D c-MOF of Ni2[CuPcS8] (Fig. 8e) that displayed a high capacitance of 312 F g−1 in non-aqueous supercapacitors with EDLC-type pseudocapacitive behavior.80 The redox-active NiS4 nodes maintain a localized lowest unoccupied molecular orbital (LUMO), which enhances the injected electrons responsible for the high capacitance.
 | ||
| Fig. 8 (a) The unit-cell structures of Cu-HHB derived using AB slipped-parallel stacking models. (b) CV curves. (c) Specific capacitances of electrodes calculated from GCD curves as a function of current density. (d) Schematic illustration of the synthesis process for p-HHB-Cu and np-HHB-Cu. Unit cell structures of HHB-Cu derived using AA and AB stacking models. (e) Schematic synthesis of the CuPc(SH)8 ligand and Ni2[CuPcS8] and the space-filling model of Ni2[CuPcS8]. (a)–(c) were reproduced with permission from ref. 78. Copyright 2023 Wiley. (d) was reproduced with permission from ref. 79. Copyright 2024 Wiley. (e) was adapted with permission from ref. 80. Copyright 2024 American Chemical Society. | ||
Chang et al. designed and reported a 2D c-MOF of Ni-TTC, showing about 1.0 S m−1 electrical conductivity, in which TTC possess redox activity with D4 symmetry.81 The semiquinone ligand radicals together with Ni2+ sites, resulting in strong antiferromagnetic properties, ensured a gravimetric capacitance of 249 F g−1 at 0.2 A g−1. Good electrical conductivity and a large surface area are beneficial for enhancing the EDL capacitance of 2D c-MOFs. The pseudocapacitance of 2D c-MOFs can be improved by embedding diverse redox-active substrates from ligands and metal ion nodes.
4.2 Batteries
Rechargeable batteries with high energy densities include lithium-ion batteries (LIBs) and the gradually rising generation of cost-effective and abundant metal ion batteries, such as sodium-ion batteries (SIBs), potassium-ion batteries (PIBs), zinc-ion batteries (ZIBs), next-generation lithium–sulfur (Li–S) batteries, and sodium iodine (Na–I2) batteries. Electrode materials play a core role in batteries because of their reversible charge–discharge processes and complex electrochemical redox reactions. The outstanding merits of 2D c-MOFs, including multiple redox-active sites, superior electrical conductivity, regular channels, and remarkable durability, make them superior electrode materials for various batteries.In 2020, we synthesized a redox-active Cu-HTQ MOF as a cathode material for LIBs.31 Cu-HTQ exhibited a large reversible capacity of 387 mA h−1 g−1 and a high energy density of 775 W h kg−1 by the Li+ insertion/extraction mechanism. Comprehensive spectroscopic techniques revealed redox reactions of one electron per Cu2+ ion and three electrons per coordinated unit. Prominent redox activity of both HTQ and copper ions occurred in the charge and discharge process, as shown in Fig. 9a, suggesting the rational design of effective 2D c-MOF-based cathode materials. Subsequently, we developed a pyrazine-based TPQG-Cu-MOF as the cathode material for LIBs (Fig. 9b).83
 | ||
| Fig. 9 (a) The evolution of electronic states of the repeating coordination unit of the 2D Cu-THQ MOF during the charge/discharge process. (b) Schematic illustration of the synthesis method of the TPQG-Cu-MOF. (c) The ex situ FT-IR TPQG-Cu-MOF electrode at different states for the lithium storage mechanism. (d) A schematic diagram of the lithium ion storage mechanism. (a) was reproduced with permission from ref. 31. Copyright 2020 Wiley. (b)–(d) were adapted with permission from ref. 83. Copyright 2023 Wiley. | ||
The TPQG-Cu-MOF displayed a reversible specific capacity of 150.2 mA h−1 g−1 at 20 mA g−1 with nearly 100% coulombic efficiency. The Li+ insertion/extraction mechanism driven by the CuO2 and pyrazine units was characterized by comprehensive in situ spectroscopic investigations, as displayed in Fig. 9c. During the charge and discharge process, six lithium atoms and six electrons were extracted and acquired by the TPQG-Cu-MOF (Fig. 9d).
Correlation studies employing redox-active ligands to construct 2D c-MOFs as cathode materials have also been reported. The highly conductive Cu-BHT revealed a capacity of 175 mA h−1 g−1via a reversible four-electron reaction.84 Cu-HHTQ is linked to nitrogen-rich tricycloquinazoline (TQ) and CuO4 units, for which the synergistic effect results in a high capacity (657.6 mA h−1 g−1 at 600 mA g−1). For the first time, TQ has been demonstrated to be a redox-active component of Li+ ions.43 Binary organic ligands with redox activity were used to construct a 2D c-MOF, Cu3(HHTP)(HTQ), as reported by Bu and coworkers.85 Aromatic rings and CuO4 units were used for Li+ ion storage, achieving a high specific capacity of 725.1 mA h−1 g−1 at 50 mA g−1. Using CuO4 units and CN sites of the TAC (triazacoronene) ligand for Li+ ion storage, Bu and coworkers reported a 2D c-MOF of Cu-TAC as an anode material, achieving a remarkable reversible capacity of 772.4 mA h g−1 at 300 mA g−1, along with outstanding rate performance and long-term cyclability.86 Fe-TABQ with two redox active centers from Fe ions and ligands delivers a high capacity of 251.1 mA h g−1 at 50 mA g−1, as reported by the Li group.87 Furthermore, to construct dense redox-active sites, the Li group also reported a one-dimensional Cu-MOF (DDA-Cu, where DDA = 1,5-diamino-4,8-dihydroxy-9,10-anthracene-dione) as a cathode material for LIBs. The Cu–O3 N units, through π-d conjugation, ensure structural robustness and high conductivity. The CN bonds and Cu–O3 N units serve as redox-active sites for Li+ storage, delivering a high-rate capability of 192 mA h g−1 at 1 A g−1. Furthermore, DDA-Cu demonstrates excellent performance when tested under lean electrolyte conditions with high mass loading.88
Hexaazatriphenylene (HATN), with the redox activity of a nitrogen-rich heteroaromatic molecule, was utilized to synthesize a Cu-based 2D c-MOF of Cu-HATN.89 The CO bonds in CuO4 units and CN bonds in HATN establish the maximum number of active sites, together with regular channels for fast mass transfer and favorable electrical conductivity. Cu-HATN demonstrates enhanced specific capacity of 763 mA h−1 g−1 at 300 mA g−1. Similarly, the 2D c-MOF of Cu-TAC with a trazacoronene core displays an excellent reversible capacity (772.4 mA h−1 g−1 at 300 mA g−1).90 The CN and CO bonds from the TAC and CuO4 units synergistically served as active sites for Li+ ion storage.
