
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectra-descriptor-based machine learning for predicting protein–ligand interactions†
Cheng
Chen‡
a,
Ledu
Wang‡
a,
Yi
Feng‡
a,
Wencheng
Yao
b,
Jiahe
Liu
a,
Zifan
Jiang
a,
Luyuan
Zhao
a,
Letian
Zhang
a,
Jun
Jiang
 a and
Shuo
Feng
*a
a and
Shuo
Feng
*a
aState Key Laboratory of Precision and Intelligent Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: sfeng18@ustc.edu.cn
bMOE Key Laboratory of Resources and Environmental System Optimization, College of Environmental Science and Engineering, North China Electric Power University, Beijing 102206, China
First published on 13th March 2025
Abstract
Machine learning models have emerged as powerful tools for drug discovery of lead compounds. Nevertheless, despite notable advances in model architectures, research on more reliable and physicochemical-based descriptors for molecules and proteins remains limited. To address this gap, we introduce the Fragment Integral Spectrum Descriptor (FISD), aimed at utilizing the spatial configuration and electronic structure information of molecules and proteins, as a novel physicochemical descriptor for virtual screening models. Validation demonstrates that the combination of FISD and a classical neural network model achieves performance comparable to that of complex models paired with conventional structural descriptors. Furthermore, we successfully predict and screen potential binding ligands for two given protein targets, showcasing the broad applicability and practicality of FISD. This research enriches the molecular and protein representation strategies of machine learning and accelerates the process of drug discovery.
Introduction
In the field of drug discovery, particularly during the lead compound identification phase, traditional methods for assessing protein–ligand interactions are often time-consuming and resource-intensive.1,2 As computer science and artificial intelligence (AI) advance, particularly with the increasing application of machine learning in drug design,3–5 researchers are now actively exploring the utilization of AI models for virtual screening (VS), with the goal of further enhancing screening efficiency and precision.6–9 Despite the remarkable performance of current AI-based virtual screening models on multiple benchmark datasets,10–14 the pharmaceutical industry remains cautious; for in real-world drug discovery scenarios, the most frequently encountered situation is that only scarce active ligand molecules can be referred, and it is insufficient for adequately training AI models.15 The limitations in generalization ability of these models have led the industry to keep on favouring physical-principle-based molecular docking approaches when assessing the protein–molecule interaction.16,17The performance of VS models depends not only on the quantity and quality of the training dataset, but also on the model architecture and the molecular representation strategies adopted. Nevertheless, in recent research progress, while significant achievements are primarily attributed to the evolution of machine learning models, these models still rely heavily on traditional structural descriptors to represent molecules and proteins.18,19 To enhance the precision of predictions through the development of more informative descriptors, we endeavour to integrate additional useful information into the descriptors. The spectrum, as a typical carrier of chemical information, offers an indirect glimpse into crucial molecular properties such as spatial configuration and electronic structure information,20,21 potentially bolstering our predictive capabilities.22 Naturally suited for representing molecules, the spectrum is also inherently suitable as an input for machine learning (ML) models due to its controllable and uniform dimensionality. Consequently, the spectrum can be a valuable tool for enhancing the efficiency of molecular and protein representation.
We propose a spectra-descriptor-based approach, which can be used for protein–molecule interaction prediction and ligand screening. To begin with, our study defines one kind of spectra-descriptor, based on infrared (IR) spectra, as the input representation for virtual screening models, with sufficient physicochemical information for both molecules and proteins. We also develop a model named MLMS (ML Molecular Spectra model) capable of rapidly calculating and extracting molecular spectra-descriptors, which are then applied together with MLPS (ML Protein Spectra model),20,21 a model capable of outputting protein spectra-descriptors. MLMS and MLPS can generate input features for the virtual screening model named VS2Net (Vibrational Spectra & Virtual Screening Net). Furthermore, we design case studies to test the ability of our proposed descriptors and models in screening unknown ligands. Through case studies, we successfully showcase the potential of this approach in addressing specific drug design problems, which are identifying potential ligand molecules for new target proteins, thereby providing valuable clues and insights for subsequent lead optimization and synthesis procedure. The detailed illustration of the ligand-finding process can be seen in Fig. 1.
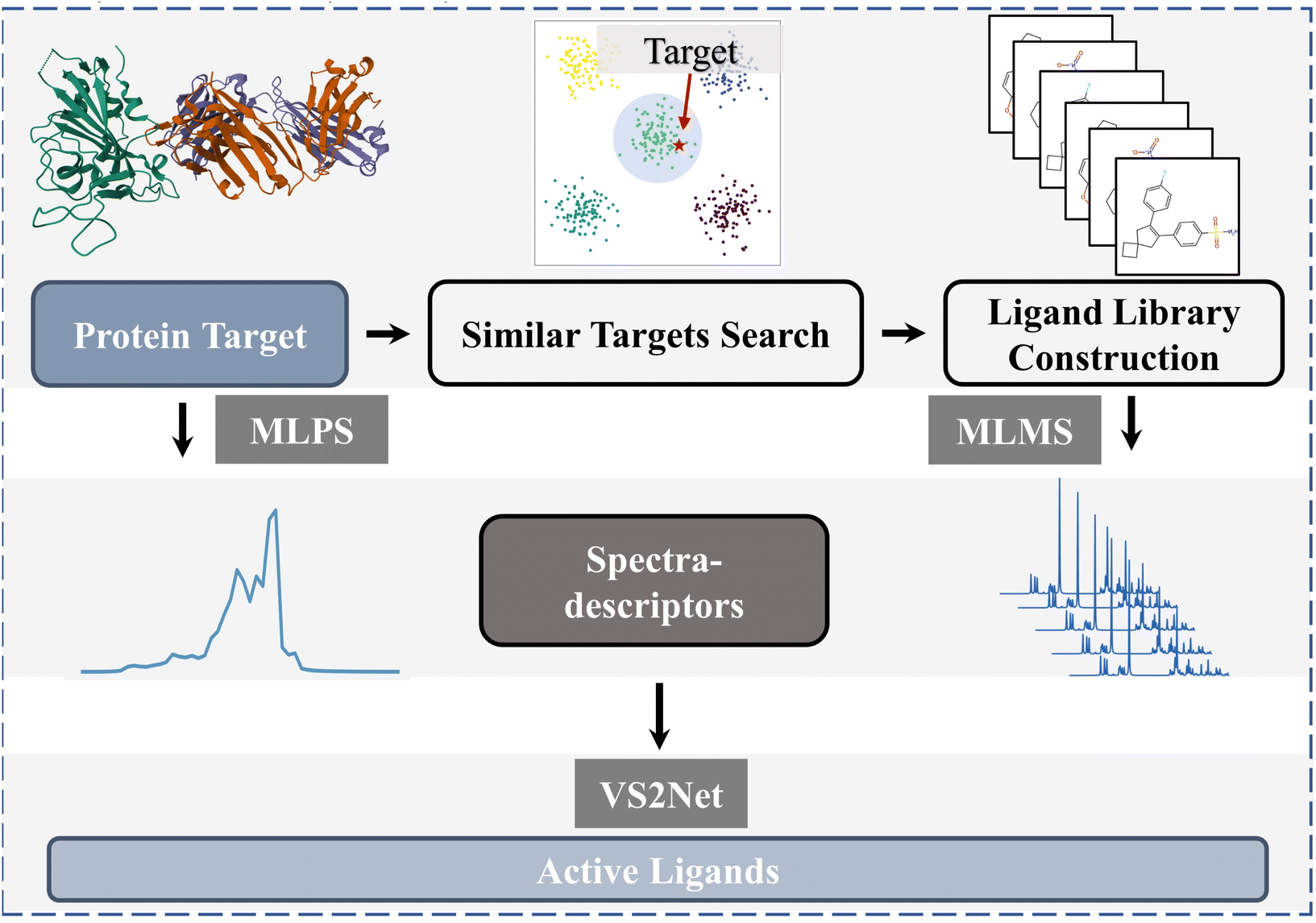 | ||
| Fig. 1 A detailed flowchart illustrating the systematic procedure for the identification of active ligands targeting specific proteins, employing machine learning (ML) models grounded in spectra-descriptor methodologies. | ||
Results and discussion
Predicting fragment integral spectrum descriptor (FISD)
