
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic proton coupled electron transfer quenching as a sensing modality in fluorescent probes†
Rasmus K.
Jakobsen‡
 ,
Stine G.
Stenspil‡
,
Junsheng
Chen
and
Bo W.
Laursen
*
,
Stine G.
Stenspil‡
,
Junsheng
Chen
and
Bo W.
Laursen
*
Nano-Science Center & Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: bwl@chem.ku.dk
First published on 18th March 2025
Abstract
Fluorescent off–on probes based on a modular design where an analyte sensitive PET moiety is attached to a fluorophore are extremely successful. Here we report a new modular fluorescence probe design switched by dynamic quenching due to proton coupled electron transfer (PCET) mediated by collision with weak bases in solution. The fluorescence lifetime of this probe directly reports on the rate of deprotonation by the weak bases in the solution. We investigate the probe design, mechanism of response, and sensitivity to various abundant weak bases/metabolites including acetate, glutamate, phosphate, valine, and amines. We find that this modular PCET based probe design, in contrast to traditional PET probes, can work efficiently with a fluorescence lifetime readout providing a calibration free probe for weak bases. Upon further development we envision such dynamic PCET probes as sensitive tools for studies of cellular buffer systems and metabolite pools.
Introduction
Fluorescent probes with the ability to report on their local environment and concentrations of various analytes are key tools in biology and biomedical research.1–4 Consequently, the development and further improvement of various probe designs have become a fertile field of research, where molecular photophysics and synthetic organic chemistry go hand in hand providing rational design guidelines and means.6–11 One of the most successful probe designs of all time is the off–on probes based on photoinduced electron transfer (PET).12,13 For these probes a fluorophore is linked to a PET quencher moiety that changes redox properties, and thus quenching efficiency, upon binding of an analyte. Hundreds if not thousands of such off–on PET probes have been reported and many are commercially available and extensively used in biology, medicine and nanoscience. Prime examples are: the pH probe LysoSensor Blue (1) and the calcium probe Rhod-2 (2), both shown in Fig. 1A.5,14,15 As the PET quenching moiety does not require direct conjugation with the fluorophore, the design is highly modular, and the fluorophore and quencher/receptor can to a large degree be optimized independently, which is a major reason for the success of these PET probes.16–19 | ||
| Fig. 1 Photoinduced electron transfer probes and mechanism. (A) 1 (LysoSensor Blue), a pH sensitive probe in which protonation of the two morpholine side groups inhibits PET quenching of the anthracene unit. Adapted from de Silva et al.52 (Rhod-2), an example of a calcium sensitive probe. Adapted from Minta et al.15 (B) Schematic representation of a molecular PET probe displaying fluorescence when bound to an analyte or undergoing PET quenching when no analyte is bound. | ||
The readout for PET probes is generally based on the fluorescence emitted from the fraction of probes that are bound to analytes. These probes will display similar photophysical properties to the chromophore scaffold without a quenching moiety (Fig. 1B). Probes not bound to an analyte do not contribute to the fluorescence signal, since PET quenching is much faster than the fluorescence process. Consequently, the readout of the probe only varies in fluorescence intensity and not in fluorescence lifetime, thus corresponding to static quenching.20 The fluorescence intensity will be proportional to the ground-state equilibrium between probes bound and not bound to an analyte. While such turn-on of fluorescence upon analyte binding is excellent for qualitative or relative measurements, this simple one-channel intensity readout is also a drawback and makes quantitative measurements dependent on the use of internal standards and/or comprehensive calibration.20,21 PET quenching that instead takes place upon collision of the freely diffusing analyte and the excited fluorophore results in a shortening of the observed fluorescence lifetime and a proportional loss of fluorescence intensity (dynamic quenching).20 Since the fluorescence lifetime readout fundamentally is independent of the concentration of the probe it provides a simple and absolute measure of analyte concentration.20,22 The advantageous effect of dynamic quenching and fluorescence lifetime readout is successfully demonstrated by the quantitative imaging of Cl− ions in biological systems.23–26 However, this is a special case, only feasible due to the high abundance and relatively low oxidation potential of Cl−, allowing this analyte to act directly as the PET quencher without binding to a PET active receptor unit.
Here we demonstrate how a modular PET probe design with a fluorophore and a covalently linked PET quencher can be utilized in the dynamic quenching regime, and via the fluorescence lifetime readout, report on biologically abundant analytes such as glutamate and phosphates. The sensing mechanism relies on the analytes' ability to be proton acceptors in proton coupled electron transfer (PCET) quenching of the excited fluorophore.27,28
Results and discussion
General concept of the modular PCET probe
The investigated probe, the phenol substituted DiAzaOxaTriAngulenium (phenol-DAOTA, Fig. 2) has in the past been used as a simple PET based off–on pH probe rationalized according to the general PET probe model with static quenching shown in Fig. 1B (X = H+).29 However, we became aware of unexpected deviations in the observed fluorescence lifetime as a function of the applied buffer, which led us to the present study and proposal of a new probe design principle based on dynamic PCET quenching. The function and response of the PCET probe is described by the state diagram in Fig. 2. The key difference between this PCET probe and traditional PET probes is that the response of the PCET probe relies on dynamic quenching of the excited protonated probe (phenol-DAOTA) and not on the ground state equilibrium with the deprotonated form (phenolate-DAOTA). | ||
| Fig. 2 Schematic representation of the PCET process for phenol-DAOTA. Molecular structures of the probe in its acidic and basic forms. In the excited state a basic analyte, B:, can deprotonate the fluorophore, which leads to fast electron transfer and formation of a PET state, thus quenching the fluorescence. | ||
For the PCET probe quenching occurs by a diffusion controlled deprotonation of the phenol group by a basic analyte (B:) and transfer of an electron from the phenol/phenolate to the excited DAOTA leading to the formation of the dark PET state. This PCET quenching can either take place stepwise by proton transfer (PT) followed by electron transfer (ET) or by a concerted proton electron transfer (CPET) process (highlighted as the blue colored processes in Fig. 2). Regardless of the mechanism, the quenching rate (kq) will depend on the concentration of the base assisting the PCET process; kq = kPCET[B:]. This dynamic quenching process leads to a reduction of the observed fluorescence lifetime of the probe, proportional to the quenching rate constant (kq) and thus the concentration of the analyte/base. This opens up the possibility to determine analyte abundance from one simple fluorescence lifetime measurement independent of probe concentration.