Recently, Dong et al. designed a 2D c-MOF of Cu2(Nx-OHPTP) (x = 0, 1, 2) for dual-ion storage based on a nitrogen-variable π-conjugated octahydroxyphenanthrotriphenylene (OHPTP).92 Cu2(N2-OHPTP), as a cathode, exhibited a large specific capacity of 53.8 mA h−1 g−1. The nitrogen atoms in the skeleton of Cu2(Nx-OHPTP) can regulate the electrical properties of narrower bandgaps and faster charge mobilities. Nitrogen can also serve as additional Li+ ion adsorption sites and redox-active centers, favoring the CuO4-nodes to adsorb Li+ and Cl−, resulting in a synergistic effect for enhanced dual-ion storage.
2D c-MOFs of M-TH5 (M = Mn, Co) with 3D extended structures featuring flower-like morphology were synthesized, in which TH5 contained a redox-active core of triptycene.91 Mn-TH5 exhibits a high initial capacity of 486.3 mA h−1 g−1 at 100 mA g−1 based on the TH5 and MnO4 as dual redox centers. For future development, new ligands with rich redox-active sites integrated into 2D c-MOFs will induce particularly satisfactory performance, together with improved elucidation of the Li storage mechanism.
600 ppm) with cheap price and low electrochemical potential (−2.71 V vs. SHE). A heavier molar mass and larger ionic radius are critical requirements for the pore structure and mass-transport ability of electrode materials. 2D c-MOFs, possessing adjustable pore and channel structures based on the size and symmetry of organic linkers, together with high electrical conductivity as well as highly dense redox-active sites from metal units and organic linkers, can effectively address the above problems.
The first attempt of using 2D c-MOFs was Co-HAB, in which HAB featured the highest density of redox centers for a six-electron reaction.93 Co-HAB can store three electrons and three sodium ions per HAB, achieving full availability of the redox centers in the HAB. The Co-HAB electrode exhibited a rate capability of 214 mA h−1 g−1. The Ni-TABQ reported by Wang et al. showed a 2D layer structure, providing two pathways along the polymer chain through Ni–N bonds and hydrogen bond direction for Na+ ion diffusion and electron conduction.94 The imines and carbonyls in the conjugated benzene rings act as dual redox-active sites to insert and extract Na+ ions. Ni-TABQ show excellent capacities of 469.5 and 345.4 mA h−1 g−1 at 100 mA g−1 and 8 A g−1, respectively.
Using inactive Zn2+ ions and the redox-active ligand HHTP, Wang et al. synthesized a 2D c-MOF of Zn-HHTP.40 Through successive redox reactions to store cations and anions, Zn-HHTP illustrated a highly reversible capacity (ca. 150 mA h−1 g−1 at 100 mA g−1). After 1000 cycles of manipulation, the capacity retention was maintained at 90%, which was superior to that of Cu-HHTP. For multi-redox sites, the Li group synthesized 2D c-MOFs of HATN–XCu (X = O/S) based on hexaazatrinaphthylene as an anode material for SIBs.95 The dual redox sites of the repeating [CuX4] units and CN groups can lead to continuous multi-electron reactions, in which HATN-OCu has a reversible capacity of 500 mA h−1 g−1 at 100 mA g−1 and a rate performance of 151 mA h−1 g−1 at 5 A g−1.
Our group introduced a large conjugated ligand of octahydroxyteterabenzoanthracene (8OH-TBA) with a narrow energy gap to construct a 2D c-MOF of Cu-TBA with rhombic topology (Fig. 10a and b).96 As displayed in Fig. 10c, Cu-TBA exhibits a metallic band structure with a high specific surface area of 613 m2 g−1. In Na+ ion storage, Cu-TBA demonstrates a decent reversible capacity of 153.6 mA h−1 g−1 at 50 mA g−1 and superior cyclability (Fig. 10d–f), outperforming the hexagonal counterpart of Cu-HHTP. The mechanism of Na+ ion storage involves a three-step reaction, including a single electron reduction of Cu2+/Cu+ and a successive two electron process of CO/C–O reduction, shown in Fig. 10g–i. Subsequently, anion and cation co-storage in SIBs was revealed for the first time using Ni3(HATQ)2 as a bipolar active material, based on a p-type ligand rich in nitrogen and deficient in electrons.97 Ni3(HATQ)2 showed a Brunauer–Emmett–Teller (BET) surface area of 513 m2 g−1 with a mesopore size of 2.24 nm and a conductivity of 2.12 mS m−1. The reversible capacity of Ni3(HATQ)2 is 115.1 mA h−1 g−1 with a fine rate performance of 77.1 mA h−1 g−1 at 2 A g−1 and long-time cycling durability. Comprehensive characterization techniques revealed that the CN bonds of the coordinated center tended to store Na+ ions, and PF6− ions were more easily bonded to the nitrogen atoms of the TQ ligand. Ultimately, the release of synergistic effects on the maximum degree of different redox-active components is pivotal, for which the rational design of 2D c-MOFs is essential.