For the purpose of efficiently embedding physicochemical information into the model, we introduced FISD, which is obtained by segmenting the infrared vibrational spectrum of a molecule into multiple fragments at specific intervals and integrating the vibrational intensities within each fragment, and the comprehensive details of which are provided in the Methods. To efficiently generate FISD for small molecules and accelerate the virtual screening process, we trained a Graph Convolutional Network (GCN) model named MLMS using the QM9 dataset.23 The model takes the Simplified Molecular-Input Line-Entry System (SMILES) representation of molecules as input features and directly outputs the corresponding FISD. Upon evaluation on the test set, the model exhibited fine predictive performance, achieving a coefficient of determination (r2) of 0.80, which robustly validates its accuracy in predicting FISD. The performance of the MLMS model on the test set is illustrated in Fig. 2a, demonstrating its satisfactory simulation capability for FISD.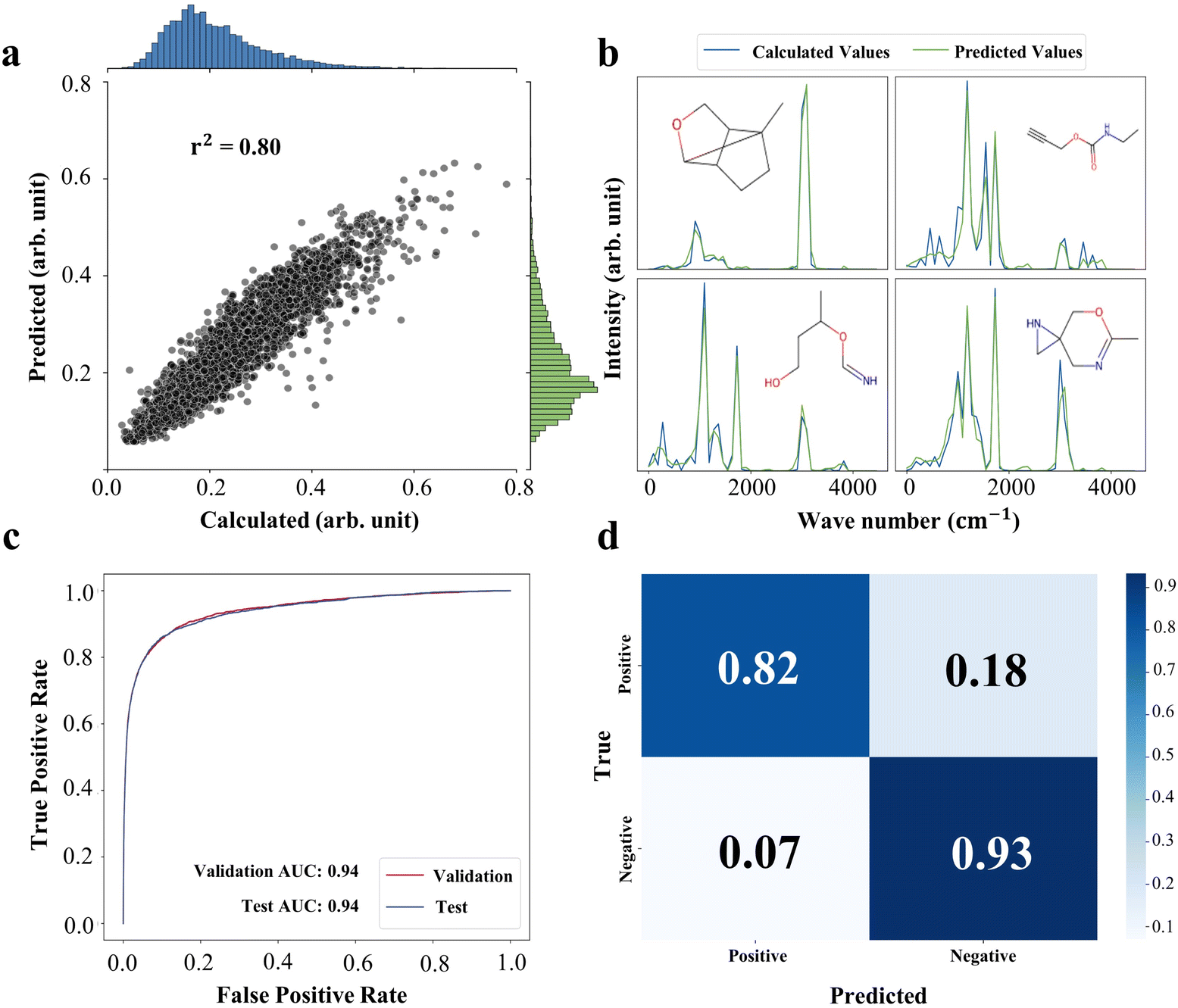 | ||
| Fig. 2 Comprehensive evaluation and visualization of performance for prediction. (a) Comparison of mean values of Density Functional Theory (DFT) calculated FISD and MLMS predicted FISD for the QM9 test set. (b) Four illustrative examples of MLMS predicted FISD (green line) and their corresponding DFT calculated FISD (blue line). (c) Receiver Operating Characteristic (ROC) curve analysis of the VS2Net for the DUD-E validation set (red line) and test set (blue line). (d) Visualization of the confusion matrix of the VS2Net for the DUD-E test set. | ||
To intuitively present the ability of the model, we also randomly selected four representative data points for detailed comparison (as shown in Fig. 2b). In these instances, the blue lines represent the DFT calculated FISD, while the green lines showcase the MLMS predicted FISD. Notably, they exhibit excellent consistency in shape trends and specific values, providing strong evidence for the MLMS model's accuracy and robustness in mimicking FISD characteristics. Additionally, we utilize the MLPS model derived from work by Ye et al. to predict the FISD of proteins.20,21
Performance of VS2Net
VS2Net can be used to determine whether small molecules and protein can bind together via their FISD. To comprehensively evaluate the effectiveness of our proposed FISD method and VS2Net model in virtual screening tasks, we employed 82 protein targets from the DUD-E database.24 Based on these targets and their corresponding ligand and decoy molecules, a training set comprising 156![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 849 compounds was constructed using a stratified sampling strategy to train the VS2Net model. The training process aimed to optimize the model's ability to recognize interactions between target proteins and potential ligands. Subsequently, a validation set of 31042 molecules was selected with a similar strategy to the training set, and the model's performance was evaluated on a test set containing 1010564 molecules. The AUC value is the area enclosed by the ROC curve and the coordinate axis, which can be used to measure the performance of classification models. The larger the AUC value, the better the performance of the classifier. As shown in Fig. 2c, results indicated that the model achieved AUC values of 0.941 and 0.940 on the validation and test sets, respectively, demonstrating the robustness of its classification capability. Further analysis revealed a recall rate of 0.82 and an overall accuracy of 0.93 on the test set's confusion matrix (Fig. 2d), confirming the model's stability and generalization ability. To ensure that no outlier targets have an impact on the overall prediction results, we tested the AUC values of the data corresponding to each target predicted by the model separately, which can be seen in Fig. S1.† These results indicate that the FISD-based VS2Net model efficiently identifies target ligands in virtual screening tasks, while obtaining few false positives and false negatives. Notably, on the test set, the model exhibited strong early enrichment capabilities, which can be valued in Enrichment Factor (EF, detailed description can be seen in the ESI†), with EF0.5% at 94.27, EF1.0% at 59.77, EF2.0% at 35.29, and EF5.0% at 16.01, which can be seen in Table 1. These metrics not only highlight the model's ability to rapidly locate highly active molecules during the initial screening phase but also underscore its competitiveness in the field of virtual screening. When compared to models using DUD-E data but conventional structural descriptors for virtual screening tasks, our model performed above average across multiple evaluation metrics, highlighting its significant advantages in enhancing virtual screening efficiency and accuracy. The performance of a model is significantly influenced by the choice of its architectural design and representation strategy. Although the classical Deep Neural Network (DNN) model adopted in our study does not exhibit significant structural advantages over models such as Random Forest (RF), Convolutional Neural Networks (CNN) or Graph Neural Networks (GNN), the unique part of our research lies in the abandonment of traditional structural descriptors and adoption of spectra-descriptors. This innovative representation approach directly leads to satisfying performance of the model. Feature engineering based on physical principles not only effectively enhances the prediction capability of the model but also significantly simplifies model complexity, highlighting its crucial role in optimizing model performance. Therefore, it can be inferred that spectra-descriptors, due to their rich physicochemical information, are suitable to serve as a powerful tool for predicting protein–ligand interactions.
849 compounds was constructed using a stratified sampling strategy to train the VS2Net model. The training process aimed to optimize the model's ability to recognize interactions between target proteins and potential ligands. Subsequently, a validation set of 31042 molecules was selected with a similar strategy to the training set, and the model's performance was evaluated on a test set containing 1010564 molecules. The AUC value is the area enclosed by the ROC curve and the coordinate axis, which can be used to measure the performance of classification models. The larger the AUC value, the better the performance of the classifier. As shown in Fig. 2c, results indicated that the model achieved AUC values of 0.941 and 0.940 on the validation and test sets, respectively, demonstrating the robustness of its classification capability. Further analysis revealed a recall rate of 0.82 and an overall accuracy of 0.93 on the test set's confusion matrix (Fig. 2d), confirming the model's stability and generalization ability. To ensure that no outlier targets have an impact on the overall prediction results, we tested the AUC values of the data corresponding to each target predicted by the model separately, which can be seen in Fig. S1.† These results indicate that the FISD-based VS2Net model efficiently identifies target ligands in virtual screening tasks, while obtaining few false positives and false negatives. Notably, on the test set, the model exhibited strong early enrichment capabilities, which can be valued in Enrichment Factor (EF, detailed description can be seen in the ESI†), with EF0.5% at 94.27, EF1.0% at 59.77, EF2.0% at 35.29, and EF5.0% at 16.01, which can be seen in Table 1. These metrics not only highlight the model's ability to rapidly locate highly active molecules during the initial screening phase but also underscore its competitiveness in the field of virtual screening. When compared to models using DUD-E data but conventional structural descriptors for virtual screening tasks, our model performed above average across multiple evaluation metrics, highlighting its significant advantages in enhancing virtual screening efficiency and accuracy. The performance of a model is significantly influenced by the choice of its architectural design and representation strategy. Although the classical Deep Neural Network (DNN) model adopted in our study does not exhibit significant structural advantages over models such as Random Forest (RF), Convolutional Neural Networks (CNN) or Graph Neural Networks (GNN), the unique part of our research lies in the abandonment of traditional structural descriptors and adoption of spectra-descriptors. This innovative representation approach directly leads to satisfying performance of the model. Feature engineering based on physical principles not only effectively enhances the prediction capability of the model but also significantly simplifies model complexity, highlighting its crucial role in optimizing model performance. Therefore, it can be inferred that spectra-descriptors, due to their rich physicochemical information, are suitable to serve as a powerful tool for predicting protein–ligand interactions.
| Method | Algorithm | AUC | EF0.5% | EF1.0% | EF2.0% | EF5.0% |
|---|---|---|---|---|---|---|
| NN score25 | DNN | 0.58 | 4.16 | 2.98 | 2.46 | 1.89 |
| RF score26 | RF | 0.62 | 5.62 | 4.27 | 3.49 | 2.67 |
| 3D-CNN27 | CNN | 0.86 | 42.55 | 26.65 | 19.36 | 10.71 |
| PocketGCN28 | GCN | 0.88 | 44.40 | 29.74 | 19.40 | 10.73 |
| GCN29 | GCN | 0.94 | — | — | — | — |
| VS2Net (our model) | DNN | 0.94 | 86.55 | 57.22 | 33.56 | 15.71 |
| AttentionSiteDTI30 | Transformer | 0.97 | 101.74 | 59.92 | 35.07 | 16.74 |
Ablation experiments
To fully validate the superiority of FISD in protein–ligand interaction prediction tasks, we designed and conducted ablation experiments. In these experiments, we attempted to compare FISD with other means of conventional representations, including graph representation directly encoded using GCN, and chemical information descriptors provided by RDKit (Cheminfo-D). The dataset was partitioned according to the standards previously described, and the model was trained for 500 epochs, with the best-performing model on the validation set selected for testing.The graph representation method follows a similar logic to MLMS but skips the process of obtaining FISD. Instead, the molecular graph is encoded into an unrestricted 50-dimensional vector using a GCN. As for the representation method of Cheminfo-Descriptor, we used the MLDescriptors module in RDKit for calculation, resulting in 206-dimension descriptors. Thus, each molecule can be represented by 206 chemical information descriptors (Table S3†), which are then concatenated with the FISD of the protein and input into the DNN for classification just like the FISD case and the graph case. The classification results of the three models are presented in Table 2.
| Method | AUC | Recall | EF0.5% | EF1.0% | EF2.0% | EF5.0% |
|---|---|---|---|---|---|---|
| FISD | 0.94 | 0.82 | 86.55 | 57.22 | 33.56 | 15.71 |
| Graph | 0.65 | 0.71 | 85.83 | 55.47 | 29.15 | 12.39 |
| Cheminfo | 0.68 | 0.67 | 84.73 | 44.49 | 23.08 | 9.53 |
When comparing model performance, in addition to utilizing the AUC value to reflect the overall model performance and the EF value to indicate the model's early enrichment capability, we have also incorporated the recall metric to assess the model's ability to identify all positive samples comprehensively. Given the highly imbalanced distribution of positive and negative samples in the DUD-E dataset, where one positive sample roughly corresponds to 200 negative samples, the capacity to correctly identify positive samples is of greater importance than identifying negative ones. Therefore, the recall value is employed to evaluate the model's performance in this regard.
The FISD method demonstrates robust performance, achieving the best results across all six metrics, as shown in Table 2. In contrast, the performance of models employing the Graph method and those using the Cheminfo-D method is worse than that of models employing the FISD, mainly reflected in the two metrics of the AUC value and Recall value. For the EF values, they all exhibit similar results.
