
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe doping intensifies the built-in electric field for tailoring the reconstruction of sulfides towards efficient oxygen evolution†
Kun
Wang
,
Chunmei
Ni
,
Lei
Jin
,
Xingyue
Qian
,
Hui
Xu
 *,
Haiqun
Chen
* and
Guangyu
He
*
*,
Haiqun
Chen
* and
Guangyu
He
*
Key Laboratory of Advanced Catalytic Materials and Technology, Advanced Catalysis and Green Manufacturing Collaborative Innovation Center Institution, Changzhou University, 21 Gehu Lake Road, Changzhou, 213164, China. E-mail: xuhui006@cczu.edu.cn; chenhq@cczu.edu.cn; hegy@cczu.edu.cn
First published on 19th March 2025
Abstract
The traditional view of sulfides as stable active centers has hindered the development of a clear structure–activity relationship and the rational design of high-performance oxygen evolution reaction (OER) catalysts. In this study, we focus on regulating sulfide reconstruction and have synthesized a Fe–Ni3S4/Cr2O3 pre-catalyst. Under the combined influence of the built-in electric field (BIEF) at the heterogeneous interface and Fe doping, both the sulfide reconstruction process and the electronic structure of the reconstructed product, namely Fe–NiOOH, were effectively tuned. The enhanced BIEF induced by Fe doping generated electron-rich regions on the sulfide surface, stabilizing the reconstruction process. Fe doping into the sulfide induced the incorporation of Fe into NiOOH, modulating the electronic states near the Fermi level of the metal–oxygen bond and subsequently activating the lattice oxygen mediated mechanism (LOM) of Fe–NiOOH, which serves as the true active center. Additionally, the BIEF optimized OH− diffusion dynamics and the energy consumption of hydroxyl deprotonation, reducing the energy barrier of the rate-limiting step of the LOM process, further enhancing OER activity. Remarkably, Fe–Ni3S4/Cr2O3 demonstrated excellent OER activity and commercial viability. This work offers a new perspective on the regulation of reconstruction products of pre-catalyst, providing fresh insights for the design of efficient OER catalysts.
1. Introduction
The slow kinetics of the oxygen evolution reaction (OER) remain a critical challenge for the development of energy conversion and storage systems, such as water electrolysis for hydrogen production, regenerative fuel cells, and rechargeable metal–air batteries.1,2 Consequently, the development of high-performance OER electrocatalysts to optimize reaction kinetics is essential.3 Although non-precious metal sulfides offer advantages over commercial precious metal catalysts, such as abundant raw materials, low cost, and high structural stability, they still exhibit lower catalytic activity.4,5 Based on the assumption that the catalytic reaction interface is stable during the OER process, with the metal active sites on the sulfide surface acting as the primary active centers, under the guidance of the adsorbate evolution mechanism (AEM), modification strategies such as defect engineering, heterostructure construction, and morphology regulation can optimize the adsorption of oxygen-containing intermediates on metal sites (OH* → O* → OOH* → O2), thereby enhancing the OER activity of sulfides.6–8However, in recent years, with the ongoing advancement of research, it has been gradually revealed that sulfides used as electrocatalytic anode catalysts exhibit thermodynamic instability at high anodic potentials, leading to structural evolution (reconstruction) during the OER process.9 This has led to a shift in perspective, where sulfides are considered as “pre-catalysts”, with the true catalytically active centers being the oxygen-containing species at higher metal oxidation states, such as metal oxyhydroxide (MOOH), amorphous metal-sulfur-oxide layers, and metal hydroxides, which are generated through surface reconstruction of the pre-catalysts.10,11 Under the influence of its unique high metal oxidation states and the interactions with the pre-catalyst, these oxygen-containing species typically exhibit excellent OER activity, resulting in catalytic performance that far exceeds that of the pre-catalyst.4,12
Thus, focusing solely on regulating the adsorption energies of oxygen-containing intermediates based on the traditional AEM overlooks the dynamic reconstruction behavior of sulfides during catalytic processes.13,14 Furthermore, the constraints imposed by the scaling relationship of the AEM prevent the regulation of adsorption energies of different intermediates without mutual interference, which makes the 370 mV OER limiting overpotential predicted using AEM theory insufficient to explain the enhanced OER performance of sulfides reported in the literature.15 Consequently, the traditional view of sulfides as stable active centers has impeded the development of a robust structure–activity relationship and the rational design of high-performance OER catalysts. Our research focuses on the regulation of the true catalytically active centers: the reconstruction products of sulfides. While much research has concentrated on understanding and characterizing the reconstruction process, there has been limited focus on controlling the reconstruction process and its products. The inherent properties of the pre-catalyst (including its electronic structure, chemical composition, crystallinity, and defects) and reaction conditions (such as applied voltage, pH, and electrolyte concentration) can all impact the reconstruction behavior.11,16
Based on this, by combining built-in electric field (BIEF) construction and doping strategies, we effectively tuned both the degree of reconstruction of the rationally designed pre-catalyst Fe–Ni3S4/Cr2O3 and the electronic structure of the true active site, Fe-doped nickel oxyhydroxide (Fe–NiOOH), formed during the reconstruction (Scheme 1). Due to the influence of the enhanced BIEF from Fe doping on the asymmetric charge distribution at the heterogeneous interface of Fe–Ni3S4/Cr2O3, the reconstruction of the electron-rich component Fe–Ni3S4 is stabilized, and the ion precipitation in the reaction process is reduced. Notably, the doping of Fe in the sulfide induces Fe atoms to be doped into the reconstructed product NiOOH, modulating the electronic states near the metal–oxygen bond Fermi level of Fe–NiOOH, thereby activating lattice oxygen and triggering the lattice oxygen mediated mechanism (LOM). This mechanism overcomes the limitations imposed by the scaling relationship of the traditional AEM. The Fe–Ni3S4/Cr2O3 catalyst exhibited an overpotential of 282 mV at a current density of 10 mA cm−2 in both 1 M KOH and simulated seawater electrolytes, and the assembled commercial electrochemical stack operated stably for 40 h. Moreover, the BIEF at the heterointerface further optimizes the diffusion kinetics of OH− and hydroxyl (OH*) deprotonation in the LOM process, further boosting OER activity. This work presents a new approach to the activity regulation of transition metal non-oxide materials, focusing on the active control of the reconstruction process and products.
 | ||
| Scheme 1 Schematic diagram of the reconstruction degree and products regulated by the Fe doping-enhanced BIEF. | ||
2. Materials and methods
Cr2O3 nanoparticles are obtained by calcining Cr(OH)3 hydrate. Under magnetic stirring, 150 mL of 0.3 M NaOH is gradually added dropwise to 100 mL of 0.1 M Cr(NO3)3·9H2O solution. The solid is then collected by centrifugation, followed by multiple washings with deionized water and ethanol. The resulting solid is dried to obtain Cr(OH)3. Next, Cr(OH)3 is calcined in air at 650 °C for 2 h with a heating rate of 2 °C min−1 to produce Cr2O3. Finally, the Cr2O3 and sulfide composite was prepared using a hydrothermal method.In this step, 300 mg of the prepared Cr2O3 was dispersed in 40 mL of a solution containing 0.3 mmol of FeSO4·7H2O, 2.7 mmol of NiCl2·6H2O, 15 mmol of urea, 4 mmol Na2S2O3·5H2O, 20 mL H2O, and 20 mL ethylene glycol using ultrasonication. The dispersion was transferred to a 100 mL Teflon-lined stainless-steel autoclave and reacted at 180 °C for 15 h. After the reaction, the mixture was washed and filtered, and the obtained solid was vacuum-dried to yield the Fe–Ni3S4/Cr2O3 composite.
Using the same preparation method, Fe–Ni3S4 was synthesized without adding Cr2O3, Ni3S4/Cr2O3 was synthesized without adding FeSO4·7H2O, and Ni3S4 was synthesized without adding Cr2O3 and Fe(SO4)3·7H2O. The aforementioned reagents were all procured from Sinopharm Group Reagent Co. Ltd, China.
3. Results and discussion
An Fe–Ni3S4/Cr2O3 pre-catalyst co-modified using Fe doping and the BIEF was prepared by a simple two-step method (Fig. 1a). First, Cr(OH)3 was synthesized via a precipitation reaction between Cr salts and an alkali, followed by calcination to obtain Cr2O3 nanoparticles (Fig. S1†). Next, a solvothermal reaction was conducted using Fe salts, Ni salts, a sulfur source, and urea to prepare the Fe–Ni3S4/Cr2O3 heterojunction, with Cr2O3 nanoparticles encapsulated by Fe–Ni3S4. This unique encapsulated morphology was confirmed by TEM images (Fig. 1b and c). To investigate the effects of doping and the BIEF on the catalyst's structure and performance, the same method was used to prepare Ni3S4/Cr2O3 heterojunctions without Fe doping, as well as single-component Ni3S4 and Fe–Ni3S4 without Cr2O3. The crystal structures of Fe–Ni3S4/Cr2O3, Ni3S4/Cr2O3, and the single components were verified by X-ray diffraction (XRD) patterns shown in Fig. 1d. Notably, the enlarged XRD patterns reveal that, compared to single-component Ni3S4, the diffraction peaks of Fe–Ni3S4/Cr2O3, Ni3S4/Cr2O3, and Fe–Ni3S4 shifted to varying extents. This shift was attributed to Fe atom doping and the successful formation of the heterojunction, which rules out the possibility of a simple mechanical composite between Fe–Ni3S4 and Cr2O3 formed via the hydrothermal method (Fig. S2†). The crystal structure of Fe–Ni3S4/Cr2O3 was further confirmed by selected area electron diffraction (SAED), high-resolution TEM (HRTEM), and corresponding Fourier transform images (Fig. 1e and f). Scanning tunneling microscopy (STM) and elemental mapping revealed the uniform distribution of various elements in the Fe–Ni3S4/Cr2O3 composite (Fig. 1g). | ||
| Fig. 1 (a) Schematic illustration of the synthesis of Fe–Ni3S4/Cr2O3. (b and c) TEM image of Fe–Ni3S4/Cr2O3. (d) XRD patterns of Ni3S4, Ni3S4/Cr2O3, Fe–Ni3S4, and Fe–Ni3S4/Cr2O3. (e) SAED pattern, (f) HRTEM image and corresponding Fourier transform images of Fe–Ni3S4/Cr2O3. (g) HAADF-STEM image and corresponding elemental mapping images of Fe–Ni3S4/Cr2O3. | ||
Then we investigated the impact of heterojunction construction and Fe doping on the electronic structure of the pre-catalyst. In the Fe 2p X-ray photoelectron spectroscopy (XPS) spectrum of Fe–Ni3S4/Cr2O3 (Fig. 2a), the deconvolution peaks corresponding to Fe–S bonds, Fe3+, and Fe2+ shifted to lower binding energies compared to those of Fe–Ni3S4. Similarly, in the Ni 2p XPS spectra of Fe–Ni3S4/Cr2O3 and Ni3S4/Cr2O3 (Fig. 2b), the deconvolution peaks for Ni3+ and Ni2+ also shifted to lower binding energies compared to those of Fe–Ni3S4, with those of Fe–Ni3S4/Cr2O3 exhibiting the greatest shift. A shift of XPS peaks to lower binding energies generally indicates a reduction in the binding energy of electrons on atomic orbitals, implying electron gains by the atoms. These shifts in the Fe 2p and Ni 2p XPS peaks suggest that the heterojunction construction induces electron enrichment in the sulfides of Fe–Ni3S4/Cr2O3. Furthermore, the S 2p XPS spectra of Fe–Ni3S4/Cr2O3, Fe–Ni3S4, and Ni3S4/Cr2O3 (Fig. 2c) show deviations in the deconvolution peaks for S 2p3/2 and S 2p1/2, indicating that doping and heterostructure construction affect the strength of the metal–sulfur bonds in the sulfides.
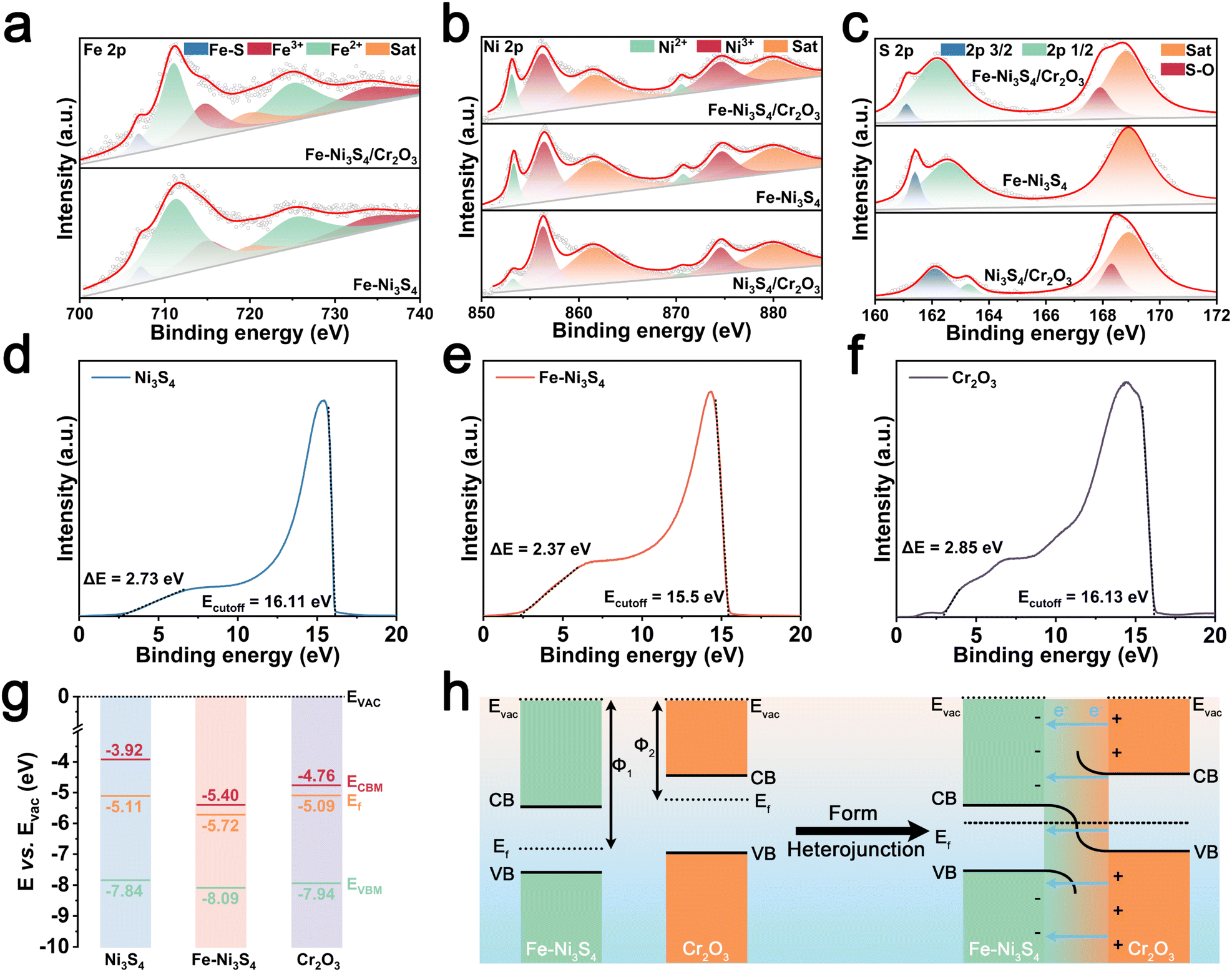 | ||
| Fig. 2 High-resolution XPS patterns of (a) Fe 2p, (b) Ni 2p, and (c) S 2p. UPS spectra of (d) Ni3S4, (e) Fe–Ni3S4, and (f) Cr2O3. (g) Energy band diagram of Ni3S4, Fe–Ni3S4, and Cr2O3. (h) Schematic diagram of heterojunction formation of Fe–Ni3S4/Cr2O3. | ||
To further elucidate the electronic structure regulation, we characterized the energy band structure of the single component of the pre-catalyst. Mott–Schottky testing revealed that single component Fe–Ni3S4, Ni3S4, and Cr2O3 were all n-type semiconductors, and thus Fe–Ni3S4/Cr2O3 and Ni3S4/Cr2O3 are n–n junctions (Fig. S3†). Solid ultraviolet-visible (UV-vis) testing showed that the band gaps of Fe–Ni3S4, Ni3S4, and Cr2O3 are 2.69 eV, 3.92 eV, and 3.18 eV, respectively, suggesting that Fe doping reduces the band gap of the sulfides and enhances their conductivity (Fig. S4†). Subsequently, UPS measurements revealed that the work function (Φ) of Fe–Ni3S4, Ni3S4 and Cr2O3 is 5.72 eV, 5.11 eV and 5.09 eV, respectively, along with valence band parameters (Fig. 2d–f). Finally, the energy band structure diagram of the catalyst shown in Fig. 2g was obtained.
The difference in Φ between the sulfides and Cr2O3 drives the directional movement of electrons at the heterojunction interface until the Fermi levels of the two semiconductors are equalized, resulting in the spontaneous formation of a BIEF.17–19 Since the Φ values of Fe–Ni3S4 and Ni3S4 are both lower than that of Cr2O3, the BIEF is directed from Cr2O3 to the sulfides, leading to charge transfer at the sulfide–Cr2O3 interface. This generates electron-rich regions on the sulfide surface and electron-deficient regions on the Cr2O3 surface, which explains the shift in the XPS deconvolution peaks of Fe and Ni in Fe–Ni3S4/Cr2O3 compared to those of Fe–Ni3S4. Moreover, Fe doping increases the Φ difference between the sulfides and Cr2O3, indicating a stronger BIEF in Fe–Ni3S4/Cr2O3 than that in Ni3S4/Cr2O3. Fe doping can effectively enhance the BIEF intensity between sulfides and Cr2O3, and the increase of the BIEF intensity will bring about more severe effects on the electron distribution at the heterogeneous interface (Fig. 2h).
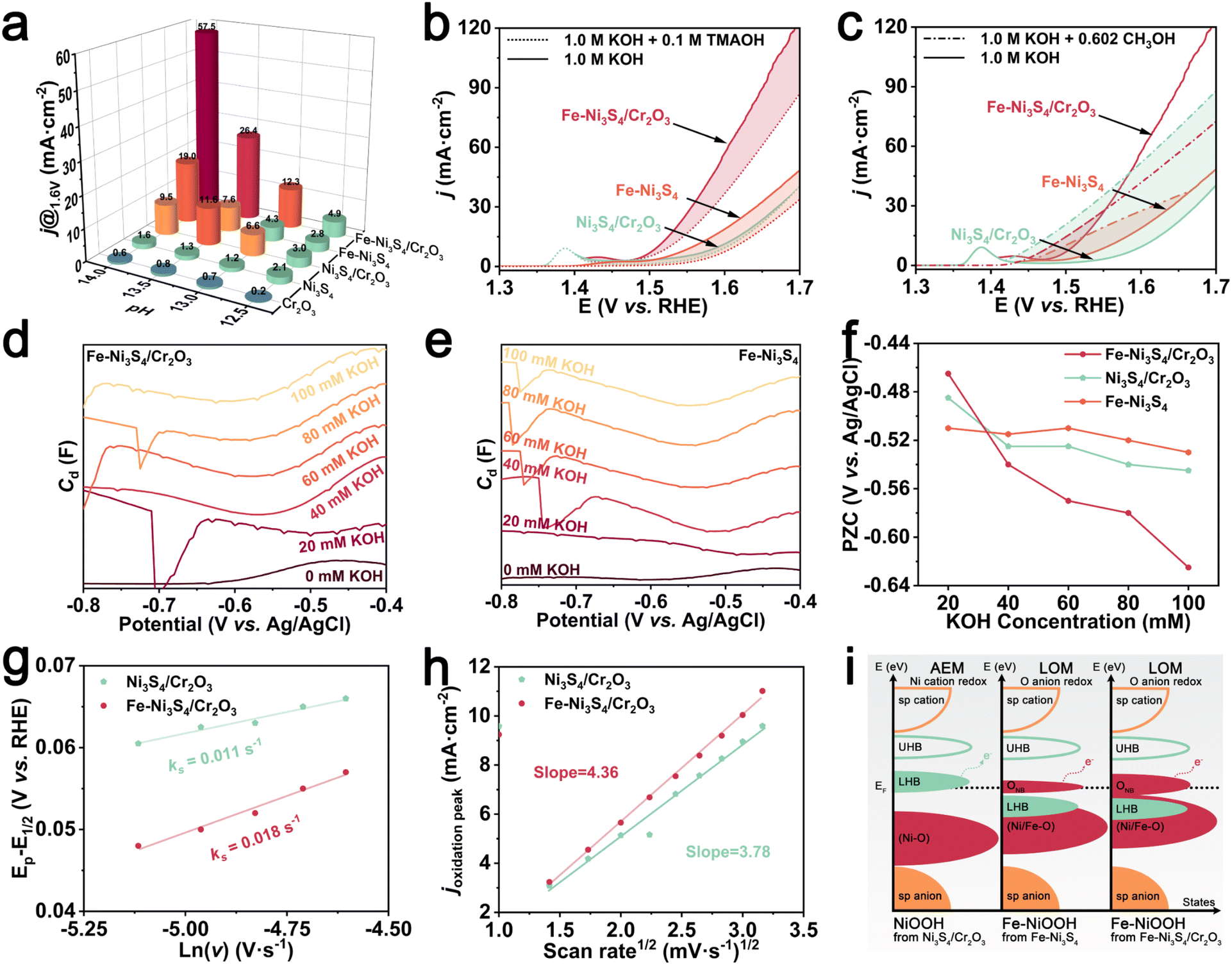
The Fe doping enhanced BIEF effectively regulates the electronic structure of the sulfides. To assess the catalytic performance, we evaluated the electrochemical properties of the catalyst in a three-electrode system. First, we investigated the OER activity of the catalyst in a 1 M KOH electrolyte. The linear sweep voltammetry (LSV) results (Fig. 3a) and Tafel plots (Fig. 3b) demonstrate that Fe–Ni3S4/Cr2O3 exhibits optimal OER performance, showing an overpotential of only 282 mV at a current density of 10 mA cm−2 and a Tafel slope of 72 mV de−1. Additionally, Fe–Ni3S4/Cr2O3 demonstrates a larger double-layer capacitance (Cdl) in the non-faradaic region, indicating a larger electrochemically active surface area (Fig. 3c and S5†). The Nyquist plot at 1.55 V shows that Fe–Ni3S4/Cr2O3 has the lowest charge transfer resistance (Rct), further demonstrating its excellent reaction kinetics (Fig. 3d). It can be observed that the activity of Fe–Ni3S4/Cr2O3 is much higher than that of Fe–Ni3S4 and Cr2O3, and the activity of Ni3S4/Cr2O3 is much higher than that of Ni3S4 and Cr2O3, which have almost no OER activity (Fig. 3e). This indicates that the construction of the BIEF will significantly enhance the activity of sulfides and chromium oxide. Moreover, the activity of Fe–Ni3S4/Cr2O3 is better than that of undoped Ni3S4/Cr2O3, showing the optimization of activity by Fe doping. When compared to similar reports in the literature, Fe–Ni3S4/Cr2O3 still demonstrates highly competitive OER activity (Fig. 3f and Table S1†). In stability tests, both multi-current chronopotentiometry (CP) (Fig. 3g) and a 100 h long-term stability test at a current density of 10 mA cm−2 (Fig. 3h) revealed that Fe–Ni3S4/Cr2O3 exhibited excellent reaction stability.
 | ||
| Fig. 3 OER activity in 1 M KOH solution. (a) LSV polarization curves, (b) Tafel plots, (c) Cdl values, and (d) Nyquist plots at 1.5 V versus RHE of Ni3S4, Ni3S4/Cr2O3, Fe–Ni3S4, and Fe–Ni3S4/Cr2O3. (e) Radar chart for comparison of comprehensive catalytic performance. (f) A comparison with other recently reported catalysts. (g) CP curve of Fe–Ni3S4/Cr2O3 at different current densities. (h) CP curve of Fe–Ni3S4/Cr2O3 at 10 mA cm−2. | ||
Given the scarcity of freshwater resources, we further evaluated the catalyst's performance in a simulated seawater electrolyte (Fig. S6 and S7†).20,21 Fe–Ni3S4/Cr2O3 exhibited the same low overpotential of 282 mV and Tafel slope of 72 mV dec−1 at 10 mA cm−2 for OER in the simulated seawater electrolyte, significantly outperforming the referenced samples. Additionally, Fe–Ni3S4/Cr2O3 showed superior Cdl and Rct values compared to the references. Notably, the OER activity of Fe–Ni3S4/Cr2O3 in simulated seawater was almost unaffected, while the activity of the comparison samples decreased significantly. To this end, we investigated the corrosion effect of Cl− on catalysts in simulated seawater mitigation through corrosion polarization curve tests. A higher corrosion potential (Ecorr) and more negative corrosion current (Icorr) typically indicate better corrosion resistance. The results showed that, although the Ecorr of Fe–Ni3S4/Cr2O3 was intermediate between those of Ni3S4, Fe–Ni3S4, Ni3S4/Cr2O3, and Cr2O3, its Icorr was the smallest, demonstrating its superior resistance to Cl− corrosion in seawater electrolysis. Fe–Ni3S4/Cr2O3 also exhibited excellent OER stability in simulated seawater, as evidenced by multi-current CP tests and 100 h CP tests. Finally, to evaluate the practical application potential of the catalyst, we used Fe–Ni3S4/Cr2O3 as the anode membrane electrode, commercial Pt/C as the cathode membrane electrode, and a proton exchange membrane as the separator to assemble a commercial-scale alkaline electrolyzer (AE) stack (Fig. S8a†). LSV testing and a 40 h stability test in 1 M KOH electrolyte confirmed the excellent commercial potential of the catalyst (Fig. S8b and c†).
To uncover the origin of the excellent catalytic activity of Fe–Ni3S4/Cr2O3, we investigated the influence of the Fe doping enhanced BIEF on the reconstruction behavior of the pre-catalyst and the formation of active sites during reconstruction. A 20 h chronoamperometry test at 1.6 V ensured sufficient reconstruction of the pre-catalyst (Fig. S9†). The reconstructed pre-catalysts (denoted as Fe–Ni3S4/Cr2O3-A, Fe–Ni3S4-A, and Ni3S4/Cr2O3-A) were analyzed by XPS (Fig. 4a–d). Shifts in the binding energies of Fe, Ni, O, and S were observed, indicating the formation of new active species due to anodic reconstruction. Notably, the XPS spectra of S exhibited significant changes, with the disappearance of the S 2p3/2 and S 2p1/2 peaks after reconstruction, suggesting that no metal–sulfur bonds remained in the newly formed active species following sulfurization. Additionally, changes in metal oxidation states and oxygen-containing bonds were analyzed by fitting the relevant XPS peaks. As shown in Fig. 4g, after the chronoamperometry test, the proportion of high-valence metals and S–O electronic states significantly increased in Fe–Ni3S4/Cr2O3, Fe–Ni3S4, and Ni3S4/Cr2O3, indicating the formation of high-valence metal MOOH species and amorphous S–O metal species on the surface, signifying obvious reconstruction. In addition, we note that the proportion of high-valence metals and S–O electronic states in Fe–Ni3S4/Cr2O3-A is lower than those of Fe–Ni3S4-A and Ni3S4/Cr2O3-A. This is because the sulfide component of Fe–Ni3S4/Cr2O3 possesses more electrons than the sulfide in Ni3S4/Cr2O3 and single component Fe–Ni3S4 under the influence of the Fe doping enhanced BIEF. The electron-rich state of the sulfide from Fe–Ni3S4/Cr2O3 prevents the structural oxidation at high anode potential, which results in the inhibition of the reconstruction behavior in the OER.
 | ||
| Fig. 4 High-resolution XPS patterns of the catalyst before and after stability testing. (a) Fe 2p (b) Ni 2p (c) O 1s, and (d) S 2p. Bode phase plots of in situ EIS on (e) Ni3S4/Cr2O3 and (f) Fe–Ni3S4/Cr2O3. (g) Histograms of the ratios of Ni2+, Ni3+, Fe2+, Fe3+, OL, OOH, Ssat, and SS–O in Ni3S4/Cr2O3, Fe–Ni3S4, and Fe–Ni3S4/Cr2O3 before and after reconstruction. Operando Raman spectra of (h) Ni3S4/Cr2O3 and (i) Fe–Ni3S4/Cr2O3. | ||
This was further confirmed by inductively coupled plasma (ICP) analysis. As is well known, the reconstruction of sulfides, especially the formation of active MOOH species, is typically accompanied by the leaching of S ions. The amount of S leaching in the electrolyte after the OER is the lowest, which also indicates the inhibited reconstruction of sulfide in Fe–Ni3S4/Cr2O3 (Fig. S10†). In addition, the reduced Fe and Ni leaching from Fe–Ni3S4/Cr2O3 highlights the catalyst's improved structural stability during reconstruction. The suppressed reconstruction of sulfides in Fe–Ni3S4/Cr2O3 helps prevent conductivity loss and mitigate the weakening of synergistic effects between the true active sites and the pre-catalyst.
The Bode phase diagrams of Fe–Ni3S4/Cr2O3, Ni3S4/Cr2O3, and Fe–Ni3S4 all show a peak signal in the high frequency region, which indicates that the reconstruction occurs (Fig. 4e and f and S11†).22,23 Additionally, the slightly lower reconstruction peak signal in Fe–Ni3S4/Cr2O3 further suggests that the Fe doping enhanced BIEF inhibits the reconstruction of the sulfide component. In situ Raman spectroscopy (Fig. 4h, i and S12†) also revealed the active species formed after reconstruction of the pre-catalyst. Characteristic peaks at 479 cm−1 and 558 cm−1, corresponding to MOOH eg bending and A1g stretching vibrations, were observed in Fe–Ni3S4/Cr2O3, Fe–Ni3S4, and Ni3S4/Cr2O3, confirming that the reconstruction products were predominantly MOOH.24,25 Based on the elemental composition of the pre-catalyst, we concluded that the reconstruction products of Fe–Ni3S4/Cr2O3 and Fe–Ni3S4 were Fe–NiOOH, while the reconstruction product of Ni3S4/Cr2O3 was NiOOH. The presence of Fe–NiOOH peaks within a narrow voltage range in Fe–Ni3S4/Cr2O3 further supports the conclusion that the enhanced BIEF suppresses the degree of reconstruction. In conclusion, the Fe-doping enhanced BIEF interferes with the reconstruction behavior of Fe–Ni3S4/Cr2O3, suppresses the excessive reconstruction of the sulfide component, and improves the catalyst's structural stability. Moreover, it successfully modifies the true active sites, leading to the formation of Fe-doped NiOOH active species on the catalyst surface, which enhances OER activity.
The reaction mechanism of the OER catalyst directly influences its catalytic activity. Therefore, we further explored the OER mechanism of Fe–NiOOH, which is the true catalytic center of Fe–Ni3S4/Cr2O3 restructures under the Fe-doped enhanced BIEF. We first evaluated the OER activity and pH dependence of the catalyst in KOH electrolytes with varying pH values (Fig. S13†). As shown in Fig. 5a, Fe–Ni3S4/Cr2O3 and Fe–Ni3S4 exhibited significant fluctuations in the reaction currents at the rated OER operating voltage, indicating strong pH dependence. This is because the O as the redox center in the LOM can tolerate a certain degree of charge accumulation in the reaction process, and the non-concerted proton-electron transfer decoupling in the process of OH* deprotonation makes the OH* deprotonation sensitive to the pH of the electrolyte, which leads to the dependence of the active pH of the OER.26 The marked pH dependence of Fe–Ni3S4/Cr2O3 and Fe–Ni3S4 confirms that their true active center Fe–NiOOH triggers the LOM. In contrast, the conventional AEM, where metals act as the redox center and involve concerted electron and proton transfer during OH* deprotonation, shows negligible pH dependence in the catalyst's activity. Thus, the true active center NiOOH of Ni3S4/Cr2O3 and Ni3S4 follows the AEM.
 | ||
| Fig. 5 (a) Current densities of Cr2O3, Ni3S4, Ni3S4/Cr2O3, Fe–Ni3S4, and Fe–Ni3S4/Cr2O3 at 1.6 V vs. RHE as a function of pH value. (b) Polarization curves for Ni3S4/Cr2O3, Fe–Ni3S4, and Fe–Ni3S4/Cr2O3 in 1.0 M KOH with and without TMAOH. (c) Polarization curves for Ni3S4/Cr2O3, Fe–Ni3S4, and Fe–Ni3S4/Cr2O3 in 1.0 M KOH with and without CH3OH. Under different concentrations of KOH solutions, the differential capacitance (Cd) curves of (d) Fe–Ni3S4/Cr2O3, (e) Fe–Ni3S4. (f) PZC of Ni3S4/Cr2O3, Fe–Ni3S4, and Fe–Ni3S4/Cr2O3 in 20, 40, 80, and 100 mM KOH solutions. (g) Ks of Ni3S4/Cr2O3 and Fe–Ni3S4/Cr2O3. (h) Variations of anodic peak current densities for Ni3S4/Cr2O3 and Fe–Ni3S4/Cr2O3 electrodes as a function of the square root of scan rates. (i) Schematic energy bands relative to the Fermi level in consideration of Mott–Hubbard splitting. | ||
TMAOH intermediate trapping experiments can further confirm the activation of the LOM in Fe–Ni3S4/Cr2O3 and Fe–Ni3S4, where Fe–NiOOH is the true active site. In LOM-based catalysts, oxygen from the catalyst lattice couples with surface-adsorbed oxygen to form a unique peroxo-like negative species (O22−). The tetra-methylammonium cation (TMA+) can trap O22−via strong electrostatic interactions, inhibiting the further release of O2.27,28 As shown in Fig. 5b and S14,† the OER activity of Fe–Ni3S4/Cr2O3 and Fe–Ni3S4 was significantly suppressed upon TMAOH addition, while that of Ni3S4/Cr2O3 and Ni3S4 remained nearly unaffected. This also indicates the triggering of the LOM at the true active site Fe–NiOOH from Fe–Ni3S4/Cr2O3 and Fe–Ni3S4. Due to the influence of Fe doping on the electronic structure of the pre-catalyst and the reconstructed product, the triggered LOM of Fe–NiOOH (the true catalytic activity center after Fe–Ni3S4/Cr2O3 and Fe–Ni3S4 reconstruction) avoids the formation of high binding energy OOH* from the AEM of NiOOH (the true catalytic activity center of Ni3S4/Cr2O3 and Ni3S4 after reconstruction), thereby breaking the limitation of the scaling relationship and significantly enhancing the OER activity.
Based on electrochemical performance testing results, we found that although both Fe–Ni3S4/Cr2O3 and Fe–Ni3S4 activate the LOM, the OER activity of Fe–Ni3S4/Cr2O3 far exceeds that of single-component Fe–Ni3S4. Research indicates that the OER mechanism switches from the AEM to the LOM, where the electron transfer in the OH* deprotonation process changes from delocalized metal orbitals to higher energy localized oxygen non-bonding states. As a result, OH* deprotonation becomes the rate-determining step of the LOM, thereby influencing OER activity. To reveal the impact of the BIEF on the reaction mechanism, we conducted methanol molecular probe experiments.
The nucleophilic reagent methanol can capture the electrophilic species OH*, creating strong competition for OH* adsorption between the methanol oxidation reaction and the OER. Therefore, methanol, used as a probe, can detect the adsorption ability of OH*.29Fig. 5c shows that the current density of Fe–Ni3S4/Cr2O3 changed less than that of Fe–Ni3S4 after adding methanol, indicating that Fe–Ni3S4/Cr2O3 has a lower OH* adsorption energy. This is attributed to the BIEF at the heterogeneous interface of Fe–Ni3S4/Cr2O3, which also regulates the electronic structure of FeOOH. The reduced adsorption energy of OH* thermodynamically favors the subsequent OH* deprotonation, thereby accelerating LOM.
Additionally, we also studied the effect of the Fe doping-enhanced BIEF on the catalyst's surface microenvironment. The minimal differential capacitance of the dilute electrolyte can determine the catalyst's potential of zero charge (PZC), which is defined as the potential at which no excess charge exists on the electrode surface (Fig. 5d, e and S15†).30 PZC is typically used to assess whether specific adsorption occurs and to evaluate the adsorption capacity.31 By gradually adding small amounts of KOH to the KCl solution, we obtained the PZC curve shown in Fig. 5f, which reflects the change in the PZC with varying amounts of KOH addition. As KOH concentration increased, the PZC values of Fe–Ni3S4/Cr2O3, Fe–Ni3S4 and Ni3S4/Cr2O3 became more negative, indicating specific OH− adsorption on the catalyst's interphase (IPH) according to the Esin–Markov effect. Notably, Fe–Ni3S4/Cr2O3 showed a more pronounced shift in PZC with increasing KOH, suggesting that the Fe doping enhanced BIEF strengthens the catalyst's OH− adsorption, enriching OH− on the surface.
Further CV analysis at different scan rates (Fig. S16†) was performed to assess OH− diffusion behavior on the electrode surface. The redox constant (Ks) was determined using the Laviron method, revealing that Fe–Ni3S4/Cr2O3 exhibited a higher Ks than Ni3S4/Cr2O3, indicating faster OH− diffusion from the electrolyte to the electrode surface (Fig. 5g).32,33 Additionally, the diffusion coefficient of OH− on Fe–Ni3S4/Cr2O3 was calculated to be 1.33 times that of Ni3S4/Cr2O3 using the Randles–Sevcik equation, supporting the conclusion that the Fe doping-enhanced BIEF accelerates OH− diffusion kinetics (Fig. 5h).34 These accelerated OH− diffusion dynamics, combined with enhanced OH* deprotonation at the Fe–Ni3S4/Cr2O3 interface, optimize the OER activity of Fe–NiOOH via the LOM.
Using orbital hybridization theory, we explore how the Fe doping enhanced BIEF activates and optimizes the LOM of Fe–NiOOH, which is crucial for designing efficient OER catalysts (Fig. 5i).6 The electronic states near the Fermi level are critical for determining the OER mechanism of a catalyst. The hybridization between the O 2p orbitals and Fe, Ni 3d orbitals, driven by differences in atomic electronegativity, results in the formation of bonding (M–O) and antibonding (M–O)* bands.35 In the antibonding (M–O)* band, electrons are further localized by strong d–d Coulomb interactions, splitting into an empty upper Hubbard band and a filled lower Hubbard band (LHB). The bonding M–O orbitals primarily exhibit oxygen-like characteristics, while the LHB in the antibonding orbitals exhibits metallic characteristics.36 Oxygen and sulfur, being highly electronegative, cause significant charge transfer between metal and non-metal atoms.27 Therefore, as a typical Mott–Hubbard insulator, the M–S or M–O electronic states near the Fermi level of Ni3S4/Cr2O3 and its reconstructed product NiOOH serve as the LHB. Thermodynamically, the metal acts as the primary redox center, with electron transfer occurring between the metal and the surface-adsorbed oxygen intermediates, following the AEM.
In Fe–NiOOH formed by the reconstruction of Fe–Ni3S4 or Fe–Ni3S4/Cr2O3, Fe doping optimizes the electronic states of Ni atoms, reduces the electronegativity difference between Ni and O atoms, lowers the energy required for charge transfer between atoms, and simultaneously enhances the d–d Coulomb interaction between electrons in the anti-bonding band. This allows the LHB to penetrate into the O 2p band, generating non-bonding oxygen states near the Fermi level.7,37 This activates the redox behavior of oxygen atoms in the lattice, making electron transfer between lattice oxygen and oxygen intermediates possible during the OER process, thereby triggering the LOM. The coupling of lattice oxygen with adsorbed oxygen generates O22−, avoiding the formation of the high-energy OOH* species in the AEM, breaking the scaling relationship limit, and accelerating the OER.38,39 Furthermore, under the influence of the Fe doping enhanced BIEF at the heterogeneous interface, the surface of Fe–NiOOH formed from Fe–Ni3S4/Cr2O3 creates a localized high-efficiency electronic region. This further optimizes the hybridization between the metal and oxygen atomic orbitals, enhancing the covalency of metal–oxygen bonds and increasing the concentration of non-bonding oxygen states. The optimized metal d-band and oxygen p-band reduce energy consumption during the deprotonation process, thereby significantly enhancing OER activity.
4. Conclusion
In summary, we have significantly enhanced the OER activity of Fe–NiOOH, a reconstruction product of Fe–Ni3S4/Cr2O3, through doping modification and the construction of BIEF. In a 1 M KOH electrolyte, the catalyst exhibits an overpotential of 282 mV at a current density of 10 mA cm−2 and maintains stable performance for 100 h. Additionally, the catalyst demonstrates excellent resistance to oxidation and corrosion from Cl− ions in simulated seawater, preserving high OER activity. It also operates efficiently and stably for 40 h in a commercially scaled alkaline electrolyzer. Various characterization techniques have revealed the source of the exceptional catalytic performance. pH-dependence and TMAOH intermediate trapping experiments indicate that Fe doping optimized Fe–NiOOH metal–oxygen orbital hybridization activates the LOM. The activated lattice oxygen participates in the coupling of adsorbed oxygen, bypassing the formation of the high energy OOH* species typically generated in the traditional AEM. Band structure characterization and ICP testing indicate that the Fe doping enhanced BIEF stabilizes the reconstruction of Fe–Ni3S4 with an electron-rich region at the heterogeneous interface, helping mitigate the decline in structural stability caused by ion leaching during the reaction process. Moreover, surface microenvironment characterization and methanol probe experiments demonstrate that the BIEF facilitates the OH− diffusion dynamics and HO* deprotonation process, which is the rate-limiting step in the LOM, further enhancing OER activity. This study highlights the effective structural regulation of the pre-catalyst reconstruction product using multiple modification strategies and establishes a structure–activity relationship between the pre-catalyst and OER performance.Data availability
The data supporting this article have been included in the main text and the ESI.†Author contributions
K. Wang, H. Xu and G. He conceived and designed this work. K. Wang and C. Ni carried out the synthesis and electrochemical measurements. All authors participated in the analysis of the data, discussed the results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 52472043, 22305025, and 22078028), the Natural Science Foundation of Jiangsu Province (BK20230640), and the China National Petroleum Corporation (CNPC) Innovation Found (2024DQ02-0310). We also thank the Analysis and Testing Center of Changzhou University for assistance in characterization studies.References
- L. Su, H. Wu, S. Zhang, C. Cui, S. Zhou and H. Pang, Adv. Mater., 2025, 37, 2414628 Search PubMed.
- Z. Qiu, X. Guo, S. Cao, M. Du, Q. Wang, Y. Pi and H. Pang, Angew. Chem., Int. Ed., 2025, 64, e202415216 CrossRef CAS PubMed.
- H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, A. Kravchenko, T. Liu, Y. Yang and Y. Fang, Nat. Catal., 2022, 5, 414–429 CrossRef CAS.
- T. Wu, Y. Sun, X. Ren, J. Wang, J. Song, Y. Pan, Y. Mu, J. Zhang, Q. Cheng and G. Xian, Adv. Mater., 2023, 35, 2207041 CrossRef CAS PubMed.
- J. Liang, K. Li, F. Shi, J. Li, J. n. Gu, Y. Xue, C. Bao, M. Guo, J. Jia and M. Fan, Angew. Chem., Int. Ed., 2024, 63, e202407870 CrossRef CAS PubMed.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- H. Wang, T. Zhai, Y. Wu, T. Zhou, B. Zhou, C. Shang and Z. Guo, Adv. Sci., 2023, 10, 2301706 CrossRef CAS PubMed.
- X. Wang, H. Zhong, S. Xi, W. S. V. Lee and J. Xue, Adv. Mater., 2022, 34, 2107956 CrossRef CAS PubMed.
- C.-X. Zhao, J.-N. Liu, C. Wang, J. Wang, L. Song, B.-Q. Li and Q. Zhang, Energy Environ. Sci., 2022, 15, 3257–3264 RSC.
- Y. Hu, Y. Zheng, J. Jin, Y. Wang, Y. Peng, J. Yin, W. Shen, Y. Hou, L. Zhu and L. An, Nat. Commun., 2023, 14, 1949 CrossRef CAS PubMed.
- X. Liu, J. Meng, J. Zhu, M. Huang, B. Wen, R. Guo and L. Mai, Adv. Mater., 2021, 33, 2007344 CrossRef CAS PubMed.
- H. Xu, L. Yang, Y. Liu, L. Jin, K. Wang, G. He and H. Chen, Fuel, 2024, 377, 132796 CrossRef CAS.
- K. Wang, H. Xu, H. Xing, L. Jin, X. Qian, G. He and H. Chen, Chem. Eng. J., 2024, 500, 157316 CrossRef CAS.
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed.
- M. J. Craig, G. Coulter, E. Dolan, J. Soriano-López, E. Mates-Torres, W. Schmitt and M. García-Melchor, Nat. Commun., 2019, 10, 4993 CrossRef PubMed.
- Y. Lyu, J. Zheng, Z. Xiao, S. Zhao, S. P. Jiang and S. Wang, Small, 2020, 16, 1906867 CrossRef CAS PubMed.
- H. Xu, L. Yang, L. Jin, Y. Liu, K. Wang, J. Chen, G. He and H. Chen, J. Colloid Interface Sci., 2025, 677, 158–166 CrossRef CAS PubMed.
- S. Zhang, C. Tan, R. Yan, X. Zou, F. L. Hu, Y. Mi, C. Yan and S. Zhao, Angew. Chem., Int. Ed., 2023, 135, e202302795 CrossRef.
- W. Li, L. Zhao, X. Jiang, Z. Chen, Y. Zhang and S. Wang, Adv. Funct. Mater., 2022, 32, 2207727 CrossRef CAS.
- N. Wang, P. Ou, S. F. Hung, J. E. Huang, A. Ozden, J. Abed, I. Grigioni, C. Chen, R. K. Miao and Y. Yan, Adv. Mater., 2023, 35, 2210057 CrossRef CAS PubMed.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- P. Zhao, S. Fu, Y. Luo, C. Peng, L. Cheng and Z. Jiao, Small, 2023, 19, 2305241 CrossRef CAS PubMed.
- Y. Lu, C. L. Dong, Y. C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- H. Liu, W. Shen, H. Jin, J. Xu, P. Xi, J. Dong, Y. Zheng and S. Z. Qiao, Angew. Chem., Int. Ed., 2023, 62, e202311674 CrossRef CAS PubMed.
- H. Lei, L. Ma, Q. Wan, S. Tan, B. Yang, Z. Wang, W. Mai and H. J. Fan, Adv. Energy Mater., 2022, 12, 2202522 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- X. Li, C. Deng, Y. Kong, Q. Huo, L. Mi, J. Sun, J. Cao, J. Shao, X. Chen and W. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202309732 CrossRef CAS PubMed.
- C. Yang, O. Fontaine, J. M. Tarascon and A. Grimaud, Angew. Chem., Int. Ed., 2017, 129, 8778–8782 CrossRef.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
- B. Huang, J. Yan, Z. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2024, e202409419 Search PubMed.
- C. Liu, G. Zhang, W. Zhang, Z. Gu and G. Zhu, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2209979120 CrossRef CAS PubMed.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 100, 263–270 Search PubMed.
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao and Z. Shu, J. Am. Chem. Soc., 2020, 142, 12087–12095 Search PubMed.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS PubMed.
- Z. Wang, W. A. Goddard III and H. Xiao, Nat. Commun., 2023, 14, 4228 CrossRef CAS PubMed.
- H. Wu, Q. Huang, Y. Shi, J. Chang and S. Lu, Nano Res., 2023, 16, 9142–9157 CrossRef.
- S. Xin, Y. Tang, B. Jia, Z. Zhang, C. Li, R. Bao, C. Li, J. Yi, J. Wang and T. Ma, Adv. Funct. Mater., 2023, 33, 2305243 CrossRef CAS.
- H. Jin, X. Liu, P. An, C. Tang, H. Yu, Q. Zhang, H.-J. Peng, L. Gu, Y. Zheng and T. Song, Nat. Commun., 2023, 14, 354 CrossRef CAS PubMed.
- R. Chen, Z. Wang, S. Chen, W. Wu, Y. Zhu, J. Zhong and N. Cheng, ACS Energy Lett., 2023, 8, 3504–3511 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08789e |
| This journal is © The Royal Society of Chemistry 2025 |