 | ||
| Fig. 10 (a) Structural model of Cu-TBA. (b) Experimental patterns, Pawley refined patterns, the difference between experimental and refined patterns, calculated pattern, and positions of reflections. (c) N2 sorption isotherms. (d) CV curves at 0.1 mV s−1. (e) GCD profiles at 50 mA g−1. (f) Cycling stability at 0.2 A g−1. (g–i) FT-IR spectra and high-resolution XPS spectra of O 1s and Cu 2p at different states of the initial cycle. Reproduced with permission from ref. 96. Copyright 2024 Wiley. | ||
The first use of 2D c-MOFs in ZIBs was for Cu3(HTTP)2, as reported by the Stoddart group.99 Cu3(HTTP)2 delivers a capacity of 124.4 mA h−1 g−1 at 4000 mA g−1, originating from a main pseudocapacitive charge-storage mechanism from two redox centers of Cu2+ and quinoidal units. Liang et al. reported a 2D c-MOF of Cu-BTA-H as the cathode material for ZIBs.100 The dual redox mechanisms of Cu2+/Cu+via a one-electron redox reaction and CN/C–N via a two-electron redox reaction resulted in an enhanced reversible capacity of 330 mA h−1 g−1 at 2000 mA g−1.
To integrate multiple redox-active components into 2D c-MOFs, we tailored a multitopic catechol ligand based on an anthraquinone core to synthesize a novel Cu-TBPQ MOF.101 Cu-TBPQ was synthesized using a flexible nonplanar ligand of 8OH-TBPQ and Cu2+ under solvothermal conditions through a one-pot cyclodehydrogenation and coordination reaction of 8OH-TBPQ and Cu2+ simultaneously. The Cu-TBPQ MOF exhibited good stability in water, common organic solvents such as DMF and acetone, and 3 M NaOH aqueous solution. As a ZIB cathode, the Cu-TBPQ MOF achieved a high specific capacity of 371.2 mA h−1 g−1 at 50 mA g−1 with superior rate performance and stability, with nearly 88% retention of the initial capacity after 500 cycles at 2 A g−1. The quinone moieties largely induced dense redox active sites, including [CuO4] units and quinone moieties, enabling the storage of Zn2+ ions in the Cu-TBPQ MOF.
A large surface area and appropriate pore size are essential for electrode materials in aqueous dual-ion batteries. To address this need, we developed two 2D c-MOFs: PA-TAPA-Cu-MOF and PA-PyTTA-Cu-MOF.102 PA-TAPA-Cu-MOF exhibits the largest pore size of 3.9 nm among the reported 2D c-MOFs. These two MOFs acted as cathode materials for polyiodide storage for the first time. The PA-TAPA-Cu-MOF displays higher specific capacity (423.4 mA h g−1 after 100 cycles at 1.0 A g−1) than that of the PA-PyTTA-Cu-MOF. In addition to the large pores that facilitate the storage and transport of I−/I3−, the nitrogen atoms and CuO4 centers also play a crucial role in anchoring I3−.
Conjugation and topology engineering are essential for adjusting the electrochemical properties of 2D c-MOFs. Wang et al. utilized the 2D c-MOF of Cu-TBA-MOF with different topologies but the same conjugated tetrabenzoanthracene (TBA) aromatic core for PIBs using a topology engineering strategy.103 The rhombus topological sql-Cu-TBA-MOF shows extended π-conjugation, larger specific surface area and better structural stability than the kagome analogue (kgm-Cu-TBA-MOF), which can expose more active centers and faster diffusion of K+ ions, resulting in better electrochemical performance. sql-Cu-TBA-MOF reveals a high specific discharge capacity of 178.4 mA h g−1 at 0.2 A g−1 and an excellent long-term cycle stability (more than 9000 cycles at 10.0 A g−1). Full PIBs constructed using dipotassium terephthalate as the anode and sql-Cu-TBA-MOF as the cathode, deliver a reversible discharge specific capacity of 146.6 mA h g−1 at 0.1 A g−1, which in series can charge the smart phone. Moreover, sql-Cu-TBA-MOF exhibited good performance at a low temperature of −20 °C. The reversible redox reactions of CO/C–O–K and Cu2+/Cu+ were verified as K+ storage mechanisms. This work sticks out the importance of topological structures and conjugation for 2D c-MOFs in energy storage devices.
Multiple active sites from the coordination units and unsaturated bonds of ligands can be integrated into 2D c-MOFs with promising electrochemical performance for PIBs. The Zhu group reported the first application of a 2D c-MOF (Cu-HATNH) with dual active centers combined with carbon nanotubes (Cu-HATNH@CNT) as a cathode material for PIBs.104 Cu-HATNH@CNT shows a core–shell structure with Cu-HATNH grown in situ on CNTs, as shown in Fig. 11a and b. Carbon nanotubes can not only improve the conductivity but also enlarge the layer space and pore size, accelerating the electron transfer and K+ migration and diffusion. Cu-HATNH@CNT displays a high initial capacity of 317.5 mA h g−1 at 0.1 A g−1, remarkable rate capacity of 147.1 mA h g−1 at 10 A g−1, and outstanding long-term cycling durability. Moreover, Cu-HATNH@CNT displayed a high invertible capacity of 290.4 mA h g−1 and capacity retention of 91.6% after 180 cycles (Fig. 11c). The unsaturated bonds of CN were verified as the active sites that bind K+. The [CuO4] units can also store K+ through redox reactions (Fig. 11d and e). The abundant CN sites from HATNH and unsaturated [CuO4] units act synergistically to store K+ ions at a high utilization rate. This study proposes a novel strategy to design 2D c-MOFs with high capacity as organic cathode materials in secondary batteries.
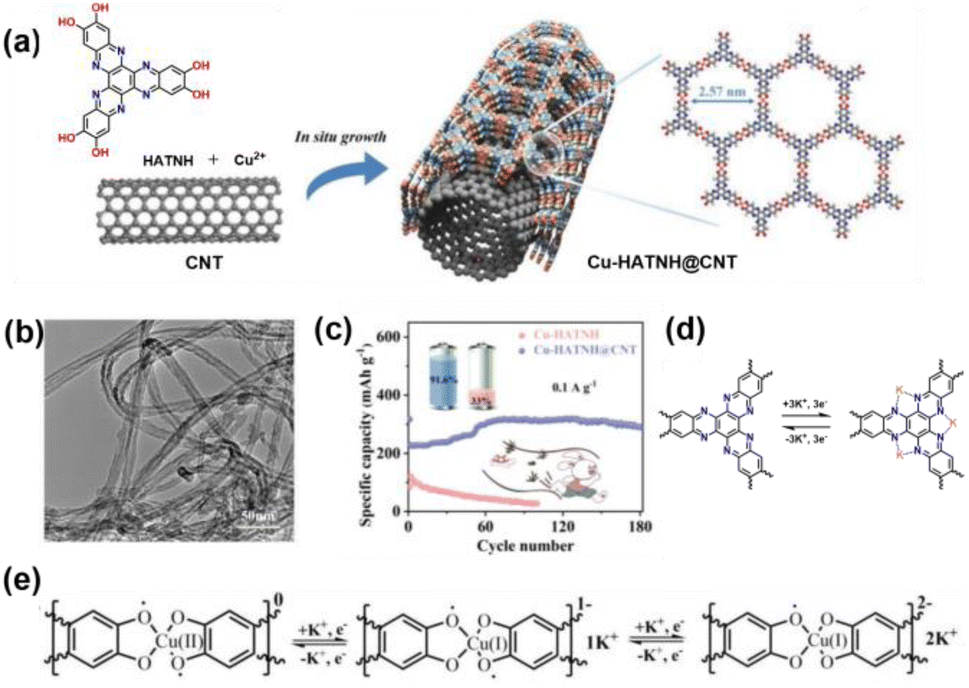 | ||
| Fig. 11 (a) Schematic of the synthesis of Cu-HATNH@CNT. (b) TEM images of Cu-HATNH@CNT. (c) Cycling performance of Cu-HATNH@CNT at 0.1 A g−1. (d) Schematic of the K+ storage process in the HATNH units. (e) Schematic of the K+ storage process in the [CuO4] units. Adapted with permission from ref. 100. Copyright 2024 Wiley. | ||
2D c-MOFs can also be used as anode electrode materials in PIBs, such as the HAN-Cu-MOF reported by the Li group.105 The nitrogen-rich ligands and CuO4 nodes with π-d conjugation endow the HAN-Cu-MOF with multiple redox-active sites and robust stability to efficiently store potassium ions at high temperatures. At 60 °C, the HAN-Cu-MOF delivered a high initial capacity of 455 mA h g−1 and extraordinary cyclability with 96.7% capacity retention after 1600 cycles. The CuO4 units and CN functional groups contribute the majority of redox-active sites, which are responsible for the high reversible capacity. The characteristics of periodic porous conjugated frameworks guarantee fast charge transport and excellent cycle durability. This study provides a new perspective for designing highly efficient battery electrode materials under high-temperature conditions based on coordination chemistry strategies.
Ammonium ion batteries (AIBs) represent a promising segment of low-cost energy storage systems, offering advantages such as environmental sustainability and extended lifespan. However, they face the challenge of slow diffusion kinetics. 2D c-MOFs can facilitate rapid reaction kinetics, alongside high electrical conductivity and abundant redox-active components, making them strong candidates for cathode materials in AIBs. Utilizing a heterogeneous ligand approach, Xu and colleagues synthesized the 2D Cu-HHTP-THBQ.107 Through electron delocalization regulated by the heteroligand and a hydrogen bond cage mechanism, the full battery demonstrates a high specific energy density of 211.84 W h kg−1. The presence of double redox-active sites from Cu cations and heteroligand anions contributes to a significant specific capacity.
Lithium–sulfur batteries (LSBs) are regarded as one of the most promising next-generation battery technologies due to their high theoretical energy density and the abundance of sulfur. However, the insulating nature of sulfur, coupled with the notorious shuttle effect and volume strain of the electrode structure, significantly limits the practical applications of LSBs. 2D c-MOFs, with their excellent intrinsic electrical conductivity, can serve as ideal modified separator layers in LSBs. The Zhu group employed the smallest π-conjugated hexaaminobenzene (HAB) ligand to construct the 2D c-MOF Ni-HAB, which was further hybridized with carbon nanotubes (CNT) to create Ni-HAB@CNT.108 As a modified separator, Ni-HAB@CNT achieves an areal capacity of 6.29 mA h cm−2 at a high sulfur loading of 6.5 mg cm−2, at an electrolyte-to-sulfur ratio of 5 μL mg−1. The Ni–N4 chemisorption sites, combined with micropores, effectively enhance sulfur affinity and mitigate the shuttle effect.
5. Summary and outlook
In this review, we summarize the recent key progress of 2D c-MOFs in electrochemical energy conversion and storage systems, including electrocatalysis (ORR, OER, HER, CO2RR, and NRR), supercapacitors, and metal-ion batteries (Li, Na, Zn and K). The synergistic effect of the metal nodes and ligands with redox-active sites was the focus of this article. Incorporated with redox ligands, 2D c-MOFs can achieve the maximum number of active sites, mainly involving unsaturated covalent bonds, such as CO and CN, etc. The intrinsic electrical conductivity, high porosity with regular channels, and rich active components make 2D c-MOF multifunctional materials suitable for gas sensing, electronics, electrochemical energy conversion, and storage applications. Considering the booming background of 2D c-MOFs, the remaining challenges must be addressed for their practical application.
First, more diverse novel redox ligands are expected to be introduced into 2D c-MOFs. Organic ligands can usually determine the topology, pore structures, and redox activity of 2D c-MOFs, which influence the functions of specific structures based on structure–function relationships. The rational design of innovative organic ligands can establish perfect synergy between various components, thereby significantly improving the performance of electrochemical devices. Meanwhile, more metal ions, such as Ti, V, Cr and Mo, need to be introduced in 2D c-MOFs to explore more intriguing applications. Furthermore, machine learning and DFT calculations can be used to screen and evaluate the properties of 2D c-MOFs using systematic models, which can aid the design and synthesis of tailor-made 2D c-MOFs.
Second, the crystallinity of 2D c-MOFs should be improved for large single crystals or single-crystalline thin films. Polycrystalline powders are commonly used and show inferior activity compared to theoretical expectations, in which the disordered structures largely influence the long-range pathways of charge and mass transport. However, structure–property relationships cannot be elucidated at the molecular level. The use of nonplanar ligands to construct 2D c-MOFs is a straightforward and accessible strategy for obtaining high-quality crystals with multiple functions.
Third, the applications of 2D c-MOFs in new fields should be developed. For example, there have been few reports of biomass catalysis and sensing.109–111 The application of 2D c-MOFs in interdisciplinary fields is promising but remains less exploited. Incorporating guest molecules such as carbon nanotubes, cationic clusters, and polyoxometalates to construct 2D c-MOF-based hybrids can offer more intriguing functions for various applications. For example, a 2D c-MOF (Cu-HATNH) with dual-active centers was anchored on carbon nanotubes, which was used as a cathode material for PIBs.104
Fourth, the real active sites of 2D c-MOFs in electrocatalytic reactions should be investigated in detail. In particular, high-valence metal oxides or hydroxides, such as MOOH derived from metal nodes, are considered as the real active sites for oxidation reactions. Defects and grain boundaries, which can be introduced through chemical modification, are confirmed to be highly active sites. For example, peripheral defects in Cu-HHTP are related to the enhanced catalytic performance of the CO2RR.104 Advanced in situ characteristic techniques should be developed to monitor the transformation of active components during the dynamic catalytic process and understand the mechanism of structural reconstruction.
Last but not least, for practical applications, economic feasibility must be considered and assessed to achieve mass production. Low-cost raw materials and advanced synthetic techniques for industrial scalability are prerequisites for practical applications that should be widely studied in the future. In general, 2D c-MOFs with multiple functions are still in their infancy with various emerging applications. This review is expected to arouse more research interest regarding the maximum utilization of the active components in 2D c-MOFs.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Z. M. Su and L. Chen conceptualized the review. X. Li, X. Su and T. Su wrote and edited the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22271023 and 52473211).References
- A. Schoedel, Z. Ji and O. M. Yaghi, Nat. Energy, 2016, 1, 16034 CrossRef.
- L. Chen, X. Xu, W. Yang and J. Jia, Chin. Chem. Lett., 2020, 31, 626–634 CrossRef.
- P. Zhang, F. Wang, M. Yu, X. Zhuang and X. Feng, Chem. Soc. Rev., 2018, 47, 7426–7451 RSC.
- J. Li, X. Gao, L. Zhu, M. N. Ghazzal, J. Zhang, C. H. Tung and L. Z. Wu, Energy Environ. Sci., 2020, 13, 1326–1346 RSC.
- Q. Fu, J. Han, X. Wang, P. Xu, T. Yao, J. Zhong, W. Zhong, S. Liu, T. Gao, Z. Zhang, L. Xu and B. Song, Adv. Mater., 2021, 33, 1907818 CrossRef CAS PubMed.
- I. Khan, N. Baig, S. Ali, M. Usman, S. A. Khan and K. Saeed, Energy Storage Mater., 2021, 35, 443–469 CrossRef.
- G. Wu and P. Zelenay, Nat. Rev. Mater., 2024, 9, 643–656 CrossRef CAS.
- H. Su, M. A. Soldatov, V. Roldugin and Q. Liu, eScience, 2022, 2, 102–109 CrossRef.
- T. Wang, X. Cao and L. Jiao, eScience, 2021, 1, 69–74 CrossRef.
- J. Li, H. M. Yin, X. B. Li, E. Okunishi, Y. L. Shen, J. He, Z.-K. Tang, W. X. Wang, E. Yucelen, C. Li, Y. Gong, L. Gu, S. Miao, L. M. Liu, J. Luo and Y. Ding, Nat. Energy, 2017, 2, 17111 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef PubMed.
- Z. Li, Y. J. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C. He, C. Peng, W. Cheong, Q. Li, M. Zhang, Z. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef PubMed.
- J. Zhang, G. Chen, K. Mullen and X. L. Feng, Adv. Mater., 2018, 30, 1800528 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. Zhang, J. Nai, L. Yu and X. W. Lou, Joule, 2017, 1, 77–107 CrossRef.
- D. Xiang, X. Bo, X. Gao, C. Du, P. Li, L. Zhu and W. J. Chen, J. Colloid Interface Sci., 2019, 534, 655–664 CrossRef PubMed.
- C. Du, Y. Gao, J. Wang and W. Chen, J. Mater. Chem. A, 2020, 8, 9981–9990 RSC.
- M. Wang, R. H. Dong and X. L. Feng, Chem. Soc. Rev., 2021, 50, 2764–2793 RSC.
- S. Wu, Y. Lin, J. Liu, W. Shi, G. Yang and P. Cheng, Adv. Funct. Mater., 2018, 28, 1707169 CrossRef.
- W. Hao, D. Chen, Y. Li, Z. Yang, G. Xing, J. Li and L. Chen, Chem. Mater., 2019, 31, 8100–8105 CrossRef.
- X. Yan, H. Liu, Y. Li, W. Chen, T. Zhang, Z. Zhao, G. Xing and L. Chen, Macromolecules, 2019, 52, 7977–7983 Search PubMed.
- J. Hou, M. Gómez, A. Krajnc, A. McCaul, S. Li, A. M. Bumstead, A. F. Sapnik, Z. Deng, R. Lin, P. A. Chater, D. S. Keeble, D. A. Keen, D. Appadoo, B. Chan, V. Chen, G. Mali and T. D. Bennett, J. Am. Chem. Soc., 2020, 142, 3880–3890 CrossRef PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef.
- M. Savage, Y. Cheng, T. L. Easun, J. E. Eyley, S. P. Argent, M. R. Warren, W. Lewis, C. Murray, C. C. Tang, M. D. Frogley, G. Cinque, J. Sun, S. Rudić, R. T. Murden, M. J. Benham, A. N. Fitch, A. J. Blake, A. J. Ramirez-Cuesta, S. Yang and M. Schrçder, Adv. Mater., 2016, 28, 8705–8711 CrossRef PubMed.
- M. Yu, R. H. Dong and X. L. Feng, J. Am. Chem. Soc., 2020, 142, 12903–12915 CrossRef PubMed.
- H. Zhong, M. Wang, G. Chen, R. H. Dong and X. L. Feng, ACS Nano, 2022, 16, 1759–1780 CrossRef.
- L. Lin, Q. Zhang, Y. Ni, L. Shang, X. Zhang, Z. Yan, Q. Zhao and J. Chen, Chem, 2022, 8, 1822–1854 Search PubMed.
- J. Liu, X. Song, T. Zhang, S. Liu, H. Wen and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 5612–5624 CrossRef PubMed.
- M. Zhao and S. Tong, Energy Fuels, 2024, 38, 13796–13818 CrossRef.
- L. Sun, M. G. Campbell and M. Dincǎ, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef PubMed.
- Q. Jiang, P. Liu, J. Xiong, Z. Xie, Q. Wang, X. Q. Yang, E. Hu, Y. Cao, J. Sun, Y. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 5273–5277 CrossRef.
- Z. Meng, A. Aykanat and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 2046–2053 CrossRef PubMed.
- J. Park, A. C. Hinckley, Z. Huang, D. Feng, A. A. Yakovenko, M. Lee, S. Chen, X. Zou and Z. Bao, J. Am. Chem. Soc., 2018, 140, 14533–14537 CrossRef PubMed.
- J. Liu, Y. Zhou, Z. Xie, Y. Li, Y. Liu, J. Sun, Y. Ma, O. Terasaki and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 1081–1086 CrossRef PubMed.
- R. Dong, Z. Zhang, D. C. Tranca, S. Zhou, M. Wang, P. Adler, Z. Liao, F. Liu, Y. Sun, W. Shi, Z. Zhang, E. Zschech, S. C. B. Mannsfeld, C. Felser and X. Feng, Nat. Commun., 2018, 9, 2637 CrossRef PubMed.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef.
- H. T. B. Pham, J. Y. Choi, M. Stodolka and J. Park, Acc. Chem. Res., 2024, 57, 580–589 Search PubMed.
- J. Liu, G. Xing and L. Chen, Acc. Chem. Res., 2024, 57, 1032–1045 CrossRef.
- X. Su, Z. Zhong, X. Yan, Y. Xu, T. Zhang, Y. Ma and L. Chen, J. Am. Chem. Soc., 2024, 146, 9036–9044 CrossRef PubMed.
- Y. Chen, Q. Zhu, K. Fan, Y. Gu, M. Sun, Z. Li, C. Zhang, Y. Wu, Q. Wang, S. Xu, J. Ma, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 18769–18776 CrossRef CAS.
- X. Yan, J. Chen, X. Su, J. Zhang, C. Wang, H. Zhang, Y. Liu, L. Wang, G. Xu and L. Chen, Angew. Chem., Int. Ed., 2024, 63, e202408189 CrossRef CAS.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS.
- J. Yan, Y. Cui, M. Xie, G. Yang, D. Bin and D. Li, Angew. Chem., Int. Ed., 2021, 60, 24467–24472 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- A. Schneemann, R. Dong, F. Schwotzer, H. Zhong, I. Senkovska, X. Feng and S. Kaskel, Chem. Sci., 2021, 12, 1600–1619 RSC.
- L. Guo, J. Sun, J. Wei, Y. Liu, L. Hou and C. Yuan, Carbon Energy, 2020, 2, 203–222 CrossRef CAS.
- E. M. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath and M. Dincă, Nat. Commun., 2016, 7, 10942 CrossRef CAS.
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS.
- E. M. Miner, L. Wang and M. Dincă, Chem. Sci., 2018, 9, 6286–6291 RSC.
- E. M. Miner, S. Gul, N. D. Ricke, E. Pastor, J. Yano, V. K. Yachra, T. V. Voorhis and M. Dincă, ACS Catal., 2017, 7, 7726–7731 CrossRef CAS.
- J. Park, Z. Chen, R. A. Flores, G. Wallnerström, A. Kulkarni, J. K. Nørskov, T. F. Jaramillo and Z. Bao, ACS Appl. Mater. Interfaces, 2020, 12, 39074–39081 CrossRef CAS PubMed.
- A. M. Dominic, Z. Wang, A. Kuc, P. Petkov, K. H. Ly, T. L. H. Pham, M. Kutzschbach, Y. Cao, J. Bachmann, X. Feng, R. Dong and I. M. Weidinger, J. Phys. Chem. C, 2023, 127, 7299–7307 CrossRef CAS.
- X. Wei, C. Jiang, H. Xu, Y. Ouyang, Z. Wang, C. Lu, X. Lu, J. Pang, F. Dai and X. H. Bu, ACS Catal., 2023, 13, 15663–15672 CrossRef CAS.
- L. Wei, M. D. Hossain, G. Chen, G. A. Kamat, M. E. Kreider, J. Chen, K. Yan, Z. Bao, M. Bajdich, M. Burke Stevens and T. F. Jaramillo, J. Am. Chem. Soc., 2024, 146, 13377–13390 CrossRef CAS PubMed.
- C. Li, L. Shi, L. Zhang, P. Chen, J. Zhu, X. Wang and Y. Fu, J. Mater. Chem. A, 2020, 8, 369–379 RSC.
- D. Xing, Y. Wang, P. Zhou, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Appl. Catal., B, 2020, 278, 119295 CrossRef CAS.
- J. Yang, Z. Feng, Y. Wang, J. Huang, C. Xu and H. Li, ACS Appl. Nano Mater., 2024, 7, 18881–18888 CrossRef CAS.
- Y. Lian, W. Yang, C. Zhang, H. Sun, Z. Deng, W. Xu, L. Song, Z. Ouyang, Z. Wang, J. Guo and Y. Peng, Angew. Chem., Int. Ed., 2020, 59, 286–294 CrossRef CAS PubMed.
- S. S. Shinde, C. H. Lee, J. Jung, N. K. Wagh, S. Kim, D. Kim, C. Lin, S. U. Lee and J. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC.
- L. Hu, F. F. Wang and Y. Jing, J. Phys. Chem. Lett., 2023, 14, 11429–11437 CrossRef CAS.
- X. Wei, S. Cao, S. Cheng, C. Lu, X. Chen, X. Lu, X. Chen and F. Dai, ACS Mater. Lett., 2024, 6, 3496–3504 CrossRef CAS.
- A. J. Clough, J. W. Yoo, M. H. Mecklenburg and S. C. Marinescu, J. Am. Chem. Soc., 2015, 137, 118–121 CrossRef CAS PubMed.
- P. Das, B. Ball and P. Sarkar, ACS Catal., 2023, 13, 16307–16317 CrossRef CAS.
- Y. Lu, H. Zhong, J. Li, A. M. Dominic, Y. Hu, Z. Gao, Y. Jiao, M. Wu, H. Qi, C. Huang, L. J. Wayment, U. Kaiser, E. Spiecker, I. M. Weidinger, W. Zhang, X. L. Feng and R. H. Dong, Angew. Chem., Int. Ed., 2022, 61, e202208163 CrossRef CAS.
- D. Yang and X. Wang, SmartMat, 2022, 3, 54–67 CrossRef CAS.
- H. X. Zhong, M. Ghorbani-Asl, K. H. Ly, J. C. Zhang, J. Ge, M. C. Wang, Z. Q. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. H. Dong and X. L. Feng, Nat. Commun., 2020, 11, 1409 CrossRef CAS.
- Y. Liu, S. Li, L. Dai, J. Li, J. Lv, Z. Zhu, A. Yin, P. Li and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 16409–16415 CrossRef CAS PubMed.
- Z. Zhao, J. Huang, P. Liao and X. Chen, J. Am. Chem. Soc., 2023, 145, 26783–26790 CrossRef CAS.
- W. Xiong, X. Cheng, T. Wang, Y. S. Luo, J. Feng, S. Y. Lu, A. M. Asiri, W. Li, Z. J. Jiang and X. P. Sun, Nano Res., 2020, 13, 1008–1012 CrossRef CAS.
- J. Zhang, X. Zhu, W. Geng, T. Li, M. Li, C. Fang, X. Shan, Y. Li and Y. Jing, J. Energy Chem., 2021, 61, 71–76 CrossRef CAS.
- A. Maibam, I. B. Orhan, S. Krishnamurty, S. P. Russo and R. Babarao, ACS Appl. Mater. Interfaces, 2024, 16, 8707–8716 Search PubMed.
- Z. Xiong, Y. Xu, A. Y. Bao, W. Sheng and J. Zhan, ACS Appl. Nano Mater., 2024, 7, 4220–4227 CrossRef CAS.
- X. Huang, S. Zhang, L. Liu, L. Yu, G. Chen, W. Xu and D. Zhu, Angew. Chem., Int. Ed., 2018, 57, 146–150 CrossRef CAS.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. Sun, Y. S. Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS.
- J. Zhao, T. Zhang, J. Ren, Z. Zhao, X. Su, W. Chen and L. Chen, Chem. Commun., 2023, 59, 2978–2981 RSC.
- M. Gao, Z. Wang, Z. Liu, Y. Huang, F. Wang, M. Wang, S. Yang, J. Li, J. Liu, H. Qi, P. Zhang, X. Lu and X. L. Feng, Adv. Mater., 2023, 35, 2305575 CrossRef CAS.
- A. Bagheri, S. Bellani, H. Beydaghi, Z. Wang, A. Morag, M. I. Zappia, J. K. P, S. Vaez, V. Mastronardi, A. Gamberini, S. B. Thorat, M. Abruzzese, R. H. Dong, L. Pasquale, M. Yu, X. L. Feng and F. Bonaccorso, ChemSusChem, 2024, e202401454 Search PubMed.
- P. Zhang, M. Wang, Y. Liu, Y. Fu, M. Gao, G. Wang, F. Wang, Z. Wang, G. Chen, S. Yang, Y. Liu, R. H. Dong, M. Yu, X. Lu and X. L. Feng, J. Am. Chem. Soc., 2023, 145, 6247–6256 Search PubMed.
- Z. Chang, M. Zhu, Y. Sun, F. He, Y. Li, C. Ye, Y. Jin, Z. Li and W. Xu, Adv. Funct. Mater., 2023, 33, 2301513 CrossRef CAS.
- K. Wada, K. Sakaushi, S. Sasaki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 8886–8890 CrossRef CAS.
- X. Sun, X. Yan, K. Song, T. Zhang, Z. Yang, X. Su, W. Chen and L. Chen, Chin. J. Chem., 2023, 41, 1691–1696 CrossRef CAS.
- Z. Wu, D. Adekoya, X. Huang, M. J. Kiefel, J. Xie, W. Xu, Q. Zhang, D. Zhu and S. Zhang, ACS Nano, 2020, 14, 12016–12026 CrossRef CAS PubMed.
- J. C. Yin, Y. Q. Zhang, Z. G. Li, M. R. Cheng, M. Liu, W. Li, N. Li and X. H. Bu, Sci. China Mater., 2023, 66, 4566–4574 CrossRef CAS.
- J. C. Yin, X. Lian, Z. G. Li, M. R. Cheng, M. Liu, J. Xu, W. Li, Y. H. Xu, N. Li and X. H. Bu, Adv. Funct. Mater., 2024, 34, 2403656 CrossRef CAS.
- J. R. Geng, Y. X. Ni, Z. Zhu, Q. Wu, S. N. Gao, W. B. Hua, S. Indris, J. Chen and F. J. Li, J. Am. Chem. Soc., 2023, 145, 1564–1571 CrossRef CAS.
- M. H. Yang, Y. Wang, Y. F. Huang, J. M. Xiao, G. Y. Zhu, Y. Fang, X. C. Zhou, J. H. Long, M. Xie, D. S. Bin and D. Li, Angew. Chem., Int. Ed., 2024, 64, e202421008 Search PubMed.
- J. Yin, N. Li, M. Liu, Z. Li, X. Wang, M. Cheng, M. Zhong, W. Li, Y. Xu and X. H. Bu, Adv. Funct. Mater., 2023, 33, 2211950 CrossRef CAS.
- J. Yin, X. Lian, Z. Li, M. Cheng, M. Liu, J. Xu, W. Li, Y. Xu, N. Li and X. H. Bu, Adv. Funct. Mater., 2024, 34, 2403656 CrossRef CAS.
- K. Shi, X. Liu, J. Sang, M. Zhang, D. Han and J. Gong, Chem. Eng. J., 2024, 499, 156021 CrossRef CAS.
- L. Sporrer, Q. Guo, X. Li, A. W. Lashkova, F. Reichmayr, S. Fu, H. I. Wang, M. Bonn, X. Li, P. A. L. Schmidt, M. Wang, Y. Lu, Y. Vaynzof, M. Yu, X. Feng and R. Dong, Angew. Chem., Int. Ed., 2024, 64, e202418390 CrossRef PubMed.
- J. Park, M. Lee, D. Feng, Z. Huang, A. C. Hinckley, A. Yakovenko, X. Zou, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10315–10323 CrossRef CAS.
- L. Wang, Y. Ni, X. Hou, L. Chen, F. Li and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 22126–22131 CrossRef CAS.
- B. Wang, J. Li, M. Ye, Y. Zhang, Y. Tang, X. Hu, J. He and C. C. Li, Adv. Funct. Mater., 2022, 32, 2112072 CrossRef CAS.
- M. Qi, L. Cheng, H. Wang, F. Cui, Q. Yang and L. Chen, Adv. Mater., 2024, 36, 2401878 CrossRef CAS PubMed.
- D. Chen, L. Cheng, W. Chen, H. Wang, F. Cui and L. Chen, Chem. Sci., 2024, 15, 11564–11571 RSC.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- K. W. Nam, S. S. Park, R. d. Reis, V. P. Dravid, H. Kim, C. A. Mirkin and J. F. Stoddart, Nat. Commun., 2019, 10, 4948 CrossRef PubMed.
- Z. Sang, J. Liu, X. Zhang, L. Yin, F. Hou and J. Liang, ACS Nano, 2023, 17, 3077–3087 CrossRef CAS PubMed.
- J. Liu, Y. Zhou, G. Xing, M. Qi, Z. Tang, O. Terasaki and L. Chen, Adv. Funct. Mater., 2024, 34, 2312636 CrossRef CAS.
- P. L. Bao, L. Q. Cheng, X. L. Yan, X. M. Nie, X. Su, H. G. Wang and L. Chen, Angew. Chem., Int. Ed., 2024, 63, e202405168 CrossRef CAS.
- L. Cheng, M. Qi, J. Yu, X. P. Zhang, H. G. Wang, F. Cui and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202405239 CrossRef CAS.
- J. Wang, H. Jia, Z. Liu, J. Yu, L. Cheng, H. Wang, F. Cui and G. Zhu, Adv. Mater., 2024, 36, 2305605 CrossRef CAS PubMed.
- M. Yang, X. Zeng, M. Xie, Y. Wang, J. Xiao, R. Chen, Z. Yi, Y. Huang, D. Bin and D. Li, J. Am. Chem. Soc., 2024, 146, 6753–6762 CrossRef CAS.
- Y. Mu, J. Nyakuchena, Y. Wang, J. R. Wilkes, T. T. Luo, M. Goldstein, B. Eler, U. Mohanty, J. L. Bao, J. Huang and D. W. Wang, Angew. Chem., Int. Ed., 2024, 63, e202409286 Search PubMed.
- H. Y. Lu, J. S. Hu, K. Q. Zhang, Y. Zhang, B. T. Jiang, M. Zhang, S. Z. Deng, J. X. Zhao, H. Pang and B. G. Xu, Adv. Mater., 2024, 36, 2408396 CrossRef CAS.
- T. Guo, Y. C. Ding, C. Xu, W. X. Bai, S. C. Pan, M. L. Liu, M. Bi, J. W. Sun, X. P. Ouyang, X. Wang, Y. S. Fu and J. W. Zhu, Adv. Sci., 2023, 10, 2302518 CrossRef CAS.
- S. Yang, Y. Guo, P. Zhao, H. Jiang, H. Shen, Z. Chen, L. Jiang, X. Xue, Q. Zhang and H. Zhang, ACS Catal., 2024, 14, 449–462 CrossRef CAS.
- Y. Liu, M. Liu, S. Shang, W. Gao, X. Wang, J. Hong, C. Hua, Z. You, Y. Liu and J. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 16991–16998 CrossRef CAS PubMed.
- Y. Li, J. Lv, S. Li, L. Dai, B. Wang and P. Li, J. Mater. Chem. A, 2024, 12, 24549–24555 RSC.
| This journal is © The Royal Society of Chemistry 2025 |