The Graph model yields an AUC of 0.65, a recall value of 0.71, and a good EF value, indicating its strong early enrichment capability. Initially, the model exhibited almost no convergence during training. Subsequently, we pre-trained the model using a small subset of the training set molecules until it achieved a good fit, and then utilized the entire dataset for training to achieve convergence. We hypothesize that the potential reason for this is that FISD can encode molecules in a suitable latent space while serving as a continuous structural descriptor. FISD may enhance the model's performance by restricting the result-generating space and using vibrational spectroscopy as the fitting target during MLMS training, which enables it to learn the correlation between the structure and spectrum, effectively encode molecular electronic information and spatial structure, and thus obtain better classification results. Theoretically, Graph could achieve the same effectiveness as FISD. However, in practice, without specific target space constraints, it is challenging for GCN to quickly locate the ideal feature space. Pre-training with MLMS maps molecules into the fragment integral spectrum descriptor space, improving representation efficiency and predictive capability.
The Cheminfo-D model achieves an AUC of 0.68, a recall rate of 0.67, and an EF value slightly lower than those of the other two models. We speculate that the primary reason for the performance differences is the discreteness of the cheminfo-descriptors. The description based on chemical structures is typically discrete, as atoms are discontinuous. This discreteness can lead to issues, such as the phenomenon of activity cliffs, making it challenging to capture finer differences between structures.
Case study
To validate the effectiveness of the FISD-based VS2Net in addressing practical drug design tasks, particularly in identifying potential ligands for specific protein targets, we designed two case studies. In these studies, we selected targets of significant biological importance from the authoritative Protein Data Bank (PDB), avoiding those already present in the DUD-E dataset. One of the targets we chose is the tau protein (PDB ID: 8q96),31 which is intimately linked to Alzheimer's disease pathology. The study results of finding its active ligands are presented in Fig. 3. Another target we selected is the spike protein (PDB ID: 7lm9),32 which is crucial for SARS-CoV-2 virus entry. The results of our study on this target are shown in Fig. 4.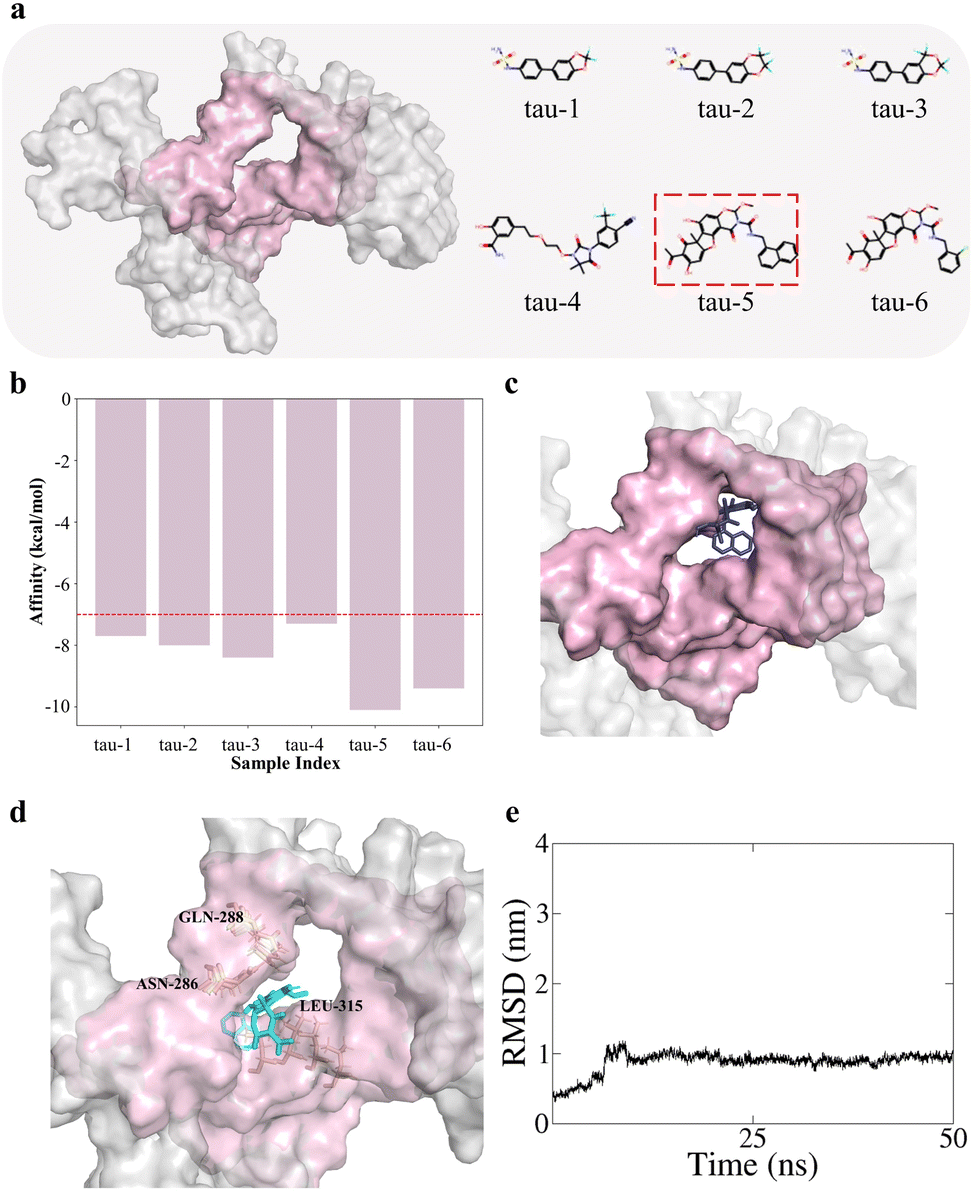 | ||
| Fig. 3 (a) Structure of tau protein with the pocket colored in purple and VS2Net-Identified hit ligands for tau protein, the molecule (tau-5) exhibiting the most desirable docking score is highlighted with a red dotted contour. (b) Docking affinity score of each molecule, and a red dotted line indicates the threshold of −7 kcal mol−1. (c) The docking conformation of the tau-5 molecule with the tau protein from the Autodock Vina, where the tau-5 molecule is colored blue. (d) The zoom-in conformation of the molecular dynamic simulation of the tau-5 molecule with the tau protein. The tau-5 molecule is colored cyan, the pocket is colored purple and residues that may interact with the ligand are annotated. (e) The RMSD changes of the tau-5 molecule during the MD process. | ||
 | ||
| Fig. 4 (a) Structure of spike protein with the pocket colored in yellow and VS2Net-Identified hit ligands for spike protein, the molecule (spike-4) exhibiting the most desirable docking score is highlighted with a red dotted contour. (b) Docking affinity score of each molecule, and a red dotted line indicates the threshold of −7 kcal mol−1. (c) The docking conformation of the spike-4 molecule with the spike protein from the Autodock Vina, where the spike-4 molecule is colored blue. (d) The zoom-in conformation of the molecular dynamics simulation of the spike-4 molecule with the spike protein. The spike-4 molecule is colored cyan, the pocket is colored yellow and residues that may interact with the ligand are annotated. (e) The RMSD changes of the spike-4 molecule during the MD process. | ||
Notably, our virtual screening model was trained solely on the dataset comprising 82 unique targets and their corresponding small-molecule ligands as well as decoys from the DUD-E database. Therefore, it is unrealistic to directly apply the model to other protein targets for ligand screening and expect excellent results. However, the FISD and model proposed still hold practical application value. Since spectra reflect the structural and interactional properties of substances, theoretically, proteins and molecules with similar FISD should also possess similar interactional characteristics. Therefore, we initially conducted a K-means clustering analysis on the 82 known targets from the DUD-E dataset, along with two additional protein targets, pre-setting the number of clusters to eight. This step aimed to categorize protein targets into clusters with similar FISDs, and the clustering results are detailed in Fig. S2.† Subsequently, we can attempt to screen ligands within clusters of similar proteins. The new targets clustered with existing protein targets from DUD-E enabled us to obtain the corresponding ligands of these DUD-E proteins as a ligand library for virtual screening. We also calculated ligand similarity to validate our proposal, as shown in Fig. S3.† The process is universal, and when given requests of new proteins, a ligand library can always be established through a similar process for further screening. After that, we applied our VS2Net model to predict protein–ligand interactions. We analysed the top-10 molecules for two target proteins and found that for tau protein and spike protein, there were 6 and 5 molecules, respectively, with model-predicted interaction probabilities higher than 0.99999. The screening results and the protein structures are illustrated in Fig. 3a and 4a.
To verify if the screened molecules serve as potential ligands for their respective protein targets, AutoDock Vina was employed for molecular docking.33 In this process, eight independent docking conformations were generated for each candidate molecule, and the one with the highest score was chosen as the representative docking result, ensuring both accuracy and representativeness.
For the tau protein, docking analysis of the six selected candidate ligand molecules was performed, with results presented in Fig. 3b. Encouragingly, all six molecules achieved a binding free energy threshold below −7 kcal mol−1, indicating effective docking to tau protein, thereby confirming the predictive accuracy and broad applicability of our screening model. Notably, tau-5 (in Fig. 3a) exhibited the lowest affinity value while maintaining effective docking, and its detailed binding conformation is shown in Fig. 3c, further elucidating the potential of our strategy in identifying ligands for given protein targets.
Regarding the five candidate ligand molecules for spike protein, their corresponding docking scores are presented in Fig. 4b. Based on the same empirical criterion for successful docking, three molecules met this condition, validating their potential to bind with spike protein. Notably, while all successfully docked molecules exhibited good affinity, spike-4 (in Fig. 4a) stood out with the lowest score, and its specific binding mode with spike protein is illustrated in Fig. 4c, visually demonstrating the interaction interface between the molecule and the protein target.
To better validate the ability of our model to identify ligands and further assess the stability and intermolecular interactions of candidate compounds binding to proteins, we conducted molecular dynamics (MD) simulations using GROMACS for the molecules with the best affinity for the two target proteins, spike-4 and tau-5. After the simulations, the trajectories were corrected, and representative structures from the equilibrium trajectories were extracted to display the binding conformations between the molecules and proteins. The binding conformations of these molecule-protein complexes are shown in Fig. 3d and 4d. It can be seen that although the contact positions of molecules are slightly different from those obtained via Autodock Vina (Fig. 3c and 4c), the molecules are also binding well to the proteins, where the residues that may interact with the ligand are annotated. We also calculated the RMSD of the ligand molecules over time to evaluate the simulation process, with the results shown in Fig. 3e, and 4e. In the later stages of the simulations, the RMSD of the ligands remained basically stable, indicating that the protein–ligand complexes were in a relatively stable state at this time.
Moreover, we extracted 10 ns trajectory files from the stable trajectories and calculated the binding free energy using the Molecular Mechanics/Poisson–Boltzmann (Generalized Born) Surface Area (MM/PBSA) method to assess the binding strength.34 The MM/PBSA values for tau-5 binding to tau protein and spike-5 binding to spike protein were found to be −56.25 kcal mol−1 and −20.85 kcal mol−1, respectively. These results indicate strong binding abilities and further prove that the method we proposed effectively identified good ligands for the given proteins, and has the potential to be widely applied.
In addition, we utilized ligands of two previously reported protein targets for model validation. Specifically, we retrieved experimentally validated ligands for tau protein from the binding DB, and acquired a set of spike protein ligands from the work conducted by Timoteo et al.35,36 Following the same selection criteria as previously mentioned, we selected the top-ranked molecules corresponding to tau/spike-like proteins and conducted MD experimental validation on them. Furthermore, we also chose the lowest-ranked molecules obtained during our case studies as representatives of inactive molecules for MD validation. The top-ranked and lowest-ranked molecules are listed in Fig. S8.† The results from VS2Net and MD align well with each other. Both in terms of quantitative validation through RMSD (Fig. S9 and S10†) and MM/PBSA (Table S4†), VS2Net's predictions are in good agreement with the MD simulations as well as the experimental data reported in the literature.
Discussion
We present an approach to enhance protein–ligand interaction predictions by introducing the FISD framework and leveraging advanced physicochemical feature engineering to overcome the limitations of traditional means of representation. Prior research has primarily focused on two aspects: leveraging sophisticated ML model structures to uncover concealed chemical insights within abstract inputs, and incorporating more appropriate chemical details into the input features. While significant progress has been made in the former, the latter aspect has garnered relatively less attention. A key limitation of traditional input features is that they often struggle to encapsulate spatial information, especially when they are just simplistic sequences (such as SMILES or amino acid sequences). Moreover, the absence of electronic structural information further increases the difficulty in predicting protein–ligand interactions. To address this limitation, we introduce the FISD framework. Our research improves protein–ligand interaction predictions by using advanced spectral descriptors that capture physiochemical nuances. Remarkably, its performance in predicting protein–ligand interactions and achieving early enrichment is comparable to that of sophisticated models utilizing conventional structural descriptors. This underscores the significance of meticulous feature engineering from a physicochemical perspective in addressing real-world challenges, demonstrating the effectiveness of our approach.While our research presents promising advancements, it is not without limitations. First, we did not focus our efforts on optimizing the FISD dimension parameter, so 50 dimensions may not necessarily be the best choice. However, based on current results, selecting a 50-dimensional sequence can accomplish the intended tasks. If there is a desire to optimize this parameter, we believe that the selected dimension has to satisfy the requirements of retaining adequate spectral information for accurate predictions by subsequent classification models, while also striving to reduce dimensionality to decrease model complexity and lower the risk of overfitting. Besides, the training data encompass a limited number of protein targets amidst the vast number of molecules, leading to a constraint in model generalizability. Although the existing models and descriptors cannot directly support zero-shot learning, we have made the following attempts to demonstrate their few-shot learning capabilities: after training the model with data corresponding to 17 targets from DUD-E, we randomly selected one remaining target (HIVPR) for transfer learning. We progressively expanded the training data to validate the model's ability to predict protein–molecule interactions. As shown in Fig. S7,† we found that with only a small amount of data (660 samples for training, including 60 active and 600 inactive samples), the model was able to achieve a certain level of performance, yielding satisfactory results. Consequently, when confronted with novel targets, retraining the model with pertinent data becomes necessary and doable to ensure accurate predictions. Fortunately, the limited open-source datasets do not hinder the application of FISD in tackling diverse tasks. In practical drug design scenarios, where the target protein is often unique and predefined, it is advisable to train a single-target model leveraging the pre-accumulated data tailored to the FISD approach, thereby maximizing the utility of our framework.
Methods
Fragment integral spectrum descriptor (FISD)
In this study, we utilize infrared spectra-based descriptors to represent molecules and proteins. The infrared spectrum reflects the microscopic vibrational patterns and spatial configurations of substances, uniquely corresponding to each material. For spectra calculated using DFT, the calculations are typically based on the optimal configuration of the molecule, i.e., the configuration after structural optimization. However, when a molecule binds to a protein, its binding conformation and configuration may differ from the lowest energy configuration and conformation obtained when optimized alone. This implies that, although the MLMS model is trained on a QM9 molecular dataset that has been optimized individually, it may not directly and accurately predict the detailed spectrum of the molecule in its bound state. Recognizing that not all structural information is necessary for predicting protein–ligand interactions, we further compress the infrared spectra to achieve a higher information density. We propose a method to convert the complete spectrum into FISD. The core idea of this method is to minimize the impact of high-frequency noise in the spectrum by segmenting and integrating it, thereby capturing the overall characteristics of the spectrum rather than the fine structural details. We pre-process the original spectral data, aimed at condensing spectral information into a unified format suitable for machine learning models. Specifically, the process involves the following steps: for molecules, we first employ DFT calculations to obtain detailed vibrational frequencies and intensities. After being broadened and going through a normalization process, the spectra are segmented into 50 equal-length fragments based on fixed intervals of 90 cm−1. Within each fragment, integration is performed to calculate the total intensity. This procedure yields an FISD for each molecule, depicted as a fixed-length 50-dimensional sequence, and the process is shown in Fig. 5. | ||
| Fig. 5 The comprehensive process of derivation of FISD from original spectral data. The initial spectral data undergo broadening, followed by normalization to standardize the scale. The data are then systematically partitioned into 50 discrete segments, with the intensities of each segment being integrated to determine the FISD value. | ||
For protein samples, we adopt the MLPS method proposed by Ye et al., which takes protein structural files as the input and outputs fragmental spectral information. Thus, we utilize MLPS to extract the spectral range from 1550 cm−1 to 1750 cm−1 in the infrared spectra of proteins, covering a total spectral range of 200 cm−1. Similarly, after being normalized, the spectral interval is divided into 50 equally spaced fragments, and each fragment undergoes integration, resulting in an FISD for the protein. The resulting FISD is also a 50-dimensional sequence, ensuring consistency and comparability in data formats.
Datasets and pre-process
In this study, we primarily leverage two widely acknowledged public datasets, DUD-E and QM9. Our study mainly involves two models: VS2Net for predicting protein–molecule interactions and MLMS for rapidly obtaining the FISD of molecules. For the training, validation, and testing processes of VS2Net, only data from DUD-E were used; for the training, validation, and testing processes of MLMS, only data from QM9 were utilized. Next, we will introduce the datasets and pre-processing procedures in sequence.As a dataset for quantum chemical properties, QM9 provides detailed quantum chemical information for 127468 small molecules containing up to 9 heavy atoms (C, N, O, F), stored in SMILES format. Aiming at obtaining vibrational spectra, molecules were optimized at the level of b3lyp/TZVP and frequencies were calculated using the Gaussian16 software program. After obtaining the vibrational frequencies and intensities of the QM9 molecules, we can pre-process them into FISD following the process mentioned above, obtaining FISD sequences for each molecule. Based on these data, we partitioned the QM9 dataset into training, validation, and test sets (100000/20000/7468) for the MLMS model.
The DUD-E dataset, a benchmark for evaluating molecular docking algorithms and model performance, comprises exhaustive data for multiple target proteins, each associated with approximately 200 active ligands and 50 decoy molecules per ligand. Despite their similarities in physicochemical properties and 2D topological structures, these molecules exhibit distinct biological activities.
For the DUD-E dataset, we selected a subset of 2200 molecules that met specific criteria. We used the Gaussian16 program to optimize their structures and calculate vibrational frequencies under identical parameters. This subset of data was used in preliminary experiments, and the relevant content can be found in the Related DUD-E Subsets part in the ESI.†
For the FISD of proteins, we employed the MLPS to attempt to obtain the FISD for the target proteins corresponding to the DUD-E dataset (encompassing 82 targets, excluding 20 targets that were incompatible with the MLPS model). For the molecules, we processed the ligands and decoy molecules corresponding to each target protein separately. Their SMILES strings were converted into a format readable by MLMS using RDKit (Graph), and the FISD of the molecules was obtained through MLMS.
For VS2Net model training, validation, and testing, we allocated 80% of the active molecules (14259 small molecules) corresponding to each available target protein, along with ten times that number of decoy molecules (142590 small molecules), as the training set. We designated 10% of the active molecules (2822 small molecules), along with ten times that number of decoy molecules (28220 small molecules), as the validation set, and the remaining molecules (2822 active molecules and 1007742 decoy molecules) were used as the test set.
While the training protein structures all originated from the DUD-E dataset, we designed two case studies to demonstrate model capabilities when facing the scenarios of introducing new target proteins. We selected two targets which are not included in the DUD-E dataset: the spike protein (PDB ID: 7lm9), and the tau protein (PDB ID: 8q96). Their FISDs are obtained using the MLPS model.
Molecular and protein representations
We integrated two distinct model architectures to tackle complex data from different dimensions and accomplish diverse tasks. First, VS2Net focuses on processing high-dimensional sequential data, taking a concatenated sequence of 100-dimensional feature vectors derived from the FISD of proteins and molecules as the input. VS2Net employs a dense neural network frame for machine learning, ultimately refining its output into a single dimension indicating binding probability. Second, the MLMS model, designed for rapid acquisition of molecular FISD, takes SMILES strings as inputs, converts them into molecular graphs using RDKit,37 and leverages the power of GNN to capture the complexity and intrinsic relationships within molecular structures.Specifically, the MLMS model processes SMILES strings into graph structures composed of atomic nodes and chemical bond edges via RDKit. Each atomic node is endowed with a 45-dimensional feature vector encompassing atom type, formal charge, hybridization state, etc., and the detailed information can be seen in Table S1.† The optimized MLMS model directly generates FISD representing input molecules.
The MLMS model makes rapid virtual screening possible, because the VS2Net's input requires two parts of features: FISD for molecules, either from DFT calculated vibrational frequency or from MLMS outputs, and corresponding FISD for protein targets, obtained by MLPS. These descriptors are concatenated into a 100-length joint feature vector, which serves as the input to VS2Net. Trained under supervised learning, the model outputs a value, which can be translated into a binary classification: values closer to 1 indicate higher likelihood of active molecule–protein interactions, while those closer to 0 suggest inactive (decoy) interactions, predicting the active or inactive status of protein–ligand interactions.
Model construction and training
To efficiently and accurately generate molecular FISD, we developed the MLMS model, comprising three pivotal components centered on GCN and DNN. The process of using MLMS to get a molecule's FISD can be seen in Fig. 6a, while the process of using MLPS is shown in Fig. 6b. | ||
| Fig. 6 (a) Diagrammatic representation of employing the MLMS model for forecasting the FISD of given molecules. (b) Schematic flow of applying the MLPS model for the prediction of FISD for proteins. (c) Illustrative flowchart outlining the utilization of VS2Net for executing virtual screening operations. | ||
The first two components of the MLMS model employ a four-layer GCN architecture, each layer utilizing the ReLU activation function to exploit the topological structural information of molecular graphs. A global max pooling layer follows the GCNs to capture the overall graph representation and facilitate subsequent prediction tasks. This is further refined through two dense layers, outputting preliminary FISD. During training, we employ Mean Squared Error (MSE) and Cosine Embedding Loss (CosEmbeddingLoss) as loss functions, training the two components separately to capture distinct aspects of the descriptors.
The third component involves fusing and optimizing the results from the previous two components. Initially, a scaling factor is derived from the numerical values of these results, and the model output trained with CosEmbeddingLoss is multiplied by this factor to align its scale with that of the MSE-trained model. Subsequently, the outputs from both models are concatenated into a 100-dimensional sequence and fed into dense neural networks, yielding the final optimized 50-dimensional FISD. This step integrates the strengths of models trained with different loss functions, enhancing the accuracy and robustness of the descriptors. For the training of the MLMS model, we utilized the QM9 dataset, specifically the training set data as delineated in the pre-processing stage. In the first two components, we used molecular graphs as the input and the processed FISD as labels for supervised learning. In the third component, we took the output values from the first two models as the input and the processed FISD as labels for further supervised learning. Ultimately, we obtained the predictive model.
Thus, the MLMS model is capable of swiftly generating FISD for molecules by inputting SMILES strings. Although the MLMS model is trained solely on the QM9 dataset, it remains capable of achieving a certain level of consistency with DFT-calculated spectra when applied to predict molecules in the larger DUD-E dataset, which can be seen in Fig. S4.†
We designed and implemented a virtual screening model named VS2Net based on a DNN, consisting of five DNN layers with ReLU as the inter-layer activation function to enhance the network's nonlinear processing capabilities. The process of using VS2Net to conduct virtual screening is shown in Fig. 6c. The model's input integrates 100-dimensional FISD (50 for molecule-FISD and 50 for protein-FISD). At the output layer, a sigmoid activation function is applied to output a probability value between 0 and 1, and based on the results, we can classify the molecules as either active molecules or inactive molecules. During training, the Binary Cross-Entropy Loss (BCEloss) is employed as the loss function to minimize the discrepancy between predicted values and true labels.
VS2Net was primarily trained and tested on the DUD-E dataset. Additionally, we conducted pre-experiments and transfer-learning cases, where the models shared a similar structure with VS2Net but were trained on different data. The detailed results and a transfer learning case result can be seen in Fig. S5–S7.† Their training processes are similar. Both take the FISD of proteins and molecules as the input and the classification results of whether the molecule is an active or inactive ligand for the protein as the output for supervised learning. The model that performs best on the validation set is saved and used for testing.
Conclusions
In this study, we emphasize and demonstrate the exceptional performance of leveraging FISD in virtual screening, achieving desirable outcomes even with a simple yet classical model architecture. For the first time, we present the application of spectra-descriptors to virtual screening. This efficient representation of physicochemical properties and spatial characteristics enhances machine learning models' ability to capture and recognize protein–ligand interactions. In the case studies, our proposed strategy successfully identified suitable ligand molecules for two specific target proteins under real-world application scenarios, further validating the practical value of our approach.In future endeavours, one can harness the predictive prowess of this methodology and seamlessly integrate it with Artificial Intelligence Generated Content (AIGC) technology, thereby enabling a more profound and efficient exploration of chemical space. The integration will facilitate the intricate design of novel ligands, advancing the frontier of drug design. Furthermore, the model's generalization capabilities and versatility can be augmented by enriching it with an even broader spectrum of high-quality data, ensuring its robustness across diverse chemical contexts. The study provides physicochemical insights that are valuable to researchers in the field of explainable AI, shedding light on novel perspectives regarding the intricate interplay between model performance and reasonable feature engineering. It not only deepens our understanding of these complexities but also serves as a foundational cornerstone for drug discovery research, inspiring the development of innovative spectroscopic descriptor methodologies that will revolutionize the drug design landscape.
Data availability
The source code and the results of this study are available at: https://github.com/KeantChen/FISD. The processed data can be found at https://doi.org/10.5281/zenodo.14600585. The DUD-E dataset can be downloaded from https://dude.docking.org/ and the QM9 dataset can be downloaded from http://quantum-machine.org/datasets/.Author contributions
C. C. and S. F. did the conceptualization. C. C., L. W. and L. Z. did the data curation. C. C. wrote the original draft, J. L. and S. F. revised the manuscript. All authors contributed to discussions regarding the results and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22303088, 22393892, 22025304, and 22033007), the Innovation Program for Quantum Science and Technology (2021ZD0303303), and the Fundamental Research Funds for the Central Universities (WK9990000129). Numerical calculations were performed at the Hefei advanced computing center and Supercomputing Center of the University of Science and Technology of China.References
- A. V. Sadybekov and V. Katritch, Computational approaches streamlining drug discovery, Nature, 2023, 616, 673–685 CrossRef CAS PubMed.
- E. N. Muratov, R. Amaro, C. H. Andrade, N. Brown, S. Ekins, D. Fourches, O. Isayev, D. Kozakov, J. L. Medina-Franco, K. M. Merz, T. Oprea, V. Poroikov, G. Schneider, M. H. Todd, A. Varnek, D. A. Winkler, A. Zakharov, A. Cherkasov and A. Tropsha, A critical overview of computational approaches employed for COVID-19 drug discovery, Chem. Soc. Rev., 2021, 50, 9121–9151 RSC.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Accurate prediction of protein structures and interactions using a three-track neural network, Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- A. A. Sadybekov, A. V. Sadybekov, Y. Liu, C. Iliopoulos-Tsoutsouvas, X.-P. Huang, J. Pickett, B. Houser, N. Patel, N. K. Tran, F. Tong, N. Zvonok, M. K. Jain, O. Savych, D. S. Radchenko, S. P. Nikas, N. A. Petasis, Y. S. Moroz, B. L. Roth, A. Makriyannis and V. Katritch, Synthon-based ligand discovery in virtual libraries of over 11 billion compounds, Nature, 2022, 601, 452–459 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- S. Zheng, Y. Li, S. Chen, J. Xu and Y. Yang, Predicting drug–protein interaction using quasi-visual questionanswering system, Nat. Mach. Intell., 2020, 2, 134–140 CrossRef.
- F. Ren, X. Ding, M. Zheng, M. Korzinkin, X. Cai, W. Zhu, A. Mantsyzov, A. Aliper, V. Aladinskiy, Z. Y. Cao, S. S. Kong, X. Long, B. H. M. Liu, Y. T. Liu, V. Naumov, A. Shneyderman, I. V. Ozerov, J. Wang, F. W. Pun, D. A. Polykovskiy, C. Sun, M. Levitt, A. Aspuru-Guzik and A. Zhavoronkov, AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor, Chem. Sci., 2023, 14, 1443–1452 RSC.
- A. Thakkar, V. Chadimová, E. J. Bjerrum, O. Engkvist and J. L. Reymond, Retrosynthetic accessibility score (RAscore) - rapid machine learned synthesizability classification from AI driven retrosynthetic planning, Chem. Sci., 2021, 12, 3339–3349 RSC.
- D. E. Graff, E. I. Shakhnovich and C. W. Coley, Accelerating high-throughput virtual screening through molecular pool-based active learning, Chem. Sci., 2021, 12, 7866–7881 RSC.
- H. Cai, C. Shen, T. Y. Jian, X. J. Zhang, T. Chen, X. Q. Han, Z. Yang, W. Dang, C. Y. Hsieh, Y. Kang, P. C. Pan, X. Y. Ji, J. F. Song, T. J. Hou and Y. F. Deng, CarsiDock: a deep learning paradigm for accurate protein-ligand docking and screening based on large-scale pre-training, Chem. Sci., 2024, 15, 1449–1471 RSC.
- J. T. Zhu, Z. H. Gu, J. F. Pei and L. H. Lai, DiffBindFR: an SE(3) equivariant network for flexible protein-ligand docking, Chem. Sci., 2024, 15, 18 Search PubMed.
- Y. P. Huang, H. Zhang, S. Y. Jiang, D. J. Yue, X. H. Lin, J. Zhang and Y. Q. Gao, DSDP: A Blind Docking Strategy Accelerated by GPUs, J. Chem. Inf. Model., 2023, 63, 4355–4363 CrossRef CAS PubMed.
- A. S. Rifaioglu, E. Nalbat, V. Atalay, M. J. Martin, R. Cetin-Atalay and T. Dogan, DEEPScreen: high performance drug-target interaction prediction with convolutional neural networks using 2-D structural compound representations, Chem. Sci., 2020, 11, 2531–2557 RSC.
- S. Moon, W. Zhung, S. Yang, J. Lim and W. Y. Kim, PIGNet: a physics-informed deep learning model toward generalized drug-target interaction predictions, Chem. Sci., 2022, 13, 3661–3673 RSC.
- H. H. Loeffler, J. Z. He, A. Tibo, J. P. Janet, A. Voronov, L. H. Mervin and O. Engkvist, Reinvent 4: Modern AI-driven generative molecule design, J. Cheminf., 2024, 16, 16 Search PubMed.
- Y. J. Zhang, S. Y. Li, K. Meng and S. R. Sun, Machine Learning for Sequence and Structure-Based Protein–Ligand Interaction Prediction, J. Chem. Inf. Model., 2024, 64, 1456–1472 CrossRef CAS PubMed.
- H. Wu, J. Liu, R. Zhang, Y. Lu, G. Cui, Z. Cui and Y. Ding, A review of deep learning methods for ligand based drug virtual screening, Fundam. Res., 2024, 4(4), 715–737 CrossRef CAS PubMed.
- T. Qin, Z. H. Zhu, X. S. Wang, J. Xia and S. Wu, Computational representations of protein-ligand interfaces for structure-based virtual screening, Expert Opin. Drug Discovery, 2021, 16, 1175–1192 CrossRef CAS PubMed.
- L. Yu, X. He, X. M. Fang, L. H. Liu and J. F. Liu, Deep Learning with Geometry-Enhanced Molecular Representation for Augmentation of Large-Scale Docking-Based Virtual Screening, J. Chem. Inf. Model., 2023, 63, 6501–6514 CrossRef CAS PubMed.
- S. Ye, K. Zhong, J. X. Zhang, W. Hu, J. D. Hirst, G. Z. Zhang, S. Mukamel and J. Jiang, A Machine Learning Protocol for Predicting Protein Infrared Spectra, J. Am. Chem. Soc., 2020, 142, 19071–19077 CrossRef CAS PubMed.
- L. Y. Zhao, J. X. Zhang, Y. L. Zhang, S. Ye, G. Z. Zhang, X. Chen, B. Jiang and J. Jiang, Accurate Machine Learning Prediction of Protein Circular Dichroism Spectra with Embedded Density Descriptors, JACS Au, 2021, 1, 2377–2384 CrossRef CAS PubMed.
- S. H. Rutherford, C. D. M. Hutchison, G. M. Greetham, A. W. Parker, A. Nordon, M. J. Baker and N. T. Hunt, Optical Screening and Classification of Drug Binding to Proteins in Human Blood Serum, Anal. Chem., 2023, 95, 17037–17045 CrossRef CAS PubMed.
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J. L. Reymond, Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17, J. Chem. Inf. Model., 2012, 52, 2864–2875 CrossRef CAS PubMed.
- M. M. Mysinger, M. Carchia, J. J. Irwin and B. K. Shoichet, Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking, J. Med. Chem., 2012, 55, 6582–6594 CrossRef CAS PubMed.
- J. D. Durrant and J. A. McCammon, NNScore 2.0: A Neural-Network Receptor-Ligand Scoring Function, J. Chem. Inf. Model., 2011, 51, 2897–2903 CrossRef CAS PubMed.
- P. J. Ballester and J. B. O. Mitchell, A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking, Bioinformatics, 2010, 26, 1169–1175 CrossRef CAS PubMed.
- M. Ragoza, J. Hochuli, E. Idrobo, J. Sunseri and D. R. Koes, Protein-Ligand Scoring with Convolutional Neural Networks, J. Chem. Inf. Model., 2017, 57, 942–957 CrossRef CAS PubMed.
- W. Torng and R. B. Altman, Graph Convolutional Neural Networks for Predicting Drug-Target Interactions, J. Chem. Inf. Model., 2019, 59, 4131–4149 CrossRef CAS PubMed.
- M. Tsubaki, K. Tomii and J. Sese, Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences, Bioinformatics, 2019, 35, 309–318 CrossRef CAS PubMed.
- M. Yazdani-Jahromi, N. Yousefi, A. Tayebi, E. Kolanthai, C. J. Neal, S. Seal and O. O. Garibay, AttentionSiteDTI: an interpretable graph-based model for drug-target interaction prediction using NLP sentence-level relation classification, Briefings Bioinf., 2022, 23, 14 CrossRef PubMed.
- M. Schweighauser, A. G. Murzin, J. Macdonald, I. Lavenir, R. A. Crowther, S. H. W. Scheres and M. Goedert, Cryo-EM structures of tau filaments from the brains of mice transgenic for human mutant P301S Tau, Acta Neuropathol. Commun., 2023, 11, 11 CrossRef PubMed.
- H. J. Liu, M. Yuan, D. L. Huang, S. Bangaru, F. Z. Zhao, C. C. D. Lee, L. H. Peng, S. Barman, X. Y. Zhu, D. Nemazee, D. R. Burton, M. J. van Gils, R. W. Sanders, H. C. Kornau, S. M. Reincke, H. Prüss, J. Kreye, N. C. Wu, A. B. Ward and I. A. Wilson, A combination of cross-neutralizing antibodies synergizes to prevent SARS-CoV-2 and SARS-CoV pseudovirus infection, Cell Host Microbe, 2021, 29(5), 806–818 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- T. Q. Liu, L. Hwang, S. K. Burley, C. I. Nitsche, C. Southan, W. P. Walters and M. K. Gilson, BindingDB in 2024: a FAIR knowledgebase of protein-small molecule binding data, Nucleic Acids Res., 2024, 53(D1), D1633–D1644 CrossRef PubMed.
- T. Delgado-Maldonado, A. González-González, A. Moreno-Rodríguez, V. Bocanegra-García, A. V. Martinez-Vazquez, E. d. J. de Luna-Santillana, G. Pujadas, G. Rojas-Verde, E. E. Lara-Ramírez and G. Rivera, Ligand- and Structure-Based Virtual Screening Identifies New Inhibitors of the Interaction of the SARS-CoV-2 Spike Protein with the ACE2 Host Receptor, Pharmaceutics, 2024, 16, 613 CrossRef CAS PubMed.
- G. Landrum, RDKit: A Software Suite for Cheminformatics, Computational Chemistry, and Predictive Modeling – Open-source cheminformatics, http://www.rdkit.org.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00451a |
| ‡ C. C. L. W. and Y. F. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |