
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-crystal chiral two-dimensional supramolecular organic frameworks for tunable circularly polarized luminescence†
Jialin
Cui
a,
Hui
Wang
a,
Hui
Liu
*a,
Hailong
Yu
a,
Wei
Wang
 b,
Yu
Wang
*b and
Yingjie
Zhao
*a
b,
Yu
Wang
*b and
Yingjie
Zhao
*a
aQingdao University of Science and Technology, 53 Zhengzhou Road, Qingdao, 266000, China. E-mail: hliu@qust.edu.cn; yz@qust.edu.cn
bShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, 3663N. Zhongshan Road, Shanghai, 200062, China. E-mail: yuwang@chem.ecnu.edu.cn
First published on 19th March 2025
Abstract
Chiral supramolecular organic frameworks (SOFs) and hydrogen-bonded organic frameworks (HOFs) remain an unexplored field, with very few reported examples. Here, three chiral SOFs with perfect two-dimensional (2D) framework structures are constructed by self-assembly between the chiral macrocyclic host molecule and different guest molecules through host–guest and hydrogen-bonding interactions. Variations in the guest structures lead to different host–guest interactions. The formation of the 2D frameworks of the chiral host and the guest molecules realizes chirality transfer and enhances the performance of circularly polarized luminescence (CPL) through strong charge transfer (CT) mechanisms, leading to the successful regulation of the CPL of the obtained SOF series. Chiral SOFs are significant enough due to their ability to combine chirality with versatile porous frameworks, leading to innovative solutions in optical devices, separations, catalysis, and beyond. Their tunability and eco-friendly synthesis further enhance their importance in chiral materials.
Introduction
Supramolecular organic frameworks (SOFs) and hydrogen-bonded organic frameworks (HOFs) are a fascinating class of porous materials that are constructed through the self-assembly of organic molecules via hydrogen bonds and non-covalent interactions.1–7 Unlike metal–organic frameworks (MOFs) or covalent organic frameworks (COFs), SOFs and HOFs rely on weaker, reversible forces such as hydrogen bonding, π–π stacking, van der Waals forces, etc.8–11 Modularity is a notable characteristic of these materials, which endows them with design flexibility.12–16 The structure and properties of SOFs and HOFs can be easily adjusted by modifying the organic components and the conditions under which the framework is assembled. This tunability at the molecular level provides a high degree of control over the materials with specific functionalities tailored to particular applications. Another notable feature of these materials is the dynamic process of constructing these materials.17–20 The dynamic nature of non-covalent interactions makes the construction process of these materials relatively easy, and it is usually possible to obtain single-crystal samples. Through X-ray single-crystal diffraction, structural information at atomic-level resolution can be obtained. This allows us to gain a deeper understanding of structure–property relationships at the atomic level, enabling us to modify structures based on their performance.Based on the aforementioned modularity and dynamic characteristics of SOF and HOF materials, it is reasonable to consider introducing chiral building blocks into the framework structure to construct chiral materials. Despite this, to our surprise, examples of chiral SOF and HOF materials are rare and precious.7,21–24 Designing chiral building blocks that can self-assemble into stable and well-defined frameworks is complex. The chiral building blocks must possess specific stereochemistry to induce and maintain chirality in the final framework structure. Chen and co-workers carried out the pioneering work on the construction of chiral HOF materials through chiral building blocks (BINOL).7 In addition, excellent examples have been reported in the construction of chiral supramolecular assembly25–28 and SOF materials through chiral building blocks.29–32 Nevertheless, the construction of chiral HOF or SOF materials still faces significant challenges.
As we know, chirality is a key concept that bridges multiple disciplines, influencing the development of new optical materials, the understanding of biological processes, and the advancement of asymmetric synthesis.33–35 Circularly polarized luminescence (CPL) active materials, as an important class of optical materials, hold significant importance in the application of three-dimensional (3D) displays and imaging, optical communication, chiral sensing, quantum cryptography, etc.36–39 SOFs offer significant advantages in the development of CPL materials due to their highly ordered structures with good structural tunability, which bring about versatile chemical functionalities. Leveraging the advantages of the tunable structures in SOF materials and the versatile host–guest chemistry as tools, the CPL signals in various SOFs can be modulated and amplified. Chiral supramolecular assemblies have demonstrated a remarkable ability to significantly amplify the circular polarization degree of CPL-active materials.40–42 In addition, due to the highly ordered structures, the aggregation-caused quenching (ACQ) effect of luminophores can be suppressed.43,44 This allows for highly efficient solid-state CPL-active SOF materials.45 Nevertheless, the examples of CPL-active materials based on SOFs are very limited, indicating that this field remains to be explored in depth. It is clear that the greatest challenge in this field lies in constructing high-quality CPL host–guest assembly frameworks that exhibit optimal chiroptical activity.
Here, a chiral macrocyclic host molecule was utilized for constructing two-dimensional (2D) chiral SOF single crystals with different guest molecules through hydrogen-bonding interactions between the 2D layers and host–guest interactions in the 2D layers. By adjusting the structures of guest molecules, the host–guest interactions in the frameworks can be adjusted, which further modulates electron coupling between the host and the guest, facilitating the regulation of CPL through host–guest assembly systems. Notably, due to the strong hydrogen-bonding interactions among the 2D layers and π–π interactions between the host and guest molecules in a single layer, the overall 2D framework structure is maintained despite changes in the structure of the guest molecules. In addition, the host–guest interactions are crucial for chiral transfer, playing a significant role in the overall chiral transfer (CT) process. These chiral SOF materials are interesting enough to validate and surpass some basic theories and provide precise models to comprehend the process of chiral transformation.
Results and discussion
The synthesis of the chiral macrocyclic host molecule is very straightforward.46–48 The introduction of chirality relies on chiral cyclohexanediamine as the reactant. By simply changing the chirality of cyclohexanediamine, enantiomeric triangular macrocycles (R-H1 or S-H1) can be obtained (Fig. 1a). In the triangular macrocyclic host molecule, the moiety of naphthalene diimide (NDI) has been widely recognized as an electron-deficient π-system due to the presence of four electron-withdrawing carbonyl groups.49,50 Therefore, the macrocyclic host molecule can be considered to have three electron-deficient planes uniformly oriented in three directions within the 2D plane. Thus, by introducing appropriately sized planar electron-rich π molecules, self-assembly should occur in a 2D plane through donor–acceptor (D–A) interactions between the host and guest molecules, forming 2D framework structures.51–53 A series of π-conjugated planar molecules, including 9,10-dichloroanthracene (DCA), pyrene, and perylene were chosen as the guest molecules (Fig. 1b–d). With the π-conjugated systems gradually increasing in size, they exhibit well-defined regularity with enhanced electron donating behavior.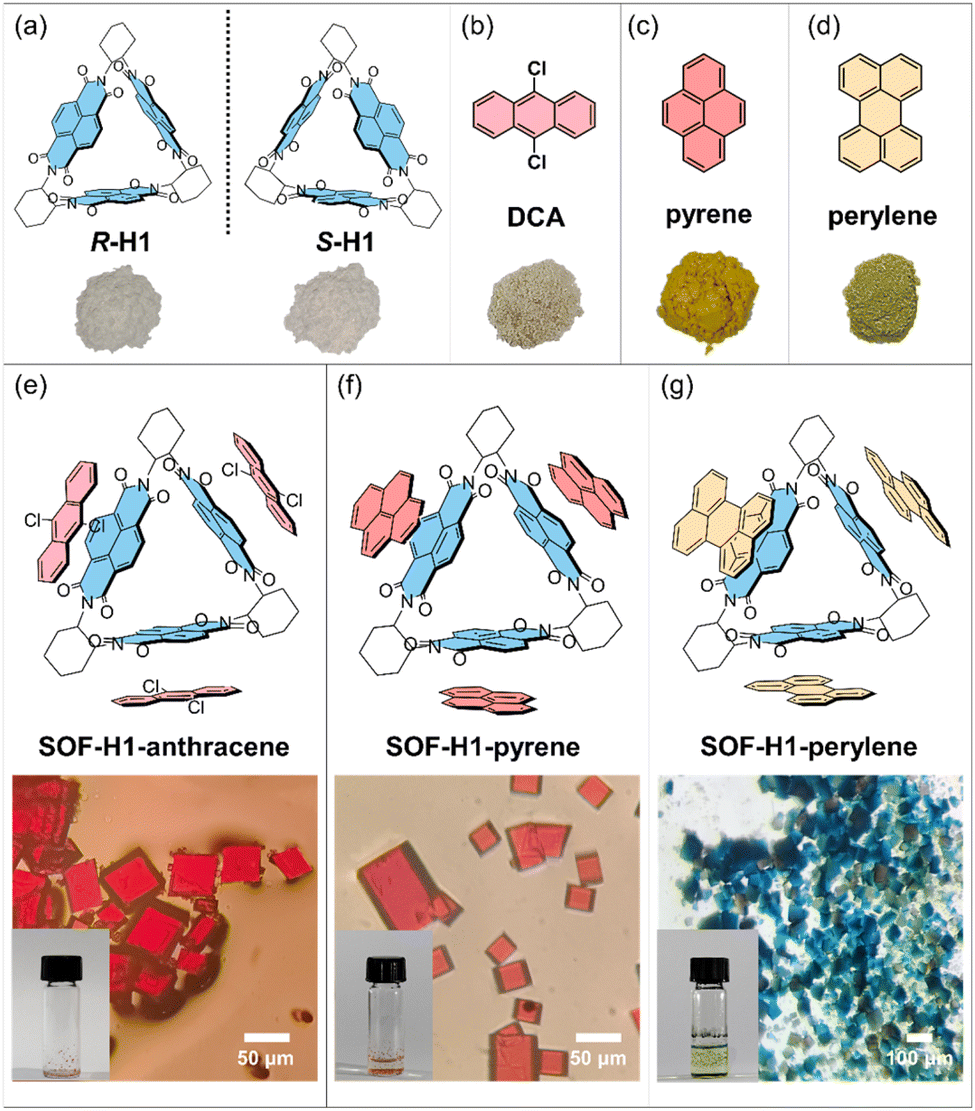 | ||
| Fig. 1 (a) The chemical structures of the enantiomers of H1 and their photographs. (b–d) The chemical structures of the guest molecules and their photographs. (e–g) The chemical structures of the three SOFs (SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene) and their optical microscope images and photographs. The R enantiomers were selected as representatives to present. | ||
Fortunately, the single crystal of the host macrocyclic molecule itself and the host–guest co-crystal can be easily grown in common solvents through the two-phase diffusion method or slow solvent evaporation. The host molecule H1, serving as the acceptor molecule, appears as a white powder, while the selected electron donors DCA, pyrene, and perylene are yellow, white, and bright yellow powders, respectively (Fig. 1a–d). The co-crystal of H1 and DCA has been reported by Stoddart and co-workers.53 Dissolving white H1 and yellow DCA in dichloromethane forms a colorless solution, but as methanol vapor slowly diffuses in, a large number of red rectangular SOF-H1-anthracene co-crystals were obtained (Fig. 1e). Dichloromethane solvent was gradually added to the mixture of pyrene and H1. The solution quickly turns red and then becomes colorless. The red quadrilateral SOF-H1-pyrene co-crystals were easily obtained by slow evaporation of the dichloromethane solution of H1 and pyrene (Fig. 1f). When yellow perylene and white H1 powder are added to chloroform, the solution turns green. High-quality dark green rectangular SOF-H1-perylene co-crystals were obtained by slowly diffusing n-hexane vapor into a chloroform solution of H1 and perylene (Fig. 1g). More interesting structural features emerge. As shown in Fig. 2, all three host–guest co-crystals exhibit 2D framework structures. The NDI moieties within the three planes of the triangular host macrocycle can interact with three guest molecules through face-to-face π–π interactions, resulting in the formation of a 2D structure in the a–b planes (Fig. 2b, e, and h). This process yields a perfectly hexagonal honeycomb porous framework, characterized by an equilateral hexagonal pore and six equilateral triangular pores in each layer (Fig. 2c, f, and i). The three NDI planes as electron acceptors can form face-to-face stacking through π–π interactions with the electron donor plane (DCA, pyrene, and perylene). The distances between the NDI moiety plane and the three donor planes are almost identical. The face-to-face π–π stacking distances between the NDI and the perylene, DCA, and pyrene are measured to be 3.32 Å, 3.38 Å, and 3.39 Å, respectively (Fig. 2b, e, and h).
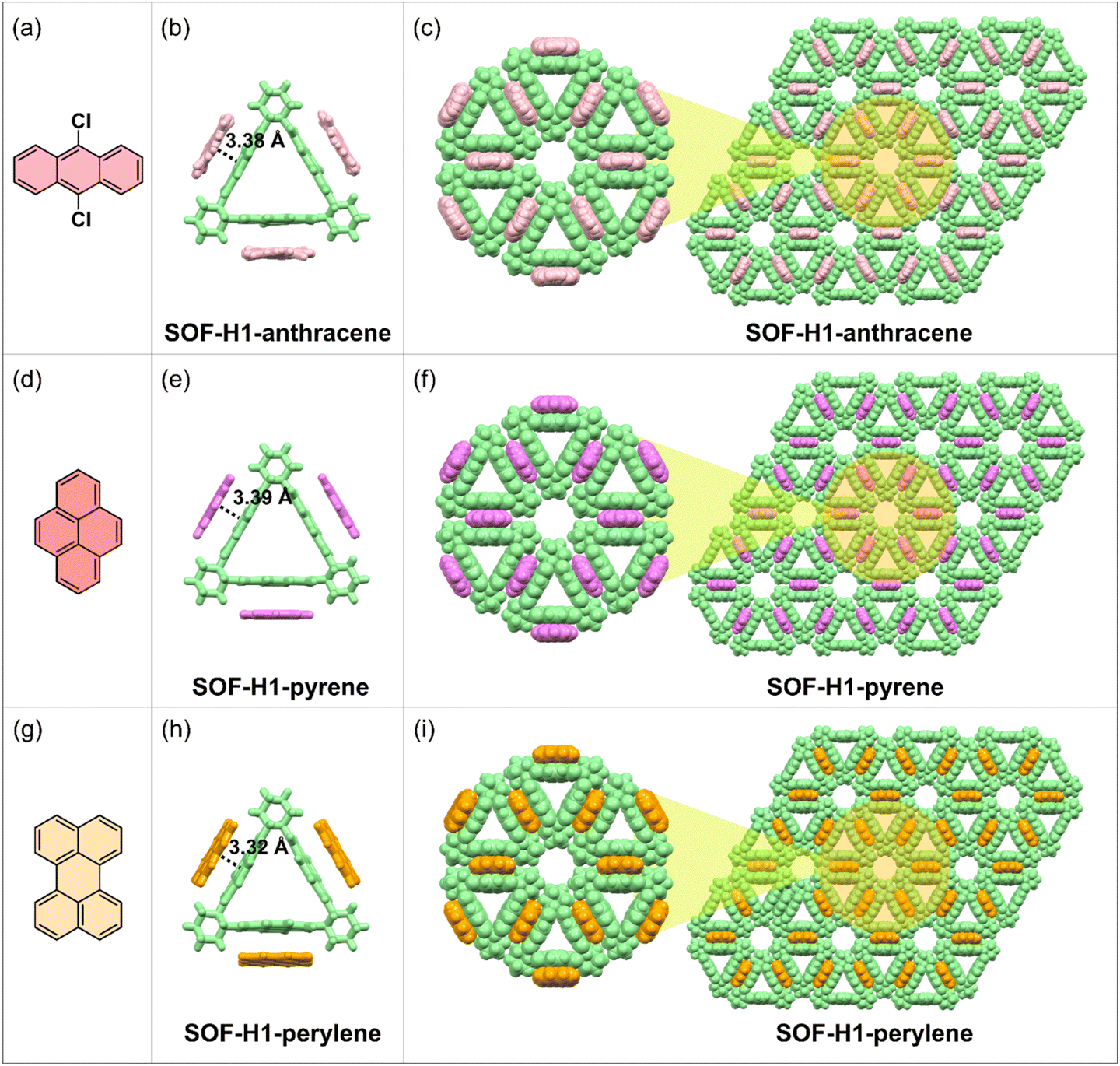 | ||
| Fig. 2 (a, d and g) The chemical structures of the three guest molecules. (b, e and h) The single-crystal structures of the SOFs showing the π–π interactions between the host and guest molecules. (c, f and i) The single-crystal 2D framework structures of the three obtained SOFs in the ab plane along the c-axis. The R enantiomers were selected as representatives. | ||
The hydrogen-bonding interactions along with the c-axis (between the 2D layers) stabilize the framework structure, endowing the material characteristics of SOFs. In the side view along the a or b-axis direction, the layered structures, which adopt ABC stacking modes, are clearly visible (Fig. 3). The self-assembly mode is identical in layers A, B, and C, differing only by a staggered shift (Fig. 3g and h). Along with the direction of the c-axis, multiple [C–H⋯O] hydrogen bonds between the H from the naphthalene and the O from the carbonyl connect each pair of triangular rings (Fig. 3a–c). The distances of the [C–H⋯O] hydrogen bonds are 2.32 Å, 2.39 Å, and 2.39 Å, respectively (Fig. 3a–c). To our surprise, the changes in the structures of the guest molecules do not affect the assembly pattern and the formation of the 2D framework structures (Fig. 3d–f). Interestingly, although the structures of the guest molecules have changed significantly, their cell parameters in the single crystals are almost unchanged. In the case of the R-SOF-H1-anthracene co-crystal, the structure corresponds to the R32 space group, with specific cell parameters as follows: a = 24.6999 Å, b = 24.6999 Å, c = 25.1460 Å, α = 90°, β = 90°, γ = 120° and volume = 13285.9 Å3. For the R-SOF-H1-pyrene co-crystal, the individual crystal structure conforms to the R32 space group, featuring specific cell parameters: a = 24.6162 Å, b = 24.6162 Å, c = 25.2652 Å, α = 90°, β = 90°, γ = 120° and volume = 13258.5 Å3. With regard to the R-SOF-H1-perylene co-crystal, the individual crystal structure also conforms to the R32 space group, featuring specific cell parameters: a = 24.0509 Å, b = 24.0509 Å, c = 25.9832 Å, α = 90°, β = 90°, γ = 120° and volume = 13016.3 Å3. The cell volumes of the SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene co-crystals are 13285.9 Å3, 13258.5 Å3, and 13016.3 Å3, respectively, showing a slight decreasing trend with increasing size of the guest molecules. The maximum variation in the volumes of the unit cell size is only 2%.
 | ||
| Fig. 3 (a–c) The single-crystal structures of the three SOFs showing the H-bonding interactions between the adjacent layers. (d–f) The top view (along the c-axis) of the single-crystal structures of the adjacent two layers in the three SOFs. (g) The side view (along the a-axis) of the single-crystal structure of SOF-H1-pyrene as a representative example with two periodic ABC stackings (6 layers). (h) The top view (along the a-axis) of the single-crystal structure of SOF-H1-pyrene as a representative example with two periodic ABC stackings (6 layers). The R enantiomers were selected as representatives. | ||
We also compared the unit cell parameters of the enantiomers. As expected, the R and S enantiomers of the three systems exhibited extremely similar parameters. The individual crystal structure of the S-SOF-H1-pyrene co-crystal accords with the R3 space group, featuring specific cell parameters of a = 24.6538 Å, b = 24.6538 Å, c = 25.2249 Å, α = 90°, β = 90°, γ = 120° and volume = 13277.9 Å3. Meanwhile, for the S-SOF-H1-anthracene co-crystal, the structure corresponds to the R32 space group, with specific cell parameters as follows: a = 24.6999 Å, b = 24.6999 Å, c = 25.1460 Å, α = 90°, β = 90°, γ = 120° and volume = 13285.9 Å3. As for the S-SOF-H1-perylene co-crystal, the individual crystal structure conforms to the R32 space group, featuring specific cell parameters as follows: a = 24.0466 Å, b = 24.0466 Å, c = 25.9712 Å, α = 90°, β = 90°, γ = 120° and volume = 13005.6 Å3.
Powder X-ray diffraction (PXRD) tests have been performed for all the co-crystals. As shown in Fig. S3,† the PXRD signals are rich and clear, and they align well with the simulated data, confirming the purity of the bulk materials. The thermogravimetric analysis (TGA) of all the SOF crystals has been performed. Similar tendencies were observed with two obvious steep drops for all three SOFs. The thermal stabilities of the three samples reach up to 250 °C (Fig. S4†).
This is really rare in the self-assembled single-crystal structure of the framework. Typically, slight adjustments in the structure of the guest molecules can cause significant changes in the entire single-crystal structure of the self-assembly. The appropriate sizes of the guest molecules and suitable D–A interactions between the host and guest are key to maintaining the 2D framework structure. The strong and suitable host–guest interactions in the plane of a single layer and the hydrogen-bonding interactions between the layers connect the host and guest molecules, finally forming the 2D framework structure.
Despite the staggered pattern mode adopted in the framework along the c-axis, the intrinsic pores are maintained due to the macrocyclic structure of the host molecules. We then studied the porous properties of the obtained SOF material. Before the gas adsorption tests, we first activated the three SOF crystals. The crystals were initially soaked in n-hexane for solvent exchange to remove the solvent molecules from the SOF crystals. The resulting samples were then further activated under a high vacuum at 50 °C for 12 hours to obtain desolvated crystals. The adsorption–desorption isotherms of this series of SOF materials were finally obtained by using CO2 molecules at 195 K (Fig. 4a–c). The CO2 isotherms show that the apparent Brunauer–Emmett–Teller (BET) surface areas of R-SOF-H1-anthracene, R-SOF-H1-pyrene, and R-SOF-H1-perylene are 90.6 m2 g−1, 106.7 m2 g−1, and 200.8 m2 g−1, respectively. The pore sizes of R-SOF-H1-anthracene, R-SOF-H1-pyrene, and R-SOF-H1-perylene were calculated using the Horvath–Kawazoe (HK) model, yielding values of 0.50 nm, 0.51 nm, and 0.58 nm, respectively (Fig. 4d–f). These results successfully demonstrated the porous nature of these SOF materials. Although the ABC stacking pattern occurs along the c-axis direction, the cavities of the host macrocyclic molecules remain interconnected. They are stacked in different layers by rotating 60° around the center of the triangle, forming a one-dimensional channel (Fig. 3h). The pores in the SOFs are originally induced by the chiral macrocyclic hosts themselves. The staggered stacking mode does not influence the formation of a through channel constructed by the host molecules themselves. Most importantly, due to the chiral cavities brought about by the host molecules, these channels possess chiral characteristics. The single-crystal structure of the macrocyclic host molecule itself has also been well investigated.47,51,54,55 In the absence of guest molecules, the self-assembly relying solely on the host molecules does not form a framework structure. It has been reported that the H1 molecule usually does not form a perfect ‘face-to-face’ stacking to finally realize the formation of 2D framework structures. The main weak interactions in the single-crystal host molecules are the π–π interactions between the NDI moieties. Either monoclinic P21 or triclinic P1 space groups, which are both non-chiral space groups, are obtained for the H1 single crystals under different crystallization conditions.
 | ||
| Fig. 4 CO2 adsorption–desorption isotherms (195 K) of (a) SOF-H1-anthracene, (b) SOF-H1-pyrene, and (c) SOF-H1-perylene. Pore size distribution profiles of (d) SOF-H1-anthracene, (e) SOF-H1-pyrene, and (f) SOF-H1-perylene. The R enantiomers are selected as representatives. | ||
To achieve CPL, a crucial strategy involves inducing the aggregation of organic fluorophores into well-ordered assemblies.56 To date, a variety of CPL-active materials have been developed. However, due to the generally low dissymmetry factor (glum) values of small organic molecules, multi-component assemblies are viewed as one of the most effective strategies for both CPL generation and the amplification of glum.57 In order to enhance the photophysical and chiroptical properties, various compounds with extended π-conjugated systems have been introduced into chiral macrocycle solutions, aiming to facilitate the formation of ordered aggregates through D–A interactions. We thus used three guest molecules, DCA, pyrene, and perylene, to assemble with R and S triangular rings, resulting in six types of crystals (three pairs of enantiomers), achieving an ordered arrangement from disorder in the triangular ring assembly. As the structure of the guest molecules regularly changes, the corresponding shifts in the color and luminescent properties of the SOFs become visible to the naked eye. The host macrocyclic molecule H1 itself is a colorless crystal, while both SOF-H1-anthracene and SOF-H1-pyrene are red. The color of the SOF-H1-perylene crystal even becomes green. Thus, the photophysical properties of this series of SOF materials were subsequently studied.
To elucidate the color changes of the co-crystals, the solid-state ultraviolet-visible (UV-vis) absorption spectra of SOF-H1-anthracene, SOF-H1-pyrene, SOF-H1-perylene, and their constituent monomers were recorded. Compared to the monomer H1, SOF-H1-perylene exhibits a significant red shift from ∼450 to ∼760 nm, which indicates strong charge transfer (CT) interactions and the formation of D–A complexes in the obtained frameworks (Fig. 5a). Similarly, strong CT interactions were also observed in the other two SOFs (Fig. S5†). As shown in Fig. 5b, SOF-H1-pyrene, SOF-H1-anthracene, and SOF-H1-perylene exhibit a gradual redshift trend with characteristic absorption edges at 630 nm, 659 nm, and 760 nm, respectively, which is attributed to the occurrence of n–π* or d–π* transitions. D–A interactions modify the molecular environment, leading to a decrease in the energy difference between the participating orbitals. The emission spectra also exhibit a similar trend of change. SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene exhibit emission peaks at 610 nm, 615 nm, and 709 nm, respectively. Notably, SOF-H1-perylene displays a pronounced redshift of ∼100 nm compared to SOF-H1-anthracene and SOF-H1-pyrene (Fig. 5c). This indicates that with an increase in the electron-donating ability of the donor molecules or an enlargement of the π plane, the emission spectrum progressively exhibits a redshift, accompanied by a reduction in the energy level spacing. The fluorescence lifetime of the compound is shown in Fig. S6–S8.† The absolute quantum yields of the three SOF crystals were also measured, which are 1.9% for R-SOF-H1-anthracene, 3.1% for R-SOF-H1-pyrene, and 2.1% for R-SOF-H1-perylene. It is evident that the optical properties of the luminescent materials can be tuned by manipulating the strong CT between the donor and acceptor.58
 | ||
| Fig. 5 (a) The solid-state UV-Vis spectra (diffuse reflectance mode) of H1, perylene, and SOF-H1-perylene. (b) The solid-state UV-Vis spectra (diffuse reflectance mode) of SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene. (c) Normalized fluorescence spectra of SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene. (d–f) CD and UV-Vis spectra (transmission mode) of co-assemblies obtained by drop-casting from CH2Cl2. (g–i) CPL and PL spectra of R/S-SOF-H1-anthracene, R/S-SOF-H1-pyrene, and R/S-SOF-H1-perylene recorded at 298 K. | ||
Chiral samples were prepared via the drop-casting method, followed by CD spectral measurements to confirm the transfer of chirality (Fig. S9†). As shown in Fig. S10,† the chiral macrocycles R-H1 and S-H1 exhibit good ground-state chirality in the thin film, with obvious Cotton effects at 333, 359, 368, and 389 nm, corresponding to the absorption peaks at 330, 349, 365, and 387 nm in the UV-Vis spectra. Further flipping of the sample also confirmed the reliability of the CD data (Fig. S11 and S12†). The CD signal peaks of R/S-SOF-H1-anthracene, R/S-SOF-H1-pyrene, and R/S-SOF-H1-perylene exhibited the Cotton effect at 360, 372, 392, and 435 nm; 327, 351, and 396 nm; 355, 390, 405, 417, 430, 440, 452, and 489 nm, respectively, showing significant changes, with a notable weakening of the Cotton effect, which basically corresponds to their UV-Vis absorption peaks (Fig. 5d–f). Compared with R/S-H1, the CD signals of the enantiomers of the three SOFs all show a significant red shift. The significant red shift of the CD signals relative to H1 demonstrates the successful transfer of chirality from H1 to the SOF skeleton. Taking the R configuration as an example, the negative Cotton effect near 400 nm is consistent with that of R-H1, clearly indicating that the chirality of the SOFs originates from the chiral macrocycle. To avoid the influence of linear dichroism (LD), we also conducted LD testing to ensure the authenticity of the CD (Fig. S13†). It is noteworthy that the original triangular macrocycle H1, despite exhibiting CD and weak fluorescence, lacked CPL (Fig. S10†). However, all three systems exhibited relatively CPL signals (Fig. 5g–i). The |glum| values for SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene are 0.003, 0.002, and 0.006, respectively (Fig. S14†). The original data of the CPL tests from multiple angles can be found in Fig. S18–S25.†
Additionally, single-crystal structures reveal that upon the introduction of guest molecules, a strong charge transfer occurs between the electron-deficient macrocycle and the electron-rich guest. Facilitated by π–π stacking interactions, this charge transfer induces a reorganization of the molecular self-assembly within the co-crystal, ultimately resulting in the formation of a chiral helix (Fig. S15†). This also indicates that through supramolecular assembly, the chirality of cyclohexanediamine in the triangular ring has been transferred to the CT supramolecular assembly, achieving chirality transfer.59 This is the famous “sergeant-and-soldier” (SaS) principle, where a small number of chiral units (sergeants) can dictate the helical orientation of non-chiral units (soldiers) and induce the entire macromolecule to adopt a biased handedness. Chirality is transferred from the point chirality of cyclohexanediamine to 2D planar chirality fixed in a three-layer sandwich structure. The long-range order in the 2D SOFs further amplifies the chirality. The SaS effect has not only been extended to supramolecular polymer systems but has also been widely studied in liquid crystal aggregate systems.60–62
When chiral macrocyclic molecules tightly stack with achiral guest molecules through π–π interactions to form an ordered 2D co-assembled CT system, the chiral molecules influence the electron cloud distribution and orbital arrangement of the system, leading to the formation of chiral exciplexes. The close packing enhances the π–π interactions, leading to changes in the dipole moment. This chiral induction allows the overall system to exhibit chiral luminescence during the emission process, thereby generating circularly polarized light. Additionally, the geometric configuration of the SOF structure is crucial for the CPL performance. The formation of a 2D stacked structure through supramolecular self-assembly enables an ordered arrangement of molecular orbitals, which is more favorable for the transfer and amplification of chiral information, consequently enhancing the emission of CPL.
The differences in the |glum| values among the three SOFs can be explained as follows: perylene possesses a larger π-plane than pyrene and DCA, which generally corresponds to higher electron density and enhanced conjugation effects. This enhancement facilitates stronger intermolecular interactions. The highest occupied molecular orbital (HOMO) energy level of perylene is higher than that of pyrene and DCA, leading to more pronounced π–π and CT interactions between perylene molecules and the macrocyclic components in the resulting co-crystals. Therefore, SOF-H1-perylene exhibits a higher |glum| value compared to SOF-H1-pyrene and SOF-H1-anthracene.
Density functional theory (DFT) calculations based on the solid superstructures of SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene were performed (Fig. 6). We employed the Gaussian 16 software package and the B3LYP method to calculate the HOMO–LUMO energy levels of the three different donor co-crystals, revealing their molecular orbital arrangements before and after cocrystallization, in order to understand the distinct photophysical properties of these three co-crystals.63 The HOMO levels of these three co-crystals are all located on the electron-donating molecules. Meanwhile, they exhibit similar lowest unoccupied molecular orbital (LUMO) energy levels, attributed to the concentration of their LUMO levels on the electron-accepting H1 (Fig. S26†). The HOMO–LUMO energy gaps of co-crystals SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene are calculated to be 2.55 eV, 2.66 eV, and 1.93 eV, and these values are consistent with their optical bandgaps. Molecules with large π planes can engage in π–π stacking interactions with the chiral molecule H1. This stacking effect modifies the electron cloud distribution and the charge transfer mechanism between the molecules, potentially inducing chirality in the excited state. These findings further reinforce our hypothesis.
 | ||
| Fig. 6 The HOMOs, LUMOs, and HOMO–LUMO energy gaps of the D–A fragments in SOF-H1-anthracene, SOF-H1-pyrene, and SOF-H1-perylene. | ||
Conclusions
A series of chiral SOFs with similar 2D framework structures were constructed through host–guest interactions in the 2D plane and H-bonding interactions between the 2D layers. The change in the guest molecule structures did not affect the self-assembly pattern, and the 2D framework structures were maintained. Interestingly, the CPL activity can be tuned by modifying the structures of guest molecules. The incorporation of guest molecules structurally guarantees the formation of 2D frameworks, facilitates chirality transfer, and enhances the performance of CPL through strong CT mechanisms. The ability to precisely control the structure and functionality of the SOFs allows for the customization of their optical properties to match specific CPL requirements. This level of customization is crucial for developing advanced optical materials. Given the diversity and intrinsic characteristics of SOFs, the synergistic effects of their structure and host–guest interactions create a promising platform for designing advanced CPL-active composites, while also providing additional opportunities to enhance chiroptical properties. Overall, considering the limited number of CPL-active SOFs based on host–guest systems developed to date, there is significant potential for further advancements in this field.Author contributions
Y. Zhao conceived the idea and designed the experiments. J. Cui carried out the experiments. H. Wang performed the X-ray single crystal analysis. H. Liu directed the synthesis and single-crystal sample growth. H. Yu guided the calculation part. W. Wang guided the CPL applications. Y. Wang performed all the CPL tests. J. Cui and Y. Zhao co-wrote the manuscript. Y. Zhao directed the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22175115, 22205128). Y. W. acknowledges financial support from the Postdoctoral Fellowship Program of CPSF under Grant Number GZC20230810.References
- Y. He, S. Xiang and B. Chen, A microporous hydrogen-bonded organic framework for highly selective C2H2/C2H4 separation at ambient temperature, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Multifunctional porous hydrogen-bonded organic framework materials, Chem. Soc. Rev., 2019, 48, 1362–1389 CAS.
- R.-B. Lin and B. Chen, Hydrogen-bonded organic frameworks: Chemistry and functions, Chem, 2022, 8, 2114–2135 CAS.
- Z. J. Lin, S. A. R. Mahammed, T. F. Liu and R. Cao, Multifunctional Porous Hydrogen-Bonded Organic Frameworks: Current Status and Future Perspectives, ACS Cent. Sci., 2022, 8, 1589–1608 CAS.
- B. Wang, R. B. Lin, Z. Zhang, S. Xiang and B. Chen, Hydrogen-Bonded Organic Frameworks as a Tunable Platform for Functional Materials, J. Am. Chem. Soc., 2020, 142, 14399–14416 CAS.
- Z. Xiong, S. Xiang, Y. Lv, B. Chen and Z. Zhang, Hydrogen-Bonded Organic Frameworks as an Appealing Platform for Luminescent Sensing, Adv. Funct. Mater., 2024, 34, 2403635 CAS.
- P. Li, Y. He, J. Guang, L. Weng, J. C. Zhao, S. Xiang and B. Chen, A homochiral microporous hydrogen-bonded organic framework for highly enantioselective separation of secondary alcohols, J. Am. Chem. Soc., 2014, 136, 547–549 CAS.
- L. Chen, B. Zhang, L. Chen, H. Liu, Y. Hu and S. Qiao, Hydrogen-bonded organic frameworks: design, applications, and prospects, Mater. Adv., 2022, 3, 3680–3708 CAS.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Designing Hydrogen-Bonded Organic Frameworks (HOFs) with Permanent Porosity, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CAS.
- P. Li, M. R. Ryder and J. F. Stoddart, Hydrogen-Bonded Organic Frameworks: A Rising Class of Porous Molecular Materials, Acc. Mater. Res., 2020, 1, 77–87 CAS.
- D. Yu, H. Zhang, J. Ren and X. Qu, Hydrogen-bonded organic frameworks: new horizons in biomedical applications, Chem. Soc. Rev., 2023, 52, 7504–7523 RSC.
- X. Zhao, Q. Yin, X. Mao, C. Cheng, L. Zhang, L. Wang, T. F. Liu, Y. Li and Y. Li, Theory-guided design of hydrogen-bonded cobaltoporphyrin frameworks for highly selective electrochemical H2O2 production in acid, Nat. Commun., 2022, 13, 2721 CrossRef CAS PubMed.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, Design Rules of Hydrogen-Bonded Organic Frameworks with High Chemical and Thermal Stabilities, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef CAS PubMed.
- X. Liu, G. Liu, T. Fu, K. Ding, J. Guo, Z. Wang, W. Xia and H. Shangguan, Structural Design and Energy and Environmental Applications of Hydrogen-Bonded Organic Frameworks: A Systematic Review, Adv. Sci., 2024, 11, e2400101 CrossRef PubMed.
- Q. Huang, W. Li, Z. Mao, L. Qu, Y. Li, H. Zhang, T. Yu, Z. Yang, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, An exceptionally flexible hydrogen-bonded organic framework with large-scale void regulation and adaptive guest accommodation abilities, Nat. Commun., 2019, 10, 3074 CrossRef.
- Y. Hou, X.-S. Huang, S.-H. Gong, C. Liu, Y. Liu and T.-F. Liu, Stable hydrogen-bonded organic frameworks and their photo- and electro-responses, Nano Res., 2024, 17, 7675–7699 CrossRef CAS.
- J. Yang, J. Wang, B. Hou, X. Huang, T. Wang, Y. Bao and H. Hao, Porous hydrogen-bonded organic frameworks (HOFs): From design to potential applications, Chem. Eng. J., 2020, 399, 125873 CrossRef CAS.
- Q. Huang, W. Li, Z. Mao, H. Zhang, Y. Li, D. Ma, H. Wu, J. Zhao, Z. Yang, Y. Zhang, L. Gong, M. P. Aldred and Z. Chi, Dynamic molecular weaving in a two-dimensional hydrogen-bonded organic framework, Chem, 2021, 7, 1321–1332 CAS.
- P. Tholen, C. A. Peeples, M. M. Ayhan, L. Wagner, H. Thomas, P. Imbrasas, Y. Zorlu, C. Baretzky, S. Reineke, G. Hanna and G. Yucesan, Tuning Structural and Optical Properties of Porphyrin-based Hydrogen-Bonded Organic Frameworks by Metal Insertion, Small, 2022, 18, e2204578 CrossRef.
- K. Zhu, X. Xu and B. Yan, Lanthanide Functionalized Hydrogen-bonded Organic Framework Hybrid Materials: Luminescence Responsive Sensing, Intelligent Applications and Biomimetic Design, Acc. Mater. Res., 2024, 5, 1401–1414 CrossRef CAS.
- W. Gong, D. Chu, H. Jiang, X. Chen, Y. Cui and Y. Liu, Permanent porous hydrogen-bonded frameworks with two types of Bronsted acid sites for heterogeneous asymmetric catalysis, Nat. Commun., 2019, 10, 600 CrossRef CAS PubMed.
- Y. Li, S. Tang, A. Yusov, J. Rose, A. N. Borrfors, C. T. Hu and M. D. Ward, Hydrogen-bonded frameworks for molecular structure determination, Nat. Commun., 2019, 10, 4477 CrossRef PubMed.
- V. V. Veselovsky, A. V. Lozanova, V. I. Isaeva, V. D. Nissenbaum, N. A. Davshan, A. A. Lobova and V. V. Chernyshev, New Chiral Hydrogen-Bonded Organic Framework Based on Substituted Diarylacetylene Dicarboxylic Acid, Cryst. Growth Des., 2020, 20, 3713–3721 CAS.
- V. V. Veselovsky, V. I. Isaeva, L. M. Kustov, G. I. Kapustin, V. D. Nissenbaum, C. Dejoie, A. N. Fitch and V. V. Chernyshev, Synthesis and Crystal Structure of a New Chiral Hydrogen-Bonded Organic Framework ZIOC-2, Cryst. Growth Des., 2022, 22, 2547–2556 Search PubMed.
- Y. Liu, L. Wang, L. Zhao, Y. Zhang, Z. T. Li and F. Huang, Multiple hydrogen bonding driven supramolecular architectures and their biomedical applications, Chem. Soc. Rev., 2024, 53, 1592–1623 CAS.
- J. Han, D. Yang, X. Jin, Y. Jiang, M. Liu and P. Duan, Enhanced Circularly Polarized Luminescence in Emissive Charge-Transfer Complexes, Angew. Chem., Int. Ed., 2019, 58, 7013–7019 CrossRef CAS.
- T. Zhao, J. Han, P. Duan and M. Liu, New Perspectives to Trigger and Modulate Circularly Polarized Luminescence of Complex and Aggregated Systems: Energy Transfer, Photon Upconversion, Charge Transfer, and Organic Radical, Acc. Chem. Res., 2020, 53, 1279–1292 CrossRef CAS PubMed.
- W. Shang, Y. Wang, X. Zhu, T. Liang, C. Du, J. Xiang and M. Liu, Helical Cage Rotors Switched on by Brake Molecule with Variable Fluorescence and Circularly Polarized Luminescence, J. Am. Chem. Soc., 2023, 145, 27639–27649 CrossRef CAS.
- Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Adaptive Chirality of an Achiral Cucurbit[8]uril-Based Supramolecular Organic Framework for Chirality Induction in Water, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS PubMed.
- C. Yan, Q. Li, X. Miao, Y. Zhao, Y. Li, P. Wang, K. Wang, H. Duan, L. Zhang and L. Cao, Chiral Adaptive Induction of an Achiral Cucurbit[8]uril-Based Supramolecular Organic Framework by Dipeptides in Water, Angew. Chem., Int. Ed., 2023, 62, e202308029 CrossRef CAS.
- C. Yan, Q. Li, K. Wang, W. Yang, J. Han, Y. Li, Y. Dong, D. Chu, L. Cheng and L. Cao, “Gear-driven”-type chirality transfer of tetraphenylethene-based supramolecular organic frameworks for peptides in water, Chem. Sci., 2024, 15, 3758–3766 RSC.
- Y. Zhou, B. Liu, X. Sun, J. Li, G. Li, Q. Huo and Y. Liu, Self-assembly of Homochiral Porous Supramolecular Organic Frameworks with Significant CO2 Capture and CO2/N2 Selectivity, Cryst. Growth Des., 2017, 17, 6653–6659 CrossRef CAS.
- N. H. Cho, A. Guerrero-Martínez, J. Ma, S. Bals, N. A. Kotov, L. M. Liz-Marzán and K. T. Nam, Bioinspired chiral inorganic nanomaterials, Nat. Rev. Bioeng., 2023, 1, 88–106 CrossRef CAS.
- X. Niu, R. Zhao, S. Yan, Z. Pang, H. Li, X. Yang and K. Wang, Chiral Materials: Progress, Applications, and Prospects, Small, 2023, 19, e2303059 CrossRef PubMed.
- X. Li, W. T. Xu, X. Q. Xu, Y. Wang, X. Q. Wang, H. B. Yang and W. Wang, Lighting Up Bispyrene-Functionalized Chiral Molecular Muscles with Switchable Circularly Polarized Excimer Emissions, Angew. Chem., Int. Ed., 2024, e202412548, DOI:10.1002/anie.202412548.
- Y. Deng, M. Wang, Y. Zhuang, S. Liu, W. Huang and Q. Zhao, Circularly polarized luminescence from organic micro-/nano-structures, Light: Sci. Appl., 2021, 10, 76 CAS.
- Y. Zhang, S. Yu, B. Han, Y. Zhou, X. Zhang, X. Gao and Z. Tang, Circularly polarized luminescence in chiral materials, Matter, 2022, 5, 837–875 CAS.
- J. Y. Zhao, F. F. Xu, Z. Q. Li, Z. L. Gong, Y. W. Zhong and J. Yao, Molecular Cocrystals with Hydrogen-Bonded Polymeric Structures and Polarized Luminescence, Materials, 2022, 15, 7247 CAS.
- A. Zheng, T. Zhao, X. Jin, W. Miao and P. Duan, Circularly polarized luminescent porous crystalline nanomaterials, Nanoscale, 2022, 14, 1123–1135 CAS.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Circularly Polarized Luminescence in Nanoassemblies: Generation, Amplification, and Application, Adv. Mater., 2020, 32, e1900110 Search PubMed.
- F. Song, Z. Zhao, Z. Liu, J. W. Y. Lam and B. Z. Tang, Circularly polarized luminescence from AIEgens, J. Mater. Chem. C, 2020, 8, 3284–3301 RSC.
- G. Zhang, X. Cheng, Y. Wang and W. Zhang, Supramolecular chiral polymeric aggregates: Construction and applications, Aggregate, 2022, 4, e262 CrossRef.
- Y. Hong, J. W. Lam and B. Z. Tang, Aggregation-induced emission: phenomenon, mechanism and applications, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Wu, Z. Mu, F. Gong, M. Qing, J. Zhou, K. Chen and L. Bai, Overcoming Aggregation-Caused Quenching by an Improved Porphyrin Hybrid and Its Application in Enhanced Electrochemiluminescence Biosensing, ACS Sustainable Chem. Eng., 2023, 11, 14124–14132 CrossRef CAS.
- K. Okubo, K. Oka, K. Tsuchiya, A. Tomimoto and N. Tohnai, Spirobifluorene-Based Porous Organic Salts: Their Porous Network Diversification and Construction of Chiral Helical Luminescent Structures, Angew. Chem., Int. Ed., 2024, 63, e202400475 CrossRef CAS PubMed.
- J. Gawroński, M. Brzostowska, K. Gawrońska, J. Koput, U. Rychlewska, P. Skowronek and B. Nordén, Novel Chiral Pyromellitdiimide (1,2,4,5-Benzenetetracarboxydiimide) Dimers and Trimers: Exploring Their Structure, Electronic Transitions, and Exciton Coupling, Chem.–Eur. J, 2002, 8, 2484–2494 CrossRef.
- S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Electron sharing and anion-pi recognition in molecular triangular prisms, Angew. Chem., Int. Ed., 2013, 52, 13100–13104 CrossRef CAS PubMed.
- Y. Wang, H. Wu and J. F. Stoddart, Molecular Triangles: A New Class of Macrocycles, Acc. Chem. Res., 2021, 54, 2027–2039 CrossRef CAS PubMed.
- S. L. Suraru and F. Wurthner, Strategies for the synthesis of functional naphthalene diimides, Angew. Chem., Int. Ed., 2014, 53, 7428–7448 CrossRef CAS PubMed.
- Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, The Emergence of Anion−π Catalysis, Acc. Chem. Res., 2018, 51, 2255–2263 CAS.
- Y. Beldjoudi, A. Narayanan, I. Roy, T. J. Pearson, M. M. Cetin, M. T. Nguyen, M. D. Krzyaniak, F. M. Alsubaie, M. R. Wasielewski, S. I. Stupp and J. F. Stoddart, Supramolecular Tessellations by a Rigid Naphthalene Diimide Triangle, J. Am. Chem. Soc., 2019, 141, 17783–17795 CAS.
- Y. Wang, H. Wu, P. Li, S. Chen, L. O. Jones, M. A. Mosquera, L. Zhang, K. Cai, H. Chen, X. Y. Chen, C. L. Stern, M. R. Wasielewski, M. A. Ratner, G. C. Schatz and J. F. Stoddart, Two-photon excited deep-red and near-infrared emissive organic co-crystals, Nat. Commun., 2020, 11, 4633 CAS.
- Y. Wang, H. Wu, L. O. Jones, M. A. Mosquera, C. L. Stern, G. C. Schatz and J. F. Stoddart, Color-Tunable Upconversion-Emission Switch Based on Cocrystal-to-Cocrystal Transformation, J. Am. Chem. Soc., 2023, 145, 1855–1865 CAS.
- Z. Liu, G. Liu, Y. Wu, D. Cao, J. Sun, S. T. Schneebeli, M. S. Nassar, C. A. Mirkin and J. F. Stoddart, Assembly of supramolecular nanotubes from molecular triangles and 1,2-dihalohydrocarbons, J. Am. Chem. Soc., 2014, 136, 16651–16660 CAS.
- A. Mizuno, Y. Shuku, R. Suizu, M. M. Matsushita, M. Tsuchiizu, D. Reta Maneru, F. Illas, V. Robert and K. Awaga, Discovery of the K4 Structure Formed by a Triangular pi Radical Anion, J. Am. Chem. Soc., 2015, 137, 7612–7615 CAS.
- Y.-X. Yuan, M. Hu, K.-R. Zhang, T.-T. Zhou, S. Wang, M. Liu and Y.-S. Zheng, The largest CPL enhancement by further assembly of self-assembled superhelices based on the helical TPE macrocycle, Mater. Horiz., 2020, 7, 3209–3216 RSC.
- S. Hu, L. Hu, X. Zhu, Y. Wang and M. Liu, Chiral V-shaped Pyrenes: Hexagonal Packing, Superhelix, and Amplified Chiroptical Performance, Angew. Chem., Int. Ed., 2021, 60, 19451–19457 CrossRef CAS PubMed.
- J. Rühe, K. Vinod, H. Hoh, K. Shoyama, M. Hariharan and F. Würthner, Guest-Mediated Modulation of Photophysical Pathways in a Coronene Bisimide Cyclophane, J. Am. Chem. Soc., 2024, 146, 28222–28232 Search PubMed.
- S. Wu, X. Song, C. Du and M. Liu, Macroscopic homochiral helicoids self-assembled via screw dislocations, Nat. Commun., 2024, 15, 6233 CrossRef CAS PubMed.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O’Leary and G. Willson, Macromolecular Stereochemistry: The Out-of-Proportion Influence of Optically Active Comonomers on the Conformational Characteristics of Polyisocyanates. The Sergeants and Soldiers Experiment, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Sergeants-and-Soldiers Principle in Chiral Columnar Stacks of Disc-Shaped Molecules with C3 Symmetry, Angew. Chem., Int. Ed., 2003, 36, 2648–2651 CrossRef.
- X. Cheng and W. Zhang, Polymerization-induced Chiral Self-assembly for the In-situ Construction, Modulation, Amplification and Applications of Asymmetric Suprastructures, Angew. Chem., Int. Ed., 2024, e202414332, DOI:10.1002/anie.202414332.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2063800, 2391866–2391870. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08811e |
| This journal is © The Royal Society of Chemistry 2025 |