Mapping out the operational principles of the PCET probe
In a first step towards understanding the analyte response and photophysical properties of the phenol-DAOTA probe the absorption and fluorescence were studied as a function of pH (Fig. 3A). The absorption spectra of the phenol-DAOTA show moderate but clear changes as a function of pH with λmax red shifting from 557 nm to 560 nm when going from acidic to basic solution when the phenolate is formed. The shift in the absorption spectra follows perfectly an acid–base Bjerrum plot with a pKa = 8.9 (Fig. 3B, black line), irrespective of the applied buffer and salt concentrations (Table S1†). The fluorescence intensity decreases drastically as pH is increased, with the solutions above pH ≈ 10 being practically non-fluorescent (Fig. 3A). All solutions show identical fluorescence emission spectra, only varying in intensity (Fig. S1†), in agreement with emission only stemming from the phenol form. However, quantitative analysis of fluorescence intensity as a function of pH shows very clear variations as a function of the buffer composition (Fig. 3B). In pure water, with no buffer present, the fluorescence intensity follows the changes in absorption spectra as expected for a ground state equilibrium between the fluorescent phenol form and the fully quenched phenolate form (blue points). | ||
| Fig. 3 pH response of phenol-DAOTA in acetate buffer. (A) Absorption and emission spectra of phenol-DAOTA (5 μM) in aqueous solution with 100 mM acetate buffer as a function of pH, from pH = 1 (red) to pH = 12 (black). (B) Black points: absorption at 580 nm. Fluorescence intensity as a function of pH in pure water (blue points) and 100 mM acetate buffer (red points). The full red line is a theoretical Bjerrum curve based on the pKa values of acetic acid and the probe, and the measured fractional drop in the fluorescence lifetime. (C) Fluorescence lifetime (τ) fitted monoexponentially as a function of pH for phenol-DAOTA in 100 mM acetate buffer (red), pure water (blue), and for anisole-DAOTA in acetate buffer (dark red circles). The full line represents a theoretical Henderson–Hasselbalch function. | ||
This follows the classical PET probe model shown in Fig. 1 and the PCET model in Fig. 2 when [B:] = 0 (and hence kq = 0). In buffered solutions the fluorescence intensity however displays an additional decrease at pH ≈ pKa of the buffer. For a 100 mM acetate buffer this results in a more complex fluorescence intensity response shown as the red points in Fig. 3B, significantly deviating from the response expected from the classical PET model and that observed in pure water (blue points). Investigation of the fluorescence lifetime (τ) as a function of pH shows similar buffer dependence, as shown in Fig. 3C.
In the acetate buffer solution τ = 14.0 ns at low pH, but decreases to τ = 12.0 ns at higher pH, the decays measured at all pH values were monoexponential (ESI §12†). According to the proposed PCET probe model (Fig. 2) this decrease in τ arises from collision of the excited probe with a base (B:), which is able to assist in the deprotonation of the phenol and thus the overall PCET process, leading to fluorescence quenching due to the formation of the PET state of the phenolate. The change in τ is proportional to the increased concentration of acetate ions (pKa = 4.76), which in this case is the base assisting the PCET process. According to the PCET model, the observed 14.5% drop in τ should be accompanied by a similar drop in fluorescence intensity. This is indeed confirmed when comparing the measured fluorescence intensity (red points in Fig. 3B) with a theoretical Henderson–Hasselbalch curve for the system based on a 14.0% quenching from a base with pKa = 4.76 and a full quenching from the ground-state deprotonation of the probe with pKa = 8.90, as shown by the red line (see ESI §4† for details). In agreement with the PCET model τ is unchanged in pure water (Fig. 3C, blue points), except for a small decrease in the most basic solutions corresponding to dynamic quenching from hydroxide ions (Fig. S2†). To exclude the possibility that any of the changes in fluorescence properties are due to direct interactions with the DAOTA fluorophore we also studied anisole-DAOTA, where the phenol group has been methylated and thus cannot take part in proton transfer processes. As expected, anisole-DAOTA showed no changes in fluorescence intensity or lifetime (Fig. 3C, S3 and Table S2†) as a function of buffer or pH. Importantly, anisole-DAOTA displays the same fluorescence lifetime as phenol-DAOTA in pure water (τ0), proving that water does not act as a base (proton acceptor) in the PCET process. According to our proposed model (Fig. 2) the observed τ, in the presence of bases, can thus easily be converted to a quenching rate constant (kq), which directly reflects the rate of base assisted PCET experienced by the excited probe (kPCET[B:]), as described by eqn (1).
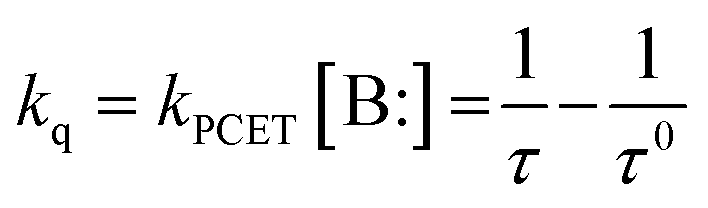 | (1) |
An important feature of both the dynamic PCET and the traditional PET sensing modalities is the very fast formation of the non-emissive PET state from the excited phenolate form, since this makes the base assisted excited state proton transfer irreversible and explains why no emission is observed from the excited phenolate. The formation of the PET state from the deprotonated probe was therefore investigated by femtosecond transient absorption spectroscopy (details in ESI §3†). Femtosecond transient absorption spectra of the phenolate-DAOTA showed two characteristic signals.
The first is an excited state absorption (ESA) at 400 nm (black data points in Fig. 4). The ESA signal has a rise time corresponding to the instrument response function (IRF) (≈100 fs) and an additional component of 0.4 ps (highlighted in pink) and decay with a 1 ps time constant. The second feature observed is a ground state bleach (GSB) of the first DAOTA absorption band at 580 nm with a rise time identical to the instrument response function (IRF) (≈100 fs), and a decay of 1.2 ps (red data). From this we deduce that the excited state of the phenolate form only has a lifetime of 0.4 ps before converting to the PET state, which itself has a short lifetime of 1 ps before returning non-radiatively (by back electron transfer) to the ground state. Transient absorption spectra of the protonated form support this interpretation with ESA and GSB signals rising with the 100 fs response time of the instrument and not decaying on the ps time scales (in agreement with the 14 ns lifetime measured from the fluorescence decay). Femtosecond transient absorption spectra of phenolate- and phenol-DAOTA are shown in Fig. S4 and S5.† Very fast formation of the PET state from the excited phenolate-DAOTA is observed in the femtosecond transient absorption measurements and confirms that base assisted proton transfer from phenol-DAOTA in the excited state will be irreversible, thus supporting the PCET model (Fig. 2).
 | ||
| Fig. 4 Femtosecond transient absorption kinetic analysis of phenolate-DAOTA. ESA (400 nm) (black circles), GSB (580 nm) (red circles), and IRF (blue dots). ESA is fitted with the IRF, a second rise component of 0.4 ps (highlighted in pink), and a fixed decay of 1 ps. The GSB is fitted with the IRF and a decay of 1.2 ps. | ||
Response to various analytes
The elementary operation of the dynamic PCET probe, phenol-DAOTA, has now been established above. We therefore investigate how the probe responds to different analytes and thereby gain more insight into the mechanism of action and potential applications of the probe.As the operation of the PCET probe relies on a diffusion controlled interaction with basic molecules, the most obvious analytes in biological systems are the abundant metabolites making up cellular buffer systems. Cellular systems contain complex mixtures of hundreds of small molecule metabolites, reaching a total concentration of approximately 300 mM.30
This metabolite pool is dominated by weak bases such as glutamate and organic phosphates reaching concentrations of ∼100 mM and ∼50 mM respectively. Also, inorganic phosphate is abundant inside cells reaching concentrations of ∼50 mM. With this in mind, we investigated the response of the phenol-DAOTA probe in a series of bases covering a range of pKa values. The probe response was investigated in solutions of acetate, formate, valine, glutamate, phosphate, glucose-6-phosphate, and ethanolamine where the concentrations of the individual bases were varied by adjusting the pH.
For a 100 mM glutamate solution the fluorescence lifetime response as a function of pH is shown in Fig. 5. At low pH τ = 14 ns, identical to the lifetime for pure water solutions, showing that the fully protonated glutamic acid (Glu+) is not facilitating the PCET quenching. At pH = 3 where the solution contains ≈100 mM Glu0, the measured fluorescence lifetime has only dropped ≈0.5% making it clear that Glu0 has very little impact on the probe and that this is on the limit of detection. However, clear drops in fluorescence lifetime are observed at pH values equal to the second and third pKa values proving that both Glu− and Glu2− indeed promote the PCET quenching, with Glu2− being more efficient than Glu−. As seen for acetate the fractional drop in τ for glutamate is in all cases followed by a corresponding drop in fluorescence intensity in full agreement with the PCET model (Fig. S8†). For all the investigated bases, we found perfect correspondence between the changes in the fluorescence lifetime of the probe and their reported pKa values. Quantitative correlation between the change in fluorescence intensity and the observed fractional drop in fluorescence lifetime was also observed (Fig. S6 to S14†). Stronger bases most efficiently promote the PCET quenching. This is clearly seen when the fluorescence lifetime as a function of concentration of each base is displayed in a Stern–Volmer plot, as shown for selected bases in Fig. 6 (plots for all bases are shown in Fig. S15–S23†). The concentrations of each base are calculated based on the pKa of their corresponding acid, pH in the solution, and the total buffer concentration. The perfect straight lines of these plots confirm that each base acts as a dynamic quencher according to the classical Stern–Volmer equation (reorganized from eqn (1)):
 | (2) |
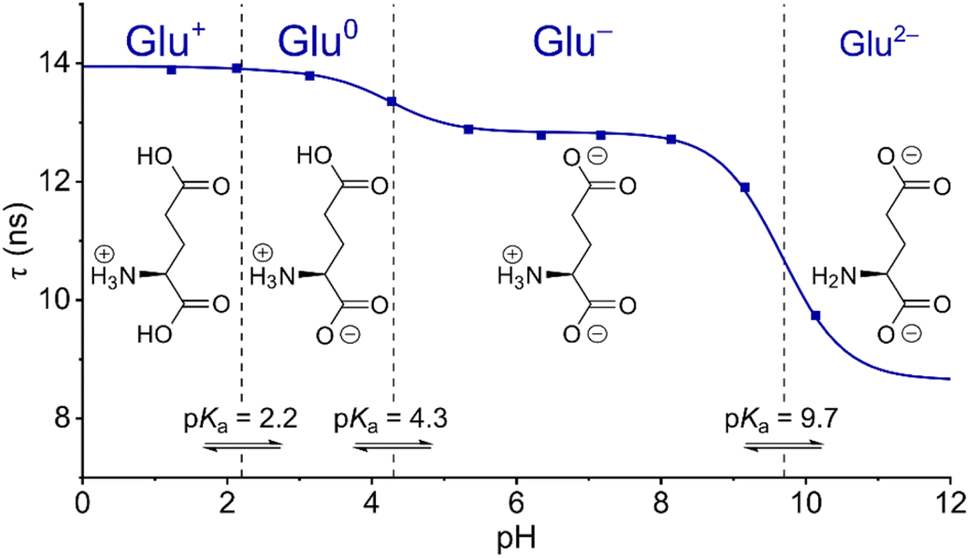 | ||
| Fig. 5 Fluorescence lifetime response of phenol-DAOTA in glutamate buffer. Fluorescence lifetime of the phenol-DAOTA as a function of pH for a 100 mM solution of glutamic acid. The solid line represents a theoretical Henderson–Hasselbalch function based on the pKa values of glutamic acid with only the fractional drop in τ as a fitting parameter. | ||
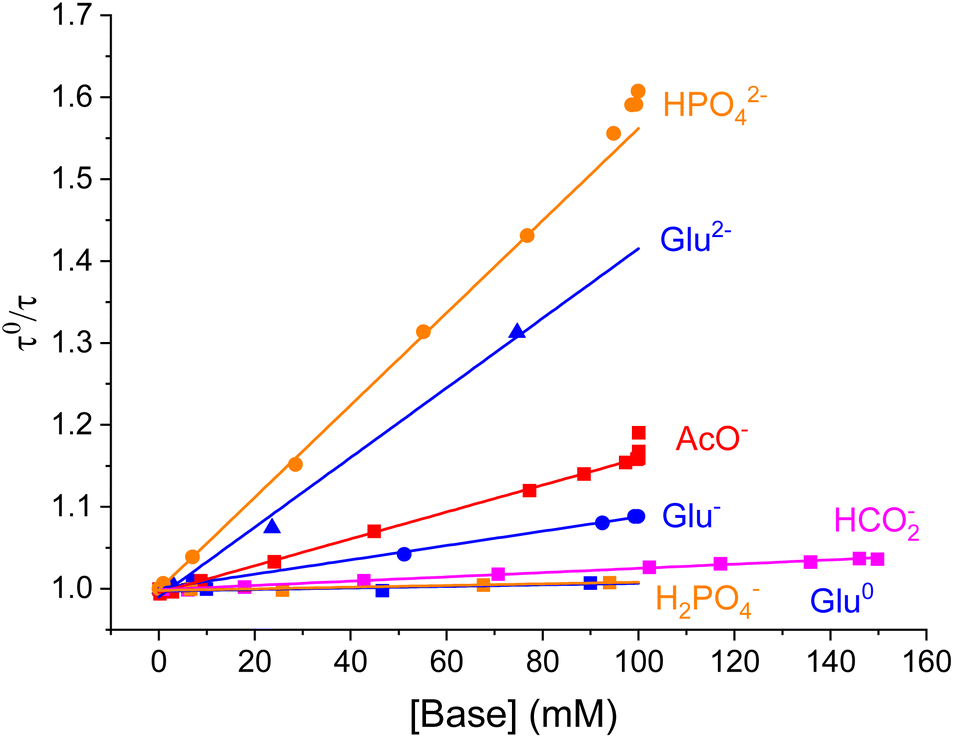 | ||
| Fig. 6 Stern–Volmer plots for selected bases. Stern–Volmer analysis of quenching rates as a function of concentration of various bases. Data for H2PO4− and HPO42− obtained in the presence of 150 mM NaCl. | ||
For hydrogenphosphate the Stern–Volmer plot shows a significant upward curvature at concentrations above 40 mM (shown in Fig. S20†), indicating a departure from an ideal solution caused by electrostatic attraction between the cationic probe and dianionic base (HPO42−), leading to a local increase of the base concentration around the probe. This effect arising for oppositely charged fluorophores and quenchers is predicted by the Debye–Hückel theory and has been reported previously.31,32 In agreement with this explanation, increased ionic strength suppresses this effect and reinstates linear Stern–Volmer plots. We demonstrate this by addition of NaCl (150 mM) as shown in Fig. S24.†
To demonstrate the potential of the phenol-DAOTA probe in more complex buffer/metabolite systems, lifetime measurements in a mixed system of acetate and phosphate showed very good correlation with expected lifetimes based on the kPCET rates established for the individual bases (see ESI §10†).
Measurements of the PCET quenching of phenol-DAOTA in a 100 mM glutamate dissolved in D2O display PCET rates 2–3 times lower than in H2O (ESI §9†). This kinetic isotope effect confirms that proton transfer is indeed involved in the rate-determining step of the quenching as expected from our model for PCET in this system (Fig. 2).27
As expected, the rate of PCET (kPCET) derived from the slopes of the Stern–Volmer plots increases with the base strength of the proton acceptors as shown in Fig. 7.27,33,34 Here log(kPCET) is plotted as a function of ΔpKa, which represents the difference between the pKa of the conjugated acid of the proton acceptor (BH) and the proton donor (phenol-DAOTA). The plot reveals several interesting features of the base promoted quenching process. Firstly, the rate of PCET reaches a plateau at kPCET ≈ 108.5 M−1 s−1 for the stronger bases (ΔpKa > −2) with the exception of hydroxide, which reaches the expected diffusion limit around 1010 M−1 s−1. This behavior is quite similar to ground state deprotonation of phenol itself (indicated by the gray line), when taking the larger size and thus slower diffusion of phenol-DAOTA into account, as shown in the diffusion corrected plot in Fig. S25 (ESI §6†).35,36
 | ||
| Fig. 7 Rate of PCET as a function of the base strength of the quencher. Log(kPCET) as a function of ΔpKa from the base strength of the quencher (given as pKa of the corresponding acid, BH) relative to the pKa of the proton donor. Data points for phenol-DAOTA quenching in blue, the black line is a guide to the eye. The gray line shows literature values measured of the ground state deprotonation rate of phenol (see ESI §6† for details). | ||
Secondly, phenol-DAOTA shows a higher sensitivity for weaker bases than phenol itself by ∼3 ΔpKa units, as seen by the horizontal shift of the full black vs. gray line. For the diffusion corrected values, this offset is close to 4 pKa units (Fig. S25†). This suggests that phenol-DAOTA is a stronger acid in the excited state (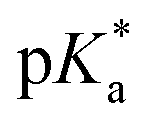 ≈ 5) than in its ground state (pKa = 8.9). The minute shift in the absorption spectra of phenol-DAOTA (Fig. 3A) upon deprotonation shows that the excited state acidity does not originate from stabilization of the phenolate form itself37 but must be a result of a concerted proton electron transfer (CPET) directly to the low energy PET state that is more favorable than the sequential PCET transfer.27,38 Thus, the additional driving force for proton transfer comes from formation of the low energy PET state (Fig. 2). From a sensor point of view, this enhanced sensitivity to weak bases is highly advantageous since the ground state pKa of 8.9 allows the probe to be fully protonated (and thus emissive) at all biologically relevant pH values while still being sensitive to (quenched by) relevant weak bases/metabolites. The sensitivity of the phenol-DAOTA probe for determining concentrations of bases in the various buffer solutions can be estimated from the Stern–Volmer equation (eqn (2)) and the accuracy of the fluorescence lifetime measurements. The minimum analyte concentration that is measurable by the phenol-DAOTA probe is 38 mM for formic acid, while acetate and phosphate can be detected at concentrations as low as 6 mM and 2 mM respectively (see ESI §7 and §8† for details on experimental reproducibility and detection limits). These detection limits may not seem impressive compared to PET probes binding to analyte with high affinity receptors in their ground state (like the calcium probe 2 in Fig. 1), however they are actually in the relevant range for the abundant and important metabolites we have tested here.30 In the tested buffer systems the fluorescence lifetimes (and intensities) are quenched by ∼4–50% (Table S4†). This range is close to ideal as this leaves sufficient signal to obtain reliable lifetime measurements, in contrast to 80–100% quenching where little to no signal is measurable resulting in large errors in lifetime values. The long fluorescence lifetime of the DAOTA fluorophore furthermore allows for time-gated measurements, eliminating interference from other fluorescent dyes and autofluorescence in biological samples.39,40 We demonstrate this by reproducing the probe lifetimes in a phosphate buffer in the presence of a cyanine fluorophore providing an overlapping fluorescence signal accounting for up to ≈25% of the total intensity (see ESI §11†). We thus envision that dynamic PCET probes, like phenol-DAOTA, can be developed into general tools for investigating complex biological buffer systems and changes in metabolite pools, even in the presence of overlapping scattering and autofluorescence.
≈ 5) than in its ground state (pKa = 8.9). The minute shift in the absorption spectra of phenol-DAOTA (Fig. 3A) upon deprotonation shows that the excited state acidity does not originate from stabilization of the phenolate form itself37 but must be a result of a concerted proton electron transfer (CPET) directly to the low energy PET state that is more favorable than the sequential PCET transfer.27,38 Thus, the additional driving force for proton transfer comes from formation of the low energy PET state (Fig. 2). From a sensor point of view, this enhanced sensitivity to weak bases is highly advantageous since the ground state pKa of 8.9 allows the probe to be fully protonated (and thus emissive) at all biologically relevant pH values while still being sensitive to (quenched by) relevant weak bases/metabolites. The sensitivity of the phenol-DAOTA probe for determining concentrations of bases in the various buffer solutions can be estimated from the Stern–Volmer equation (eqn (2)) and the accuracy of the fluorescence lifetime measurements. The minimum analyte concentration that is measurable by the phenol-DAOTA probe is 38 mM for formic acid, while acetate and phosphate can be detected at concentrations as low as 6 mM and 2 mM respectively (see ESI §7 and §8† for details on experimental reproducibility and detection limits). These detection limits may not seem impressive compared to PET probes binding to analyte with high affinity receptors in their ground state (like the calcium probe 2 in Fig. 1), however they are actually in the relevant range for the abundant and important metabolites we have tested here.30 In the tested buffer systems the fluorescence lifetimes (and intensities) are quenched by ∼4–50% (Table S4†). This range is close to ideal as this leaves sufficient signal to obtain reliable lifetime measurements, in contrast to 80–100% quenching where little to no signal is measurable resulting in large errors in lifetime values. The long fluorescence lifetime of the DAOTA fluorophore furthermore allows for time-gated measurements, eliminating interference from other fluorescent dyes and autofluorescence in biological samples.39,40 We demonstrate this by reproducing the probe lifetimes in a phosphate buffer in the presence of a cyanine fluorophore providing an overlapping fluorescence signal accounting for up to ≈25% of the total intensity (see ESI §11†). We thus envision that dynamic PCET probes, like phenol-DAOTA, can be developed into general tools for investigating complex biological buffer systems and changes in metabolite pools, even in the presence of overlapping scattering and autofluorescence.
Development of modular PCET probes
Detection of changes in metabolite concentrations by fluorescent probes sensitive to proton transfer, akin to phenol-DAOTA, is not an established sensing modality. However, some examples of probes responsive to weak bases, such as phosphate and acetate, via proton transfer have been reported earlier. These probes are however not modular and have the active proton acceptor/donor group integrated with the chromophore core. Among these are several xanthene based dyes reported by Crovetto and co-workers, where excited state proton transfer alters the fluorescence properties in response to weak bases like, e.g., acetate or inorganic phosphate.41–44 The Gryczynski group studied proton transfer to a carboxylated pyrene assisted by weak acids, including acetic and lactic acids.34 Common for these systems is however that both the protonated and deprotonated forms are fluorescent and have different spectra, quantum yields and radiative rates, which complicate the data analysis significantly. With both the protonated and deprotonated forms having significant excited state lifetimes both protonation and deprotonation rates also have to be included in the analysis and the fluorescence decays may have to be analyzed by multi-exponential models with much increased uncertainty. Dynamic (diffusional) PCET quenching of the fluorophore by solvents and weak acids/bases has also been studied for several systems to investigate the energetics and mechanisms of the PCET process.27,45Dynamic PCET probes cannot be expected to be selective for any specific analyte in a complex system, but will provide a response that is the sum of contributions from each base, weighted by its concentration and kPCET, as we showed for the acetate/phosphate mixture (ESI §10†). In cases where one analyte is all dominating it may be possible to extract its concentration, as in the case for Cl− sensing in cellular systems by PET probes.25 While the PCET probes in most cases will not provide a measure on one specific analyte/metabolite, we envision that the cumulative response of dynamic PCET probes will provide a new general descriptor of a buffer system. In conjunction with the pH value, this may provide new insights on the state of, or changes to, cellular buffer systems. For instance, it has been speculated that cells may have differential local buffers to support various biochemical processes.46 PCET probes could map out spatiotemporal variations in metabolite/buffer composition and/or concentration to elucidate such questions. Likewise, PCET probes may help us understand how different buffers and anions influence other probes and biomolecules.47,48
For the development of PCET probes, we can consider the optimal properties of such probes. The probes should fulfil the following design criteria: (1) have only one emitting state, for which the fluorescence lifetime is proportional to the rate of PCET quenching induced by collision with the analytes (eqn (1) and Fig. 2). (2) The probe should not be quenched by water, as is the case for many photoacids.49 (3) The probe should be fully ON at physiologically relevant pH values and still be effectively quenched by most relevant metabolites/bases. (4) Long excited state lifetimes (τ0) will increase sensitivity (eqn (2)). For example, the 14 ns lifetime of the DAOTA fluorophore50,51 in water enhances its sensitivity to diffusion limited quenching by a factor of 3 or more compared to most fluorophores with lifetimes below 4 ns. The long fluorescence lifetime also allows the use of time-gated detection to eliminate the interference from autofluorescence.40,52–54 (5) The modularity of the probe will allow for tuning properties of the dye component somewhat independent of the quencher group. For example, the pKa of the quencher can easily be tuned by substitution,17,18,27 while decreasing the reduction potential of the dye will increase the PCET driving force, even to the point where water will facilitate quenching.55 (6) Finally, fluorescence lifetime (τ0) and spectra of the fluorophore should be insensitive to salt and buffer variations, as is the case for phenol-DAOTA (Tables S1 and S4†).
All of the above listed properties are fulfilled by the phenol-DAOTA. However, for use in cells and other complex biological systems we will need further improvements of phenol-DAOTA. This includes introducing reactive functionalities so the probes can be conjugated to dextrans or other carrier biomolecules in order to enhance their water solubility,56 avoid undesired localization and control the probe distribution in complex biological systems.57–59
Conclusions
In this work, we have investigated the dynamic quenching of phenol-DAOTA assisted by weak bases. We have found that the quenching is directly proportional to the concentration of the bases and their base strength relative to the probe (ΔpKa) as expected for the acid–base reaction. The high sensitivity to even weak bases suggests that the quenching is driven by direct formation of the PET state via a concerted PCET process. The efficient PCET process and the long excited state lifetime of DAOTA makes the probe sensitive to mM concentrations of important metabolites such as glutamate and organic phosphates, while the simple measurements of single fluorescence lifetimes reduce complicated analysis significantly. We suggest that phenol-DAOTA constitutes a new modular probe design, where dynamic PCET quenching may be used to investigate the composition of or spatiotemporal changes to complex buffer systems, e.g., in live cells. We further discuss the optimal properties and further development of such probes.Data availability
Experimental data are available from the corresponding author upon reasonable request.Author contributions
RKJ conducted spectroscopic measurements, analyzed the TA data, and wrote the first draft with SGS who also conducted initial studies. JC conducted transient absorption measurements. BWL conceived the idea and supervised the study. All authors have contributed to the manuscript and given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Novo Nordisk Foundation (NNF20OC0062176). This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 871124 Laserlab-Europe.References
- X. Tian, L. C. Murfin, L. Wu, S. E. Lewis and T. D. James, Fluorescent small organic probes for biosensing, Chem. Sci., 2021, 12, 3406–3426 RSC.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, The Fluorescent Toolbox for Assessing Protein Location and Function, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- J. Yin, L. Huang, L. Wu, J. Li, T. D. James and W. Lin, Small molecule based fluorescent chemosensors for imaging the microenvironment within specific cellular regions, Chem. Soc. Rev., 2021, 50, 12098–12150 RSC.
- T. Ueno and T. Nagano, Fluorescent probes for sensing and imaging, Nat. Methods, 2011, 8, 642–645 CrossRef CAS PubMed.
- A. P. de Silva and R. A. D. D. Rupasinghe, A new class of fluorescent pH indicators based on photo-induced electron transfer, J. Chem. Soc. Chem. Commun., 1985, 1669, 10.1039/C39850001669.
- J. B. Grimm and L. D. Lavis, Caveat fluorophore: an insiders' guide to small-molecule fluorescent labels, Nat. Methods, 2022, 19, 149–158 CrossRef CAS PubMed.
- J. V. Jun, D. M. Chenoweth and E. J. Petersson, Rational design of small molecule fluorescent probes for biological applications, Org. Biomol. Chem., 2020, 18, 5747–5763 RSC.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Specific Covalent Labeling of Recombinant Protein Molecules Inside Live Cells, Science, 1998, 281, 269–272 CrossRef CAS PubMed.
- P. Shieh, V. T. Dien, B. J. Beahm, J. M. Castellano, T. Wyss-Coray and C. R. Bertozzi, CalFluors: A Universal Motif for Fluorogenic Azide Probes across the Visible Spectrum, J. Am. Chem. Soc., 2015, 137, 7145–7151 CrossRef CAS PubMed.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, A new generation of Ca2+ indicators with greatly improved fluorescence properties, J. Biol. Chem., 1985, 260, 3440–3450 CrossRef CAS PubMed.
- A. S. Klymchenko, Solvatochromic and Fluorogenic Dyes as Environment-Sensitive Probes: Design and Biological Applications, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- H. Niu, J. Liu, H. M. O'Connor, T. Gunnlaugsson, T. D. James and H. Zhang, Photoinduced electron transfer (PeT) based fluorescent probes for cellular imaging and disease therapy, Chem. Soc. Rev., 2023, 52, 2322–2357 RSC.
- B. Daly, J. Ling and A. P. de Silva, Current developments in fluorescent PET (photoinduced electron transfer) sensors and switches, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- I. D. Johnson, Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies, Life Technologies Corporation, 2010 Search PubMed.
- A. Minta, J. P. Y. Kao and R. Y. Tsien, Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores, J. Biol. Chem., 1989, 264, 8171–8178 CrossRef CAS PubMed.
- M. Oheim, M. van't Hoff, A. Feltz, A. Zamaleeva, J.-M. Mallet and M. Collot, New red-fluorescent calcium indicators for optogenetics, photoactivation and multi-color imaging, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843, 2284–2306 CrossRef CAS PubMed.
- C. G. Frankær, M. Rosenberg, M. Santella, K. J. Hussain, B. W. Laursen and T. J. Sørensen, Tuning the pKa of a pH Responsive Fluorophore and the Consequences for Calibration of Optical Sensors Based on a Single Fluorophore but Multiple Receptors, ACS Sens., 2019, 4, 764–773 CrossRef PubMed.
- D. Aigner, S. A. Freunberger, M. Wilkening, R. Saf, S. M. Borisov and I. Klimant, Enhancing Photoinduced Electron Transfer Efficiency of Fluorescent pH-Probes with Halogenated Phenols, Anal. Chem., 2014, 86, 9293–9300 CAS.
- A. Csomos, B. Kontra, A. Jancsó, G. Galbács, R. Deme, Z. Kele, B. J. Rózsa, E. Kovács and Z. Mucsi, A Comprehensive Study of the Ca2+ Ion Binding of Fluorescently Labelled BAPTA Analogues, Eur. J. Org Chem., 2021, 2021, 5248–5261 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer-Verlag New York Inc., New York, 3rd revised edn, 2006 Search PubMed.
- P. J. Del Nido, P. Glynn, P. Buenaventura, G. Salama and A. P. Koretsky, Fluorescence measurement of calcium transients in perfused rabbit heart using rhod 2, Am. J. Physiol., 1998, 274, H728–H741 CAS.
- F.-J. Meyer-Almes, Fluorescence lifetime based bioassays, Methods Appl. Fluoresc., 2017, 5, 042002 CrossRef PubMed.
- V. Untiet, P. Kovermann, N. J. Gerkau, T. Gensch, C. R. Rose and C. Fahlke, Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation, Glia, 2017, 65, 388–400 Search PubMed.
- H. Kaneko, I. Putzier, S. Frings, U. B. Kaupp and T. Gensch, Chloride accumulation in mammalian olfactory sensory neurons, J. Neurosci., 2004, 24, 7931–7938 CrossRef CAS PubMed.
- T. Gensch, V. Untiet, A. Franzen, P. Kovermann and C. Fahlke, in Advanced Time-Correlated Single Photon Counting Applications, ed. W. Becker, Springer International Publishing, Cham, 2015, pp. 189–211, DOI:10.1007/978-3-319-14929-5_4.
- K. Funk, A. Woitecki, C. Franjic-Würtz, T. Gensch, F. Möhrlen and S. Frings, Modulation of Chloride Homeostasis by Inflammatory Mediators in Dorsal Root Ganglion Neurons, Mol. Pain, 2008, 4, 32 CrossRef PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, Proton-Coupled Electron Transfer Guidelines, Fair and Square, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- J. C. Lennox, D. A. Kurtz, T. Huang and J. L. Dempsey, Excited-State Proton-Coupled Electron Transfer: Different Avenues for Promoting Proton/Electron Movement with Solar Photons, ACS Energy Lett., 2017, 2, 1246–1256 CrossRef CAS.
- M. Rosenberg, A. K. R. Junker, T. J. Sørensen and B. W. Laursen, Fluorescence pH Probes Based on Photoinduced Electron Transfer Quenching of Long Fluorescence Lifetime Triangulenium Dyes, ChemPhotoChem, 2019, 3, 233–242 CrossRef CAS.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli, Nat. Chem. Biol., 2009, 5, 593–599 CrossRef CAS PubMed.
- R. I. Cukier, On the quencher concentration dependence of fluorescence quenching: the role of solution dielectric constant and ionic strength, J. Am. Chem. Soc., 1985, 107, 4115–4117 CrossRef CAS.
- M. D. Newsham, R. I. Cukier and D. G. Nocera, Contribution of long-range Coulomb interactions to bimolecular luminescence quenching reactions, J. Phys. Chem., 1991, 95, 9660–9666 CrossRef CAS.
- M. Eigen, Kinetics of proton transfer processes, Discuss. Faraday Soc., 1965, 39, 7 Search PubMed.
- B. Zelent, J. M. Vanderkooi, R. G. Coleman, I. Gryczynski and Z. Gryczynski, Protonation of Excited State Pyrene-1-Carboxylate by Phosphate and Organic Acids in Aqueous Solution Studied by Fluorescence Spectroscopy, Biophys. J., 2006, 91, 3864–3871 CAS.
- M. Eigen, Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis. Part I: Elementary Processes, Angew Chem. Int. Ed. Engl., 1964, 3, 1–19 Search PubMed.
- M. Eigen, W. Kruse, G. Maass and L. Demaeyer, Rate Constants of Protolytic Reactions in Aqueous Solution, Prog. React. Kinet. Mech., 1964, 2, 278–318 Search PubMed.
- For simple excited state acids (not involving PCET)
 can be evaluated by the Förster cycle, ref. 53. Where red shift of the anion absorption/emission spectra represent the additional stabilization of the base form in the excited state and thus in-creased acidity.
can be evaluated by the Förster cycle, ref. 53. Where red shift of the anion absorption/emission spectra represent the additional stabilization of the base form in the excited state and thus in-creased acidity. - M. Kuss-Petermann and O. S. Wenger, Photoacid Behavior versus Proton-Coupled Electron Transfer in Phenol–Ru(bpy)32+ Dyads, J. Phys. Chem. A, 2013, 117, 5726–5733 CAS.
- P. A. Summers, B. W. Lewis, J. Gonzalez-Garcia, R. M. Porreca, A. H. M. Lim, P. Cadinu, N. Martin-Pintado, D. J. Mann, J. B. Edel, J. B. Vannier, M. K. Kuimova and R. Vilar, Visualising G-quadruplex DNA dynamics in live cells by fluorescence lifetime imaging microscopy, Nat. Commun., 2021, 12, 162 CAS.
- R. M. Rich, D. L. Stankowska, B. P. Maliwal, T. J. Sørensen, B. W. Laursen, R. R. Krishnamoorthy, Z. Gryczynski, J. Borejdo, I. Gryczynski and R. Fudala, Elimination of autofluorescence background from fluorescence tissue images by use of time-gated detection and the AzaDiOxaTriAngulenium (ADOTA) fluorophore, Anal. Bioanal. Chem., 2013, 405, 2065–2075 CAS.
- J. M. Paredes, D. Miguel, S. Resa, M. C. Gonzalez-Garcia, Y. Diaz-Torres, J. M. Cuerva and L. Crovetto, Design, synthesis and photophysical studies of improved xanthene dye to detect acetate, J. Photochem. Photobiol., A, 2019, 371, 300–305 CAS.
- V. Puente-Muñoz, J. M. Paredes, S. Resa, A. M. Ortuño, E. M. Talavera, D. Miguel, J. M. Cuerva and L. Crovetto, Efficient acetate sensor in biological media based on a selective Excited State Proton Transfer (ESPT) reaction, Sens. Actuators, B, 2017, 250, 623–628 Search PubMed.
- S. Resa, A. Orte, D. Miguel, J. M. Paredes, V. Puente-Muñoz, R. Salto, M. D. Giron, M. J. Ruedas-Rama, J. M. Cuerva, J. M. Alvarez-Pez and L. Crovetto, New Dual Fluorescent Probe for Simultaneous Biothiol and Phosphate Bioimaging, Chem.–Eur. J., 2015, 21, 14772–14779 CAS.
- J. M. Paredes, L. Crovetto, A. Orte, J. M. Alvarez-Pez and E. M. Talavera, Influence of the solvent on the ground- and excited-state buffer-mediated proton-transfer reactions of a xanthenic dye, Phys. Chem. Chem. Phys., 2011, 13, 1685–1694 CAS.
- O. S. Wenger, Proton-coupled electron transfer with photoexcited ruthenium(II), rhenium(I), and iridium(III) complexes, Coord. Chem. Rev., 2015, 282–283 Search PubMed.
- J. L. Ross, The Dark Matter of Biology, Biophys. J., 2016, 111, 909–916 CAS.
- S. Poddar and M. Levitus, Buffer-Dependent Photophysics of 2-Aminopurine: Insights into Fluorescence Quenching and Excited-State Interactions, J. Phys. Chem. B, 2024, 128, 2640–2651 CAS.
- K. A. Paterson, J. Arlt and A. C. Jones, Dynamic and static quenching of 2-aminopurine fluorescence by the natural DNA nucleotides in solution, Methods Appl. Fluoresc., 2020, 8, 025002 CAS.
- R. Nandi and N. Amdursky, The Dual Use of the Pyranine (HPTS) Fluorescent Probe: A Ground-State pH Indicator and an Excited-State Proton Transfer Probe, Acc. Chem. Res., 2022, 55, 2728–2739 CAS.
- I. Bora, S. A. Bogh, M. Rosenberg, M. Santella, T. J. Sørensen and B. W. Laursen, Diazaoxatriangulenium: synthesis of reactive derivatives and conjugation to bovine serum albumin, Org. Biomol. Chem., 2016, 14, 1091–1101 CAS.
- P. M. Badri, F. Rafal, R. Sangram, K. Rutika, S. Thomas Just, L. Bo Wegge, G. Zygmunt and G. Ignacy, Long-lived bright red emitting azaoxa-triangulenium fluorophores, PLoS One, 2013, e63043, DOI:10.1371/journal.pone.0063043.
- M. Y. Berezin and S. Achilefu, Fluorescence Lifetime Measurements and Biological Imaging, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- L. Kacenauskaite, N. Bisballe, R. Mucci, M. Santella, T. Pullerits, J. Chen, T. Vosch and B. W. Laursen, Rational Design of Bright Long Fluorescence Lifetime Dyad Fluorophores for Single Molecule Imaging and Detection, J. Am. Chem. Soc., 2021, 143, 1377–1385 CrossRef CAS PubMed.
- M. Rosenberg, K. R. Rostgaard, Z. Liao, A. O. Madsen, K. L. Martinez, T. Vosch and B. W. Laursen, Design, synthesis, and time-gated cell imaging of carbon-bridged triangulenium dyes with long fluorescence lifetime and red emission, Chem. Sci., 2018, 9, 3122–3130 RSC.
- R. K. Jakobsen and B. W. Laursen, New Principle for Simple Water Detection using Fluorescence Lifetime Triangulenium Probes, ChemPhotoChem, 2024, 8, e202300215 CrossRef CAS.
- N. Bisballe and B. W. Laursen, What is Best Strategy for Water Soluble Fluorescence Dyes?—A Case Study Using Long Fluorescence Lifetime DAOTA Dyes, Chem.–Eur. J., 2020, 26, 15969–15976 CrossRef CAS PubMed.
- R. Mehvar, Dextrans for targeted and sustained delivery of therapeutic and imaging agents, J. Controlled Release, 2000, 69, 1–25 CAS.
- C. L. Slayman, V. V. Moussatos and W. W. Webb, Endosomal Accumulation of pH Indicator Dyes Delivered as Acetoxymethyl Esters, J. Exp. Biol., 1994, 196, 419–438 CAS.
- G. Despras, A. I. Zamaleeva, L. Dardevet, C. Tisseyre, J. G. Magalhaes, C. Garner, M. De Waard, S. Amigorena, A. Feltz, J.-M. Mallet and M. Collot, H-Rubies, a new family of red emitting fluorescent pH sensors for living cells, Chem. Sci., 2015, 6, 5928–5937 RSC.

Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00326a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |