
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile post-synthesis of isomeric covalent organic frameworks via precise pore surface engineering†
Yuhao
Liu
*a,
Yaze
Chen
a,
Ke
Shi
a,
Haijiao
Peng
a and
Chao
Lu
 *ab
*ab
aPingyuan Laboratory, College of Chemistry, Zhengzhou University, No. 100 Science Avenue, 450001, Zhengzhou, China. E-mail: yuhaoliu@zzu.edu.edu.cn; luchao@mail.buct.edu.cn
bState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, No. 15 Beisanhuan East Road, 100029, Beijing, China
First published on 15th April 2025
Abstract
Isomeric covalent organic frameworks (COFs) have developed dramatically due to having the same chemical composition but distinct physicochemical characteristics. However, exploring novel synthetic strategies for the precise construction of COFs with isomeric pore microenvironments remains challenging and in its infancy. In this contribution, we have developed a controllable, simple, and efficient post-synthesis modification strategy to design isomeric COFs via precise pore surface engineering. The as-prepared isomeric COFs showed comparable crystallinity and porosity but significantly different pore microenvironments. Interestingly, the isomeric moieties endow the isomeric COFs with specific capture performances and excellent recycling ability. The specific interactions between these isomeric COFs and guests are verified by fluorescence spectra and theoretical calculation. This study will open a novel avenue for the construction of isomeric COFs and facilitate the development of isomeric COFs with specific properties.
Introduction
Covalent organic frameworks (COFs) are a kind of crystalline porous polymer formed through covalent bonding of organic building units.1–5 Isomeric COFs with the same chemical composition but distinct atomic organization structures and physicochemical characteristics are one of the particularly interesting categories of COFs.6–9 The small structural differences of these isomeric COFs can lead to significant changes in their optical properties, electrical performances, and adsorption behaviors, enabling them to have potential applications in recognition,10 photocatalysis11 and adsorption.12 Although the construction of positional isomers and framework isomers has been widely studied in COFs, COFs with isomeric pore microenvironments are far less explored. Therefore, it is of great importance to design isomerized porous COF materials to widen the scope of their applications.Currently, isomeric COFs could be constructed through exchanging the locations of functional groups, employing isomeric monomers, or regulating the synthetic conditions. One method is to manipulate the reaction groups (e.g., –NH2, –CH2CN, and –CHO) of building blocks for polycondensation to construct isomeric COFs.13–16 The distinctions among these isomeric COFs are typically reflected in linkages (e.g., –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– and –NCH–; –CCH(CN)– and –CH(CN)C–). The synthesized isomeric COFs exhibit the same chemical compositions but significant differences in their photoelectric characteristics. For instance, Li and co-workers demonstrated that the photocatalytic activities of imine-linked COFs significantly depended on the orientations of the imine bond.17 Recently, Feng et al. reported that two structurally isomeric vinylene-linked cationic COF films via the Knoevenagel reaction displayed anion transport selectivity.18 Another method is to choose isomeric monomers as building units to prepare isomeric COFs.19–21 For example, Zhuang and colleagues synthesized two isomeric dual-pore COFs through azulene-based and naphthalene-based isomeric monomers with C2h and C2v symmetries which exhibited higher NO2-sensing performances.22 Meanwhile, Medina's group investigated the effect of thienothiophene sulfur positions on photophysical properties by synthesizing two isomeric COFs with distinct structures (thieno[3,2-b]thiophene and thieno[2,3-b]thiophene).23 However, the uncontrollable crystallization process, inconsistent crystallinity and porosity, and complicated design of isomeric monomers restrict wide applications of positional COF isomers and study of structure–property relationships. In addition, Dai et al. successfully prepared highly crystalline isomerized COFs with eclipsed AA stacking and staggered AB stacking by adjusting the reaction conditions.24 However, the synthesis of isomeric COFs with different stacking patterns is often accidental and irregular.9,24 Moreover, most studies have predominantly focused on the positional isomers and framework isomers of COFs, and isomeric pore microenvironments of COFs remained relatively unexplored. Therefore, it is highly desired to explore facile synthetic strategies for the precise construction of COFs with isomeric pore microenvironments.
N– and –NCH–; –CCH(CN)– and –CH(CN)C–). The synthesized isomeric COFs exhibit the same chemical compositions but significant differences in their photoelectric characteristics. For instance, Li and co-workers demonstrated that the photocatalytic activities of imine-linked COFs significantly depended on the orientations of the imine bond.17 Recently, Feng et al. reported that two structurally isomeric vinylene-linked cationic COF films via the Knoevenagel reaction displayed anion transport selectivity.18 Another method is to choose isomeric monomers as building units to prepare isomeric COFs.19–21 For example, Zhuang and colleagues synthesized two isomeric dual-pore COFs through azulene-based and naphthalene-based isomeric monomers with C2h and C2v symmetries which exhibited higher NO2-sensing performances.22 Meanwhile, Medina's group investigated the effect of thienothiophene sulfur positions on photophysical properties by synthesizing two isomeric COFs with distinct structures (thieno[3,2-b]thiophene and thieno[2,3-b]thiophene).23 However, the uncontrollable crystallization process, inconsistent crystallinity and porosity, and complicated design of isomeric monomers restrict wide applications of positional COF isomers and study of structure–property relationships. In addition, Dai et al. successfully prepared highly crystalline isomerized COFs with eclipsed AA stacking and staggered AB stacking by adjusting the reaction conditions.24 However, the synthesis of isomeric COFs with different stacking patterns is often accidental and irregular.9,24 Moreover, most studies have predominantly focused on the positional isomers and framework isomers of COFs, and isomeric pore microenvironments of COFs remained relatively unexplored. Therefore, it is highly desired to explore facile synthetic strategies for the precise construction of COFs with isomeric pore microenvironments.
To advance isomeric COF research, a facile post-synthesis modification strategy was proposed to construct isomeric COFs with tunable pore microenvironments via precise pore surface engineering (Scheme 1). In this work, isomeric COFs with comparable crystallinity and porosity but significantly different pore microenvironments were constructed by introducing phenylboric acid isomers into the pores of parent donor–acceptor (D–A) COFs via the aza-Diels–Alder cycloaddition reaction. The introduction of phenylboric acid isomers not only improved the stability, crystallinity, and fluorescence characteristics of the parent COFs, but also reduced the pKa value of phenylboric acid, which was conducive to the selective adsorption of diols with similar structures. These boric acid-functionalized isomeric COFs could specifically capture nucleoside guest molecules due to their distinct pore characteristics and strong affinity with diols. In addition, the specific capture performances of these isomeric COFs were verified by fluorescence spectra and theoretical calculation. This novel strategy eliminates the requirement for the complicated isomeric monomer design and the necessity to consider the influence of synthesis conditions on the COFs' structures. On the other hand, it opens new avenues for investigating the structure–property relationships of COFs with isomeric pore microenvironments.
 | ||
| Scheme 1 Previous studies and present work on general strategies for the design and synthesis of isomeric COFs. | ||
Results and discussion
Synthesis and characterization
The D–A-COF was designed and constructed by the Schiff-base reaction at 120 °C for 72 h between monomers 2,4,6-tris(4-aminophenyl)triazine and tris(4-formylphenyl)amine, using 1-butanol and o-dichlorobenzene as a mixed solvent and CH3COOH (3 M) as the catalyst. As shown in Fig. 1, boric acid-functionalized COFs (COF-o-BA, COF-m-BA or COF-p-BA) were prepared by linking the boronic acid monomers (2-ethynylphenyl)boronic acid, (3-ethynylphenyl)boronic acid or (4-ethynylphenyl)boronic acid) with the D–A-COF framework under different synthesis conditions based on the aza-Diels–Alder cyclization reaction (Fig. S1 and Table S1†). This post-synthesis modification method endowed the isomeric COFs with different orientations of boronic acid groups in the pores. At the same time, the formation of a quinoline bond led to improved acid stability and crystallinity (Fig. S2†), because of the enhanced conjugation effect. | ||
| Fig. 1 Synthesis of isomeric COFs with tunable pore microenvironments via pore surface engineering by using phenylboronic acid. | ||
The refined crystalline structures of D–A-COF (Fig. S3†) and COF-BA (Fig. 2a–c) were determined by using the powder X-ray diffraction (PXRD) patterns. As depicted in Fig. 2d, the D–A-COF exhibited a prominent peak at 4.50° (100), along with relatively weaker peaks at 7.82° (100), 9.02° (200), 11.95° (210), 15.92° (310) and 22.91° (001). A full-profile Pawley refinement was performed to afford the unit cell parameters (a = b = 22.6 Å, c = 3.95 Å; α = β = 90°, γ = 120°, Rp = 0.23%, and Rwp = 0.34% for D–A-COF), which led to satisfactorily low residual values and acceptable profile differences. Meanwhile, the six main diffraction peaks and diffraction angles showed negligible change, suggesting that the crystal structures of the D–A-COF after post-modification were similar (Fig. 2e). The presence of boronic acid in COF-BA was confirmed by a variety of characterization techniques. Based on the Fourier transform infrared (FT-IR) spectra of these COFs (Fig. 2f), we could observe that COF-BA had four new peaks at 1587 cm−1, 1353 cm−1, 1180 cm−1 and 770 cm−1, respectively. The peak at 1587 cm−1 represented the stretch of quinoline generated during the aza-Diels–Alder reaction.25,26 The other peaks were consistent with the earlier reports, which might be assigned to the B–O stretching vibration, C–B stretching vibration, and –OH out-of-plane bending vibration.27 All of the above results suggested that the post-modifications were successful. The nitrogen adsorption–desorption isotherms at 77 K were obtained to explore the porosity of D–A-COF and COF-BA (Fig. 2g). The Brunauer–Emmett–Teller (BET) specific surface areas of the D–A-COF, COF-o-BA, COF-m-BA and COF-p-BA were 1929.9 m2 g−1, 1311.5 m2 g−1, 1172.2 m2 g−1 and 1381.4 m2 g−1, respectively. The drastic decrease in specific surface area was mainly due to the introduction of boronic acid in their pores. In addition, the pore size distribution of the structure was estimated using a quenched solid density functional theory (QSDFT) model to fit the adsorption branch of the isotherm. As shown in Fig. 2h, the pore size of the D–A-COF was 2.05 nm, whereas COF-o-BA, COF-m-BA and COF-p-BA exhibited pore sizes of 1.85 nm. Meanwhile, it was observed that the pore volume of the D–A-COF was 0.933 cm3 g−1, while the pore volume of COF-o-BA, COF-m-BA and COF-p-BA was only 0.688, 0.666, and 0.713 cm3 g−1. To sum up, compared with the D–A-COF, the specific surface area, pore size, and pore volume of all boronic acid modified COFs are decreased. This was primarily due to the addition of phenylboronic acid that blocked the pore channel. Solid-state 13C NMR was employed to further verify the incorporation of the boric acid group into D–A-COF through post-synthesis modification (Fig. 2i and S4†). COF-BA and D–A-COF exhibited the same spectral signal at ∼173 ppm, which was related to the carbon of triazine, indicating that the pristine structure of COFs was preserved after modification.28,29 The peak shape and strength of the imine bond at ∼161 ppm were significantly different after cycloaddition due to the generation of quinoline by the aza-Diels–Alder reaction. Moreover, a novel signal was observed in the COF-BA spectrum at around 142 ppm. This signal was identified as that of the carbons originating from phenylboronic acid and suggested the introduction of the functional group. In addition, the X-ray photoelectron spectroscopy (XPS) analyses were also performed before and after D–A-COF modification. As shown in Fig. S5,† a new signal appeared at about 193.9 eV for COF-BA, which was the signal of the B element. The successful conversion of imine to quinoline in COF-BA was demonstrated by an additional peak at about 402.1 eV. Moreover, two peaks emerged at about 399.7 and 398.4 eV in D–A-COF and COF-BA, which could be attributed to NC and N–C, respectively.27,30 These results indicated that D–A-COF was significantly transformed into COF-BA through the cycloaddition reaction. Thermal gravimetric analysis (TGA) revealed that the mass of D–A COF started to decline after 500 °C, whereas that of modified COF-BA began to decline at 200 °C due to the presence of phenylboronic acid groups (Fig. S6†). The above results demonstrated that the boric acid groups were successfully incorporated into D–A-COF by post-modification.
 | ||
| Fig. 2 Top views of the corresponding refined 2D crystal structures of COF-o-BA (a), COF-m-BA (b), and COF-p-BA (c). PXRD patterns of D–A-COF with the experimental profiles in red, Pawley-refined profiles in black, calculated profiles in dark green, and the differences between the experimental and refined PXRD patterns in dark blue (d). PXRD patterns (e), FTIR spectra (f), N2 adsorption–desorption isotherms (77 K) (g), and pore size distribution profiles (h) of D–A-COF and COF-BA. Solid-state 13C NMR spectra of D–A-COF and COF-o-BA (i). | ||
The morphology of D–A-COF and COF-BA could be observed by using scanning electron microscopy (SEM) micrographs and high-resolution transmission electron microscopy (HRTEM) images. The SEM results showed that the morphology of COF-BA was still irregular and sheet-like after modification (Fig. S7†). However, compared with D–A-COF, COF-BA had obvious lattice diffraction fringes due to the improved crystallinity via the aza-Diels–Alder reaction (Fig. 3a–c and S8†). As illustrated in Fig. 3a–c, the ordered porous structure of COF-BA can be clearly seen. The pore size from HRTEM of COF-o-BA, COF-m-BA and COF-p-BA is 1.78 nm, 1.80 nm, and 1.76 nm, respectively, which matches well with the PXRD and nitrogen adsorption results. Furthermore, the uniform distribution of the boron element was demonstrated by the SEM elemental mapping (Fig. S9†) and TEM elemental mapping of COF-BA (Fig. 3d–f), further indicating the effective incorporation of the boric acid group in the D–A-COF. The energy dispersive X-ray spectroscopy (EDS) analyses showed that the B content of the three isomeric COF-BA was similar (Table S2†).
 | ||
| Fig. 3 HRTEM images of COF-o-BA (a), COF-m-BA (b), and COF-p-BA (c). TEM elemental mapping images of COF-o-BA (d), COF-m-BA (e), and COF-p-BA (f). Fluorescence spectra of COF-m-BA at different pH values (2.0 to 10.0) (λex = 300 nm) (g). Fluorescence titration plots of D–A-COF before and after functionalization (h). The pKa values of the corresponding isomeric precursor (i). | ||
Selective adsorption performance
The affinity properties of boric acid materials are determined by both the boric acid ligand and matrix. The lower the pKa of the boric acid ligand, the stronger its affinity ability, which is more conducive to its application in practical samples.31,32 We then investigate the pKa values of D–A-COF and COF-BA by using fluorescence spectra at different pH values (Fig. 3g and S10†). The pKa values were deduced from the intercept of linear plots of the Henderson–Hasselbalch-type mass action equation [log[(Fmax − F)/(F − Fmin)] versus pH values, where F was the fluorescence intensity of the COFs under different pH conditions, and Fmax and Fmin were the maximum and minimum limiting values of F, respectively.33 The pKa values for D–A-COF, COF-o-BA, COF-m-BA and COF-p-BA were estimated to be 5.27, 4.66, 3.64, and 3.82, respectively, based on the fluorescence titration plots (Fig. 3h and S11†). The introduction of phenylboronic acid isomers into the channel of D–A-COF can make them have a lower pKa value. At the same time, the pKa value of COF-BA is lower than that of their corresponding isomeric precursors ((2-ethynylphenyl)boronic acid = 8.36 ± 0.58, (3-ethynylphenyl)boronic acid = 7.87 ± 0.10 or (4-ethynylphenyl)boronic acid = 7.97 ± 0.10) (Fig. 3i). The different boric acid affinity in the pores of COFs could facilitate the selective uptake of the diol compound.The reversible covalent interaction between these COF-BA and the diol compound is shown in Fig. 4a. When the pH value of the solution is ≥pKa (boronate affinity material), the boric acid in the COFs first reacts with the OH− in the solution to form the tetragonal boronate anion (sp3), and then the boronate anion can covalently bond with the diol molecules to form five-membered or six-membered cyclic esters. When the pH value of the solution is <pKa (boronate affinity material), the hybrid of boron atoms becomes the original boric acid (sp2), so that the cyclic ester is dissociated and the diol molecules are released. Nucleosides, as components of nucleic acids, play an important role in the biological activity of RNA. Due to the presence of diol groups in the nucleoside structure, the nucleosides can be effectively isolated and enriched from biological samples by using boron-affinity materials, so that they can be further analyzed and identified.34,35 Thus, comparison investigations were conducted to determine the adsorption capacities between COF-BA and different nucleoside substances (cytidine, inosine, guanosine, uridine, adenosine, and thymidine) (Fig. 4b) in NaHCO3/NaOH buffer solution (pH 10.0). Fig. 4c and S12–S17† illustrate the greatest adsorption capabilities of COF-BA for the six nucleoside substances. For these nucleosides, all the boric acid-functionalized COFs showed the largest adsorption to adenosine. The highest adsorption capacity of COF-p-BA, COF-m-BA, and COF-o-BA for adenosine was 4.98, 4.47, and 2.80 mg g−1, respectively. Because of the differences in boric acid microenvironments in the COF pores, COF-o-BA and COF-p-BA showed adsorption selectivity for adenosine/uridine and adenosine/thymidine, with selectivity factors of 8.8 and 7.2, respectively. In addition, for nucleoside substances with a cis-o-diol unit, the order of maximum adsorption was COF-p-BA > COF-m-BA > COF-o-BA. However, the order of maximum adsorption was reversed for thymidine without a cis-o-diol unit. This may be because the nucleoside substances containing a cis-o-diol unit are more easily bound to boric acid inside the COF pores by covalent bonding. We also compared the adsorption capacity of D–A-COF (Fig. S18†). The adsorption capacity was low possibly due to the physical adsorption of the pores. Moreover, the adsorption performances of COFs for the nucleoside analogs without diol (didanosine and adenine hydrochloride) were evaluated (Fig. S19 and S20†). The adsorption capacity of both D–A-COF and COF-BA was also low, and there was no obvious difference between that of D–A-COF and COF-BA, indicating that the interaction of boric acid and diol was important for this adsorption process. The adsorption process between COF-BA and adenosine was examined utilizing the pseudo-first order and pseudo-second order kinetic models (Fig. 4d, S21, and S22†). Adenosine was adsorbed onto COF-BA incredibly quickly during the first 3 hours. After that, the rate of adsorption progressively decreased until the adsorption was complete after 72 hours. The adenosine adsorption process onto COF-BA was found to be well-fitted by the pseudo-second order kinetic model, as indicated by the correlation coefficients R2 (0.999) of the fitting findings. These results suggested that chemical adsorption was the primary type of adsorption process.36,37 The desorption process and cycle experimental results are shown in Fig. S23 and S24,† and 4e. The results showed that adenosine/COF-p-BA could be dissociated in HOAc/NaOAc buffer solution (pH 3.6), and 97% of adenosine was desorbed from COF-BA within 8 hours (Fig. S25†). Meanwhile, COF-BA could maintain their structure and crystallinity in the NaHCO3/NaOH buffer solution (pH 10.0) and HOAc/NaOAc buffer solution (pH 3.6) (Fig. S25†). After the adsorbent recovered, it was used to adsorb adenosine at the same concentration. The adsorption capacity of COF-p-BA remained high even after six cycles. Moreover, COF-p-BA still maintained their original structure, morphology, good crystallinity and porosity after six runs (Fig. S26†).
 | ||
| Fig. 4 (a) Operation principle of boric acid-functionalized COFs and diols. (b) Structures of the examined diols. (c) The maximum adsorption capacities of COF-BA toward six diols. (d) The adsorption kinetics curves of COF-BA toward adenosine. (e) Recycling results of COF-p-BA for adenosine after adsorption–desorption. | ||
Mechanism investigation
The interaction process between COFs and nucleoside substances was investigated by using the fluorescence spectrum due to the excellent photophysical properties of D–A-COF. As shown in Fig. 5a, the UV/vis absorption peaks of COF-BA were redshifted by 20–40 nm after modification due to the enhanced conjugation effect. Specifically, the emission peak of COF-BA was obviously different from that of D–A-COF under 300 nm excitation (Fig. 5b). Time-resolved fluorescence spectroscopy revealed that COF-o-BA, COF-m-BA, and COF-p-BA had a lifetime (τ) of 2.23, 2.75 and 2.71 ns, which was slightly larger than the 1.97 ns of D–A-COF (Fig. 5c). Notably, the fluorescence emission peak intensity of COF-BA reduced gradually with the addition of NaOH solution (Fig. 5d and S27†). However, the fluorescence emission peak intensity of D–A-COF changed irregularly with the addition of NaOH solution (Fig. S27†). As can be seen from Fig. 5d, the fluorescence spectrum of COF-BA reached equilibrium after adding 70–80 μL 0.15 M NaOH to form the adduct COF–B(OH)3. Thus, the interaction of COF-BA with nucleosides was studied after the equilibrium reaction with NaOH solution. As shown in Fig. S28 and S29,† upon treatment of COF-o-B(OH)3 with nucleosides, a noticeable enhancement in emission at ∼580 nm was observed with different nucleosides, whereas the proportion of increased fluorescence intensity varied. Following the addition of 175 μL of nucleoside solutions, the fluorescence intensities of COF-o-B(OH)3 dispersions increased by 1.32–1.87 times. Fig. 5e displays the respective Benesi–Hildebrand plots. The determined association constants (KBH) of COF-o-B(OH)3 were 1933 M−1, 2323 M−1, 3981 M−1, 671 M−1, 8122 M−1 and 2766 M−1 for cytidine, inosine, guanosine, uridine, adenosine and thymidine, respectively. The selectivity factor [SF = KBH(adenosine)/KBH(uridine)] was 12.10 for adenosine and uridine. In addition, the fluorescence emission of COF-m-B(OH)3 could also be selectively intensified by nucleosides, with an SF of 23.33 for adenosine and thymidine (Fig. 5f, S30, and S31†). Under otherwise identical conditions, COF-p-B(OH)3 could detect adenosine and thymidine, with a relatively higher SF of 46.13 (Fig. 5f, S32, and S33†). This selectivity was consistent with the results of adsorption. The selective enhancement of fluorescence intensity of COF–B(OH)3 might be due to the static enhancement caused by the combination between the diol of nucleoside and boric acid functional groups of COF–B(OH)3. The fluorescence lifetimes (τ) of COF-p-B(OH)3 before and after bonding with adenosine and thymidine were 2.64 vs. 2.58 and 2.62 ns, respectively (Fig. S34†). These results indicated the static nature of the interactions between COFs and nucleoside, which was consistent with the literature reports.38,39 The FT-IR spectra were also obtained to understand the interaction process between COF-BA and nucleoside substances. However, there was no significant difference after adsorption and desorption with nucleoside substances by COF-BA (Fig. S35†).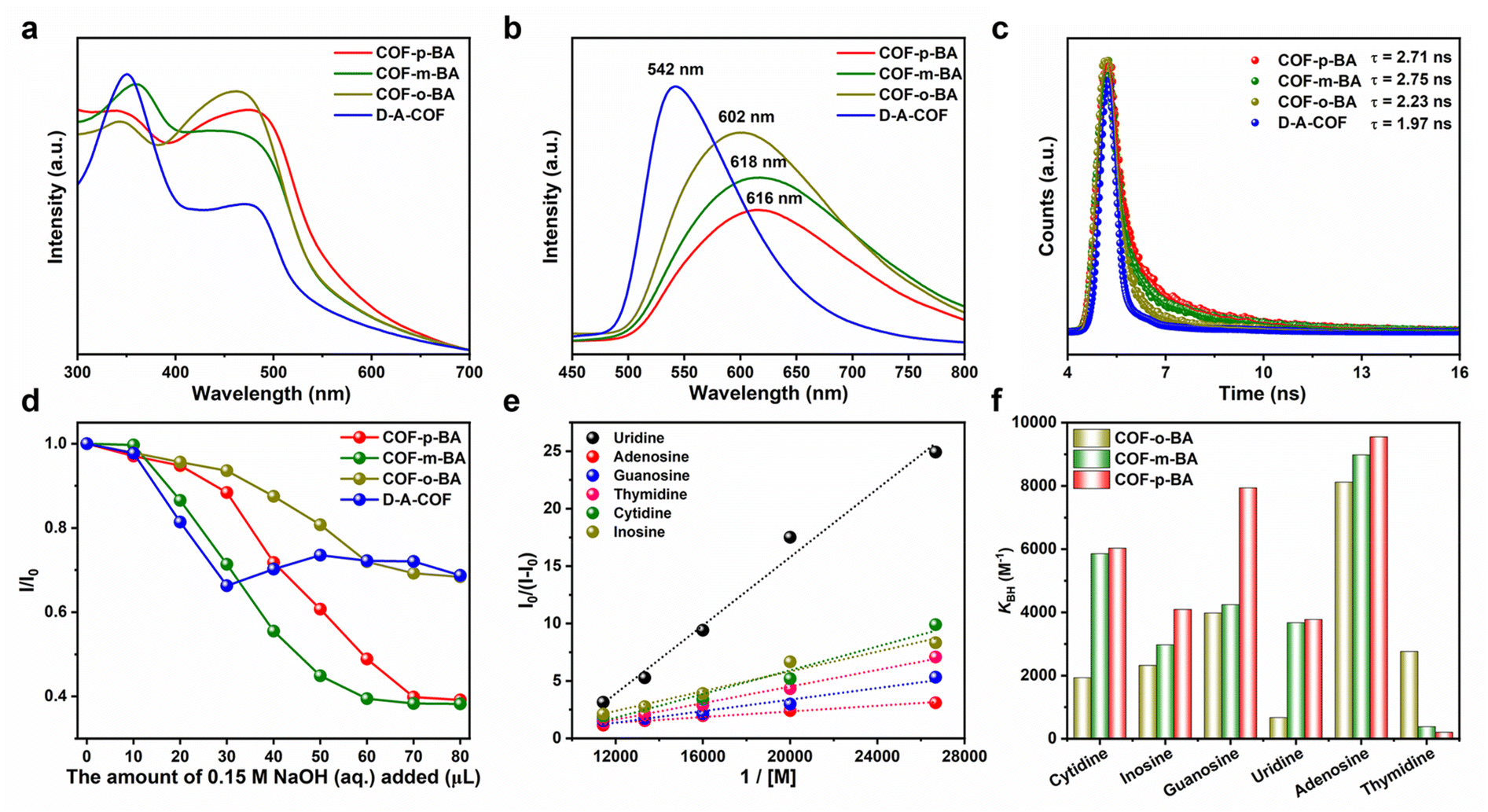 | ||
| Fig. 5 UV/vis absorption spectra (a), fluorescence emission spectra (b), and fluorescence decay curves (c) of D–A-COF and COF-BA. (d) The fluorescence quenching ratio of D–A-COF and COF-BA with increased amounts of NaOH (aq.). (e) The Benesi–Hildebrand plots of the fluorescence emissions of COF-o-BA enhanced by nucleoside substances. (f) The determined association constants of COF-BA for nucleoside substances. | ||
To further elucidate the interactions between COF–B(OH)3 and nucleoside substances and reveal the mechanism of the COF–B(OH)3-based system, preliminary investigations were conducted into the host–guest interactions using energy-minimized density functional theory (DFT) calculation. To explore the electronic distributions of COF–B(OH)3 and nucleoside substances, the electrostatic potentials (ESPs) of the COF-o-B(OH)3 fragment, the COF-p-B(OH)3 fragment, adenosine, and thymidine were calculated. The ESP results showed that the boric acid sites of COF-o-B(OH)3 and COF-p-B(OH)3 had an electron-deficient nature, which was favorable for the interactions of electron-rich molecules (Fig. 6a).40 Based on the above results, the possible bonding models were shown in Fig. 6b–d. A host model comprising one repeating unit of COF-o-B(OH)3 or COF-p-B(OH)3 was utilized for the calculation. The energies were evaluated by introducing a single adenosine or thymidine molecule into the channel of COF–B(OH)3 and conducting energy minimization for these host–guest structures. The optimized configurations revealed that both adenosine and thymidine are accommodated within the open channel of COF–B(OH)3, stabilized by the B–O bonds between the COF–B(OH)3 (host, COF-o-B(OH)3 or COF-p-B(OH)3) and hydroxyl groups (guest, adenosine or thymidine). Based on the refined host–guest structures, the calculated energy difference (ΔE) for adenosine@COF-o-B(OH)3 and adenosine@COF-p-B(OH)3 was found to be −18.32 and −32.70 kcal mol−1, respectively. These negative values indicated the favorable bonding of adenosine within the nanochannels of COF–B(OH)3. In addition, compared with COF-o-B(OH)3, the ΔE for COF-p-B(OH)3 was more negative. This suggested stronger host–guest binding between COF-p-B(OH)3 and adenosine, resulting in highly efficient adsorption. For COF-p-B(OH)3, the ΔE of thymidine@COF-p-B(OH)3 was more positive than that of adenosine@COF-p-B(OH)3, which was consistent with the experimental results that the adsorption of COF-p-BA to adenosine was greater than that of thymidine. Theoretical calculations strongly confirmed the experimental results of adsorption and fluorescence sensing, indicating that the porous structure and the isomeric boric acid microenvironment of COF-BA were crucial for the selective adsorption of diols.
 | ||
| Fig. 6 The electrostatic potential of the COF-o-B(OH)3 fragment, the COF-p-B(OH)3 fragment, adenosine, and thymidine (a). Top views of the host–guest structures of adenosine@COF-o-B(OH)3 (b), adenosine@COF-p-B(OH)3 (c) and thymidine@COF-p-B(OH)3 (d) based on energy-minimized DFT calculations. The indicated values represent the relative binding energies. | ||
Conclusions
In conclusion, we have proposed a controllable, simple, and efficient strategy to construct isomeric COFs by post-synthesis modification with phenylboric acid isomers via precise pore surface engineering. The introduction of phenylboric acid isomers in the pores of D–A-COF not only improved the stability, crystallinity, and fluorescence characteristics of the parent COFs, but also reduced the pKa value of phenylboric acid, which was conducive to the selective adsorption of diols with similar structure. As a proof-of-concept application, these obtained isomeric COFs were applied to selective enrichment of nucleosides. The o-phenylboric acid and p-phenylboric acid isomer modified COFs could specifically capture adenosine/uridine and adenosine/thymidine, respectively. The results of fluorescence spectra and theoretical calculation further validated these specific interactions. Therefore, this study not only provides a facile and novel approach for the synthesis of isomeric COFs, but also extends the application range of these functional materials with the same chemical composition but distinct physicochemical characteristics.Data availability
The data supporting the findings of this study are available within the article and ESI.† All other relevant source data are available from the corresponding authors upon reasonable request.Author contributions
Y.-H. Liu and Y.-Z. Chen synthesized the samples and participated in characterization. Y.-H. Liu and C. Lu supervised the project and wrote the manuscript. K. Shi and H.-J. Peng helped revise the manuscript. Y.-H. Liu and Y.-Z. Chen conducted research on the selective adsorption performances and mechanism. All authors commented on the data and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (U22A20397 and 22204148), Natural Science Foundation of Henan Province (242301420052), and Key R&D and Promotion Special (Scientific Problem Tackling) Project of Henan Province (242102230083).References
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chem. Soc. Rev., 2020, 49, 6248–6272 RSC.
- C. Ji, C. Kang, B. C. Patra and D. Zhao, CCS Chem., 2024, 6, 856–881 CrossRef CAS.
- X. Ren, X. Wang, W. Song, F. Bai and Y. Li, Nanoscale, 2023, 15, 4762–4771 RSC.
- Y. Li, X. Su, W. Zheng, J.-J. Zheng, L. Guo, M. Bonn, X. Gao, H. I. Wang and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202216795 CrossRef CAS PubMed.
- W. Wu, Z. Li, S. Liu, D. Zhang, B. Cai, Y. Liang, M. Wu, Y. Liao and X. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202404563 CrossRef CAS PubMed.
- Y. Chen, J. Qiu, X.-G. Zhang, H. Wang, W. Yao, Z. Li, Q. Xia, G. Zhu and J. Wang, Chem. Sci., 2022, 13, 5964–5972 RSC.
- J. Li, Y. Wan, G. Jiang, Y. Ozaki and F. Pi, Adv. Funct. Mater., 2024, 2408778 CrossRef CAS.
- S. Li, R. Ma, S. Xu, T. Zheng, G. Fu, Y. Wu, Z. Liao, Y. Kuang, Y. Hou, D. Wang, P. S. Petkov, K. Simeonova, X. Feng, L.-Z. Wu, X.-B. Li and T. Zhang, J. Am. Chem. Soc., 2022, 144, 13953–13960 CrossRef CAS PubMed.
- J.-X. Fu, Y. Liu, L.-H. Chen, W.-K. Han, X. Liu, J.-X. Shao, X. Yan and Z.-G. Gu, Small, 2023, 19, 2303897 CrossRef CAS PubMed.
- X.-H. Liu, Y.-P. Mo, J.-Y. Yue, Q.-N. Zheng, H.-J. Yan, D. Wang and L.-J. Wan, Small, 2014, 10, 4934–4939 CrossRef CAS PubMed.
- S. Ghosh, Y. Tsutsui, K. Suzuki, H. Kaji, K. Honjo, T. Uemura and S. Seki, Mol. Syst. Des. Eng., 2019, 4, 325–331 RSC.
- X.-L. Mao, Q.-X. Luo, Y.-J. Cai, X. Liu, Q.-Q. Jiang, C.-R. Zhang, R.-P. Liang and J.-D. Qiu, Anal. Chem., 2023, 95, 10803–10811 CrossRef CAS PubMed.
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. Djoko Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. Van De Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317 CrossRef CAS PubMed.
- W. Dong, Z. Qin, K. Wang, Y. Xiao, X. Liu, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202216073 CrossRef CAS PubMed.
- Y. Yang, D. Sabaghi, C. Liu, A. Dianat, D. Mücke, H. Qi, Y. Liu, M. Hambsch, Z.-K. Xu, M. Yu, G. Cuniberti, S. C. B. Mannsfeld, U. Kaiser, R. Dong, Z. Wang and X. Feng, Angew. Chem., Int. Ed., 2024, 63, e202316299 CrossRef CAS PubMed.
- Q. Wang, C. Wang, K. Zheng, B. Wang, Z. Wang, C. Zhang and X. Long, Angew. Chem., Int. Ed., 2024, 63, e202320037 CrossRef CAS PubMed.
- Q. Liao, Q. Sun, H. Xu, Y. Wang, Y. Xu, Z. Li, J. Hu, D. Wang, H. Li and K. Xi, Angew. Chem., Int. Ed., 2023, 62, e202310556 CrossRef CAS PubMed.
- Y. Li, L. Guo, Y. Lv, Z. Zhao, Y. Ma, W. Chen, G. Xing, D. Jiang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 5363–5369 CrossRef CAS PubMed.
- B. Feng, X. Chen, P. Yan, S. Huang, C. Lu, H. Ji, J. Zhu, Z. Yang, K. Cao and X. Zhuang, J. Am. Chem. Soc., 2023, 145, 26871–26882 CrossRef CAS PubMed.
- R. Guntermann, L. Frey, A. Biewald, A. Hartschuh, T. Clark, T. Bein and D. D. Medina, J. Am. Chem. Soc., 2024, 146, 15869–15878 CrossRef CAS PubMed.
- Z. Yang, H. Chen, S. Wang, W. Guo, T. Wang, X. Suo, D.-e. Jiang, X. Zhu, I. Popovs and S. Dai, J. Am. Chem. Soc., 2020, 142, 6856–6860 CrossRef CAS PubMed.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- J.-R. Wang, K. Song, T.-X. Luan, K. Cheng, Q. Wang, Y. Wang, W. W. Yu, P.-Z. Li and Y. Zhao, Nat. Commun., 2024, 15, 1267 CrossRef CAS PubMed.
- Y. Liang, T. Xia, Z. Chang, W. Xie, Y. Li, C. Li, R. Fan, W. Wang, Z. Sui and Q. Chen, Chem.–Eng. J., 2022, 437, 135314 CrossRef CAS.
- J. Yang, A. Acharjya, M.-Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed.
- J.-C. Wang, T. Sun, J. Zhang, Z. Chen, J.-Q. Du, J.-L. Kan and Y.-B. Dong, Chem. Sci., 2024, 15, 18634–18639 RSC.
- Y. Xie, T. Pan, Q. Lei, C. Chen, X. Dong, Y. Yuan, W. A. Maksoud, L. Zhao, L. Cavallo, I. Pinnau and Y. Han, Nat. Commun., 2022, 13, 2878 CrossRef CAS PubMed.
- Z. Liu and H. He, Acc. Chem. Res., 2017, 50, 2185–2193 CrossRef CAS PubMed.
- L. Ren, Z. Liu, Y. Liu, P. Dou and H.-Y. Chen, Angew. Chem., Int. Ed., 2009, 48, 6704–6707 CrossRef CAS PubMed.
- T.-B. Ren, Z.-Y. Wang, Z. Xiang, P. Lu, H.-H. Lai, L. Yuan, X.-B. Zhang and W. Tan, Angew. Chem., Int. Ed., 2021, 60, 800–805 CrossRef CAS PubMed.
- W. Zhang, W. Liu, P. Li, H. Xiao, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2014, 53, 12489–12493 CrossRef CAS PubMed.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- P. Chatterjee, A. Volkov, J. Mi, M. Niu, S. Sun, A. J. Rossini, L. M. Stanley and W. Huang, J. Am. Chem. Soc., 2024, 146, 20468–20476 CrossRef CAS PubMed.
- J. Qiu, C. Xu, X. Xu, Y. Zhao, Y. Zhao, Y. Zhao and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300459 CrossRef CAS PubMed.
- Y. Liu, L. Liu, X. Chen, Y. Liu, Y. Han and Y. Cui, J. Am. Chem. Soc., 2021, 143, 3509–3518 CrossRef CAS PubMed.
- Q. Song, J. Yang, K. Zheng, T. Zhang, C. Yuan, L.-M. Yuan and X. Hou, J. Am. Chem. Soc., 2024, 146, 7594–7604 CrossRef CAS PubMed.
- Y. Ma, Y. Han, Y. Yao, T. Zhou, D. Sun, C. Liu, G. Che, B. Hu, V. Valtchev and Q. Fang, Chem. Sci., 2024, 15, 12488–12495 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00121h |
| This journal is © The Royal Society of Chemistry 2025 |