
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd single atoms on g-C3N4 photocatalysts: minimum loading for maximum activity†
Velu
Jeyalakshmi‡
ab,
Siming
Wu‡
a,
Shanshan
Qin
 a,
Xin
Zhou
a,
Bidyut Bikash
Sarma
c,
Dimitry E.
Doronkin
d,
Jan
Kolařík
e,
Miroslav
Šoóš
b and
Patrik
Schmuki
*ae
a,
Xin
Zhou
a,
Bidyut Bikash
Sarma
c,
Dimitry E.
Doronkin
d,
Jan
Kolařík
e,
Miroslav
Šoóš
b and
Patrik
Schmuki
*ae
aDepartment of Materials Science WW4-LKO, Friedrich-Alexander-University of Erlangen-Nuremberg, Martensstrasse 7, 91058 Erlangen, Germany. E-mail: schmuki@ww.uni-erlangen.de
bDepartment of Chemical Engineering, University of Chemistry and Technology, Technická 3, Prague 160 00, Czech Republic
cLaboratoire de Chimie de Coordination (LCC), CNRS, Université de Toulouse, INPT, UPR 8241, 205 Route de Narbonne, 31077 Toulouse Cedex 4, France
dInstitute of Catalysis Research and Technology, KIT, Hermann-von Helmholtz Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
eRegional Centre of Advanced Technologies and Materials, Šlechtitelů 27, 78371 Olomouc, Czech Republic
First published on 12th February 2025
Abstract
Noble metal single atoms (SAs) on semiconductors are increasingly explored as co-catalysts to enhance the efficiency of photocatalytic hydrogen production. In this study, we introduce a “spontaneous deposition” approach to anchor Pd SAs onto graphitic carbon nitride (g-C3N4) using a highly dilute tetraaminepalladium(II) chloride precursor. Maximized photocatalytic activity and significantly reduced charge transfer resistance can be achieved with a remarkably low Pd loading of 0.05 wt% using this approach. The resulting Pd SA-modified g-C3N4 demonstrates a remarkable hydrogen production efficiency of 0.24 mmol h−1 mg−1 Pd, which is >50 times larger than that of Pd nanoparticles deposited on g-C3N4via conventional photodeposition. This significant enhancement in catalytic performance is attributed to improved electron transfer facilitated by the optimal coordination of Pd SAs within the g-C3N4 structure.
Introduction
In recent years, the demand for hydrogen as a clean renewable energy carrier has grown rapidly, driven by the need to replace traditional fossil fuels. Photocatalytic water splitting is a promising and most elegant way to directly convert solar energy into clean, renewable hydrogen (H2).1,2 Since the pioneering work of Fujishima et al., titanium dioxide (TiO2) has become the most extensively studied material for this purpose.3–5 More recently, graphitic carbon nitride (C3N4) is being increasingly explored as it shares many advantageous features with TiO2, such as high stability, abundance, and a suitable band structure for water splitting but provides the key advantage of strong visible light absorption, resulting in significantly enhanced solar light utilization.6–9Nevertheless, due to strong kinetic hindrance for the photocatalytic hydrogen evolution reaction (HER) on the C3N4 surface, an enhanced efficiency highly depends on the development of highly active co-catalysts – typically noble metals such as Pt, Pd, and Rh need to be deposited on the semiconductor surface to obtain a reasonable H2 production rate. Despite efforts made using non-precious metal HER electrocatalysts,10 noble metal-based materials remain nearly irreplaceable due to their superior activity and chemical stability. However, the scarcity and high cost of these metals pose significant challenges to the large-scale application of these noble metal-based co-catalysts. This has prompted considerable efforts to minimize noble metal usage while retaining high catalytic performance. In this context, single-atom catalysts have garnered wide attention due to their maximized atom utilization efficiency.11–13
As for many other semiconductors and also for C3N4 substrates, a wide range of noble metal SAs have been successfully deposited as nanoparticles, clusters or SA, and accordingly improvement in photocatalytic H2 production has been reported.14–21 In search of the most active SA species, Akinaga et al. conducted a remarkable study on ten transition metal elements, including Cu, Ni, Pd, Pt, Rh, Ru, Ag and Au, as SAs anchored on g-C3N4. Among the metals tested, Pd demonstrated significantly higher hydrogen evolution activity compared to other precious metals such as Pt, Rh as well as other transition metals.14 The authors ascribed the superior activity of Pd on C3N4 to the suitable electronic structure of this metal on C3N4. In their work, Akinaga et al. used a photodeposition approach to achieve a relatively high SA loading (>0.5 wt%).
However, for many semiconductors, the activity of SAs in photocatalysis is extremely dependent on the deposition approach.13,21–23 Namely, for Pt SAs on TiO2, it was reported that “reactive” deposition leads to highly active SA configurations that can provide maximized H2 production, i.e., a very high catalytic efficiency can be reached at very low noble-metal loading. This approach relies on the surface reaction of highly dilute solutions of suitable noble-metal precursors.24–27
In the present work, we first explore various Pd precursors for the feasibility of a reactive SA attachment on C3N4. We find that tetraaminepalladium(II) chloride – Pd(NH3)4Cl2 as a Pd precursor solution with C3N4 allows for an adjustable Pd SA loading with a wide range of deposition concentrations from 0.04 wt% to 0.75 wt%. Our results show that by the reaction of a minimal amount of a 0.05 mM precursor, maximum photocatalytic efficiency can be obtained. The photocatalytic hydrogen production activity of such Pd SA-decorated C3N4 achieves a normalized H2 production activity of 0.24 mmol h−1 mg−1 Pd, which is 55 times higher than that observed with Pd nanoparticle-decorated C3N4 at an effective loading that is more than 10 times lower than that typically reported in the literature for Pd on C3N4. The superior activity of Pd SAs/C3N4 is attributed to the strong coordination of Pd SAs within the C3N4 structure, forming a highly stable and catalytically effective configuration that drastically reduces the charge transfer resistance for the HER. These results illustrate how a refined anchoring of SAs on substrates can enable more cost- and production-effective use of precious metals in photocatalysis.
Results and discussion
Nanosheets of g-C3N4 were synthesized using a thermal polycondensation method starting from an equimolar mixture of melamine and dicyandiamide, followed by thermal exfoliation, as described in the literature.28–30 In order to explore the feasibility of direct deposition of a (reactive) SA such as Pd on C3N4, we examined different precursor species, namely tetraamminepalladium(II) chloride (Pd(NH3)4Cl2), palladium(II) chloride (PdCl2) and ammonium hexachloropalladate(IV) ((NH4)2[PdCl6]). To investigate the reactive deposition behavior, we used three different Pd precursors at a concentration of 2 mM to decorate Pd on C3N4. We then evaluated the general deposition behavior with electron microscopy and XPS and also evaluated the photocatalytic H2 production performance. Among the samples, XPS results reveal that both PdCl2 and (NH4)2[PdCl6] lead to relatively high Pd loadings (>1 at%) (Fig. S1a and b†); however, the strong Cl 2p signals in the XPS spectra (Fig. S1c†) indicate that most of the Pd precursor did not react with C3N4, i.e., the precursor is just physically adsorbed on the C3N4 surface. In the SEM images of these two samples (Fig. S2†), obvious Pd nanoparticles can be seen, due to the agglomeration caused by high loading. In contrast, the Pd(NH3)4Cl2 sample shows no visible metal nanoparticle formation in SEM (Fig. 1a), non-metallic Pd position in XPS (Fig. S1a†) and no detectable Cl 2p signal (Fig. S1b†), indicating a complete reaction of this particular precursor with the C3N4 surface. | ||
| Fig. 1 (a) SEM image, (b) original HAADF-STEM image, (c) the HAADF-STEM image with individual Pd SAs highlighted by red dots and yellow circles, (d) HAADF-STEM image and the corresponding EDS mapping (e) C, (f) N, and (g) Pd of Pd SAs/C3N4. | ||
Fig. 1a shows the SEM image of Pd-deposited g-C3N4 (Pd SAs/C3N4) using Pd(NH3)4Cl2 at a concentration of 0.002 mM, and Fig. S3† shows the SEM image of neat g-C3N4. The introduction of Pd SAs does not affect the morphology of C3N4 – both samples show a sheet-like structure with a thin layer thickness of approximately 16 nm. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Pd SAs/g-C3N4 is shown in Fig. 1b and c, which confirms the presence of individual Pd atoms (highlighted in Fig. 1c with red dots and yellow circles). Also, in the HAADF-STEM image (Fig. 1d), there are no observable Pd agglomeration on the g-C3N4 surface. Energy-dispersive X-ray spectroscopy (EDX) mapping (Fig. 1e–g) further proves the uniform dispersion of Pd SAs throughout the g-C3N4 structure. The density of Pd SAs was calculated as 1.6 × 106 μm−2 from HAADF-STEM images shown in Fig. S4.†
X-ray diffraction (XRD) patterns of g-C3N4 and Pd SAs/g-C3N4 are presented in Fig. 2a. Both samples display two distinct diffraction peaks at 13° and 27.6°, corresponding to the (100) and (002) crystal planes of g-C3N4, respectively.31 Notably, no diffraction peaks related to metallic Pd are observed in the Pd SAs/g-C3N4 sample (as is expected for the SA-decorated sample).14
 | ||
| Fig. 2 (a) X-ray diffraction pattern, (b–d) XPS spectra of (b) C 1s, (c) N 1s and (d) Pd 3d for C3N4 and Pd SAs/C3N4, (e) XANES spectra of Pd/C3N4 at Pd K-edge, and (f) Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of Pd SAs/C3N4. | ||
X-ray photoelectron spectroscopy (XPS) was utilized to investigate the chemical state of g-C3N4 and Pd SAs on C3N4 (Fig. 2b–d and Table S1†). The high-resolution C 1s XPS spectrum (Fig. 2b) of both samples can be fitted by three peaks at 284.7 eV, 286.2 eV, and 288.1 eV corresponding to C–C, C–N and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of the heptazine ring carbon structure, respectively.16 The fitted N 1s spectra (Fig. 2c) exhibit peaks at 398.7 eV (N1), 400 eV (N2), and 401.4 eV (N3), representing the sp2 hybridized aromatic two-coordinated (N2c) nitrogen of the triazine unit (CN–C, i.e., pyridinic N) and three coordinated (N3c) bridging N atoms connected to carbon as N–(C)3 groups and amino functional (C–NH/NHx) groups respectively.16,32,33 The Pd 3d spectrum of Pd SAs/C3N4 shows two peaks (Fig. 2d), doublets at 337.6 and 342.9 eV corresponding to Pdδ+ 3d5/2 and Pdδ+ 3d3/2 (0 < δ < 2), respectively.16 For comparison, Pd nanoparticles were deposited on g-C3N4 (Pd NPs/C3N4) using an established photodeposition method described in the literature.34,35 SEM images of Pd NPs/C3N4 (Fig. S5†) clearly show distinct Pd nanoparticles on the C3N4 surface with their typical diameter in the range of 7–15 nm. For this sample, the Pd 3d XPS spectra (Fig. S6†) of Pd NPs/C3N4 exhibit doublets at 335 eV and 340 eV, which are typically attributed to metallic Pd0.33
N of the heptazine ring carbon structure, respectively.16 The fitted N 1s spectra (Fig. 2c) exhibit peaks at 398.7 eV (N1), 400 eV (N2), and 401.4 eV (N3), representing the sp2 hybridized aromatic two-coordinated (N2c) nitrogen of the triazine unit (CN–C, i.e., pyridinic N) and three coordinated (N3c) bridging N atoms connected to carbon as N–(C)3 groups and amino functional (C–NH/NHx) groups respectively.16,32,33 The Pd 3d spectrum of Pd SAs/C3N4 shows two peaks (Fig. 2d), doublets at 337.6 and 342.9 eV corresponding to Pdδ+ 3d5/2 and Pdδ+ 3d3/2 (0 < δ < 2), respectively.16 For comparison, Pd nanoparticles were deposited on g-C3N4 (Pd NPs/C3N4) using an established photodeposition method described in the literature.34,35 SEM images of Pd NPs/C3N4 (Fig. S5†) clearly show distinct Pd nanoparticles on the C3N4 surface with their typical diameter in the range of 7–15 nm. For this sample, the Pd 3d XPS spectra (Fig. S6†) of Pd NPs/C3N4 exhibit doublets at 335 eV and 340 eV, which are typically attributed to metallic Pd0.33
The nature of Pd species was further investigated by X-ray absorption spectroscopy (XAS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements. Fig. 2e and f show the absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra of Pd SAs/C3N4. The X-ray absorption spectrum of Pd SAs/C3N4 measured at the Pd K-edge (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 350 eV) indicates the presence of Pd atoms that are non-metallic and carry a positive charge.15,16,20 The EXAFS analysis and the corresponding Fourier transformed (FT) radial distribution function of Pd SAs/C3N4 show a peak at approximately 1.5 Å (without phase correction), attributed to the Pd–N bond, and no obvious scattering is observed for the metallic Pd–Pd bonding.16,20 CO-DRIFT spectra of Pd SAs/C3N4 (Fig. S7†) show a CO vibrational peak at 2125 cm−1, which is characteristic of linearly bonded CO on a Pd single site (usually Pd2+).36,37 These results are well in line with the XPS results, i.e., Pd SAs are N-coordinated in C3N4 with an oxidation state ≈ 2.
350 eV) indicates the presence of Pd atoms that are non-metallic and carry a positive charge.15,16,20 The EXAFS analysis and the corresponding Fourier transformed (FT) radial distribution function of Pd SAs/C3N4 show a peak at approximately 1.5 Å (without phase correction), attributed to the Pd–N bond, and no obvious scattering is observed for the metallic Pd–Pd bonding.16,20 CO-DRIFT spectra of Pd SAs/C3N4 (Fig. S7†) show a CO vibrational peak at 2125 cm−1, which is characteristic of linearly bonded CO on a Pd single site (usually Pd2+).36,37 These results are well in line with the XPS results, i.e., Pd SAs are N-coordinated in C3N4 with an oxidation state ≈ 2.
We then used the above deposition approach to place Pd SAs from Pd(NH3)4Cl2 solutions in the concentration range of 0.0005 mM to 10 mM on C3N4. Fig. 3a shows the Pd 3d XPS spectra for these Pd SAs loaded on C3N4 samples. Notably, neither metallic Pd peaks nor Cl 2p peaks (Fig. S8†) are observed under any of the deposition conditions. Instead, the incorporation of Pd SAs is evident across all samples, as indicated by the Pd 3d doublet peaks at 337.6 and 342.9 eV. The XPS data align with the SEM images shown in Fig. S9,† where no Pd nanoparticles are observed in any of the samples, even at the highest precursor concentration of 10 mM.
 | ||
| Fig. 3 (a) Pd 3d XPS spectra of Pd SAs/C3N4 at different concentrations of tetraaminepalladium chloride, (b) photocatalytic H2 evolution, (c) evolved H2 at different Pd SA loadings, and (d) normalized H2 evolution rates for different concentrations Pd SAs using tetraaminepalladium chloride. | ||
The bulk loading of the samples prepared using different concentrations of Pd(NH3)4Cl2 solutions was further quantified by atomic absorption spectroscopy (AAS) – the results are shown in Table S1.† As the concentration of the Pd precursor increases, the loading of Pd SAs increases (Fig. S10†), which is consistent with the XPS data (Table S2†). For the highest precursor concentration of 10 mM, the Pd SA loading reaches 0.75 wt%.
We then examined for all samples the photocatalytic H2 evolution using a 365 nm LED light source with an intensity of 65 mW cm−2 and an aqueous solution of 10% triethanolamine (TEOA) as a hole scavenger.19,38,39 From the results shown in Fig. 3b, it is evident that the decoration of either Pd NPs or Pd SAs significantly enhances the H2 production activity compared to bare C3N4. In the concentration range of 0.0005 mM to 0.05 mM, corresponding to Pd SA loadings from 0.04 wt% to 0.26 wt% (Fig. S10†), the photocatalytic H2 production activity increases with loading (Fig. 3c). It is worth mentioning that Pd SAs deposited through our method, utilizing the Pd(NH3)Cl2 precursor, lead to the notable finding that even a minimal loading of 0.05 wt% outperformes Pd NPs synthesized via photodeposition, the latter having a much higher loading of 1.5 wt% (Table S2†). As a side note, this loading is also much more efficient than using PdCl2 and (NH4)2PdCl6 precursors (see Fig. S11†). Also, in this comparison, the photocatalytic H2 production results show that an SA deposition approach using Pd(NH3)4Cl2 leads to much higher activity, despite the significantly lower Pd loading compared with PdCl2 and (NH4)2PdCl6.
In general, the data clearly show that the H2 production amount increases with Pd precursor concentration loading until reaching a maximum at 0.05 mM (with a Pd SA loading of 0.26 wt%). Beyond this point, a further increase in Pd loading does not increase the activity any further but even leads to a slight drop in the activity and in the 10 mM case a very obvious decrease (Fig. 3c). This is likely due to the decrease in the density of Pd SAs and the formation of Pd agglomerates, which may increase charge recombination.40
To further assess and compare the effectiveness of Pd as a co-catalyst in both single-atom and nanoparticle forms, we normalized the data from Fig. 3b relative to Pd loading (Table S1†); the results are shown in Fig. 3d. The analysis reveals that the highest mass-specific photocatalytic efficiency, resulting in an H2 production rate of 0.24 mmol h−1 mg−1 Pd, is achieved with 0.002 mM Pd precursor (0.05 wt%). This efficiency is 55 times higher than that obtained through conventional photodeposition of Pd nanoparticles on g-C3N4, highlighting the superiority of our reactive deposition method for optimizing photocatalytic H2 production. The exceptional performance of low Pd SA loading on g-C3N4, prepared using our direct deposition method, is evident when compared to other Pd SA-loaded g-C3N4 structures reported in the literature for photocatalytic H2 generation. As shown in Table S3,† our work demonstrates the highest photocatalytic hydrogen evolution per Pd atom. Notably, even when compared to studies with similar or higher Pd SA loadings, the Pd SAs obtained through our reactive deposition method using Pd(NH3)4Cl2 exhibit the highest efficiency.
To better examine the origin of high activity of our SAs on C3N4, we evaluated the charge transfer properties of the Pd-decorated C3N4 photocatalysts by electrochemical impedance spectroscopy (EIS). The measurements were performed in the 0.1 M Na2SO4 electrolyte at −0.5 V vs. Ag/AgCl, i.e., close to flat band conditions (details are outlined in the ESI-Experimental section†). Fig. 4a presents the Nyquist plots for bare g-C3N4, and g-C3N4 decorated with varying amounts of Pd SAs, and g-C3N4 decorated with Pd NPs (see the zoomed-in spectra shown in Fig. S12†). The Nyquist plots were fitted using the classic Randle's equivalent circuit model (inset of Fig. 4a).41,42 The significantly smaller radius of the fitted curve for Pd SAs/C3N4, compared to bare C3N4, indicates a substantial reduction in charge transfer resistance (Rct) upon Pd SA loading. Quantitative fitting data in Table S4† show a 98-fold decrease in Rct due to Pd SA incorporation (already at a concentration of 0.05 mM). Fig. 4b shows the Rct values plotted against Pd SA loading, showing that even a minimal amount of Pd SAs (0.03 wt%) can dramatically enhance the charge transfer of C3N4 to the electrolyte. This aligns with the low loading required to achieve peak efficiency in photocatalytic H2 production. Conversely, Pd NPs on C3N4 also reduce Rct compared to bare C3N4 (Fig. 4b) but require a much higher loading (1.5 wt%) to achieve a similar reduction in charge transfer resistance, as compared to the Pd SA-loaded sample, which achieves this with just 0.03 wt%. PEIS measurements for all the samples were measured using a 365 nm LED (as described in the ESI Experimental section†). As shown in Fig. S13,† the results indicated a similar trend to the EIS data collected in the dark (Fig. 4 and Table S4†), although Rct values were different. Under illumination, Rct values decreased due to enhanced charge transfer dynamics in the presence of light (Fig. S13 and Table S5†). Notably, Pd SAs demonstrated lower Rct values compared to Pd nanoparticles, indicating the superior performance of Pd SAs. These results underscore the effectiveness of small Pd SA quantities in significantly improving the charge transfer characteristics of C3N4.
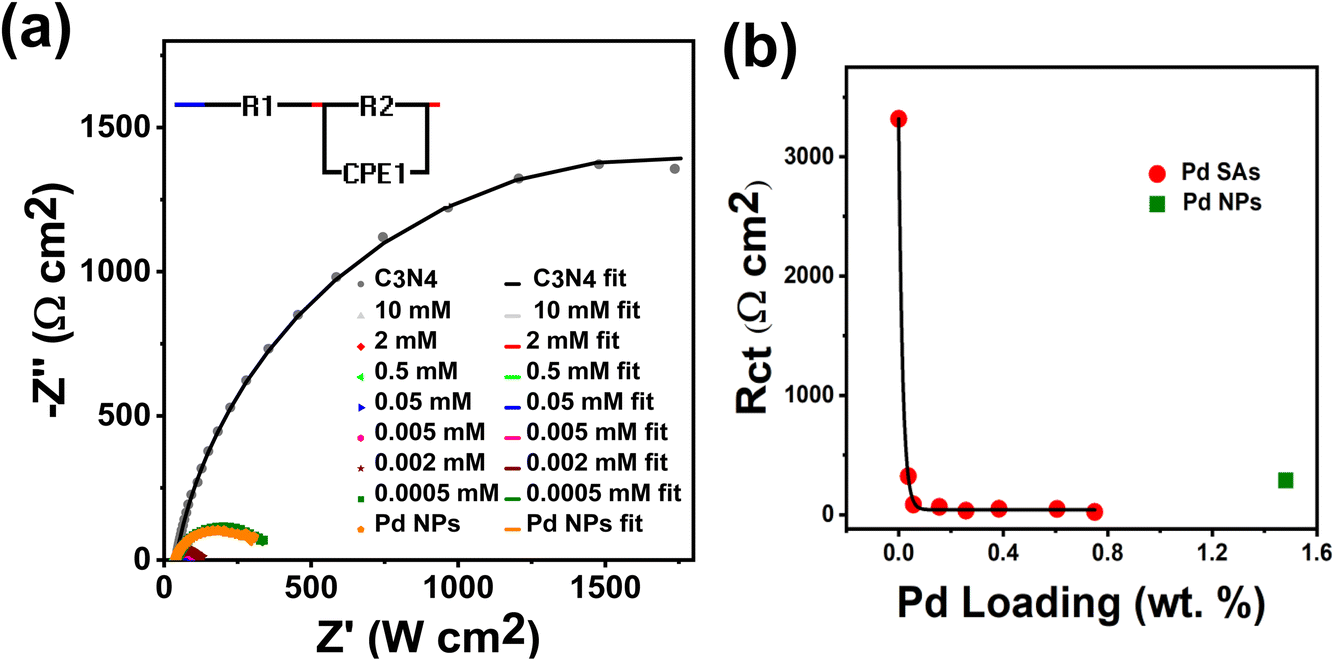 | ||
| Fig. 4 (a) EIS plots of C3N4, Pd SAs/C3N4 and Pd NPs/C3N4 at the voltage of −0.5 V (vs..Ag/AgCl) in 0.1 M Na2SO4 aqueous electrolyte. The equivalent circuit model used for fitting is depicted in the inset of (a). (b) Rctvs. Pd loading plot of Pd SAs/C3N4 samples. | ||
Incident photon-to-current conversion efficiency (IPCE) measurements were also conducted to assess the photoelectrochemical characteristics of C3N4 and Pd SAs/C3N4. Fig. S14† shows photocurrent spectra for both samples with a photocurrent onset in the visible range. The bandgap (determined from a replot of the photocurrent data according to an indirect transition, Fig. S14-inset†) was 2.7 eV, consistent with the typical bandgap of g-C3N4.14,43 Additionally, the photocurrent (Fig. S15†) increases after the decoration of Pd SAs on g-C3N4 (under near flat band conditions), which confirms the beneficial effect of Pd SAs in promoting charge transfer under illumination conditions.
The enhanced performance of Pd-SAs deposited by our decoration technique, compared with the literature, must be attributed to the used precursor Pd(NH3)42+ that leads to the direct formation of active Pd–N configurations (as confirmed by EXAFS, Fig. 2e and f) – in the literature, such sites are regarded as premier active sites in C3N4.16,20,44,45
The presence of NH3 groups in Pd(NH3)42+ ions may promote the formation of such a Pd–N coordination structure compared to the chloride-coordinated Pd precursors due to suitable ligand exchange energetics.46,47 Furthermore, the Pd2+ in Pd(NH3)4Cl2 enables stronger interactions with nitrogen atoms in C3N4 than the Pd4+ in (NH4)2PdCl6 or Pd2+ in PdCl2.48,49 Evidently, chloro-coordinated Pd precursors in the Pd2+ or Pd4+ state are either adsorbed onto the C3N4 surface at less specific sites, as shown by the XPS Pd 3d spectra (Fig. S1a†) and the significant presence of Cl detected in the XPS Cl 2p spectra (Fig. S1b†), or reduced and form metallic Pd agglomerates, as clearly observed in the SEM images (Fig. S2†). The tetraammonium complex, on the other hand, leads without any evident change in the reduction state to active Pdδ+ (δ ≈ 2) N-coordinated SAs, accompanied by the complete loss of Cl coordination during the reaction (Fig. S8†). Therefore, Pd(NH3)4Cl2 is identified as the most suitable precursor for direct reactive deposition of Pd SAs on C3N4.
Considering that many literature studies, particularly those involving DFT calculations, suggest that Pd in an N4-coordination on C3N4 exhibits the highest degree of stability and activity, one may conclude that Pd²⁺ undergoes a ligand exchange process to form this active Pd–N4 configuration. This means that the process is of self-homing nature (as described for the reactive deposition of Pt)23–25,35i.e., the Pd precursor reacts and deposits Pd SAs at most active surface sites on g-C3N4, these sites then provide a maximized electron transfer and thus are highly catalytically active. This explains why such a low loading of Pd SAs is sufficient in our work to achieve maximised photocatalytic H2 production efficiency compared to the higher Pd loadings required, as reported in most literature studies (Table S3†).14,45,49,50
The remarkable activity becomes particularly clear if the present data are compared to the work of Akinaga et al.,14 where a 0.5 wt% Pd SA loading was required to maximize the photocatalytic H2 production activity, i.e., our very low Pd SA loading of 0.05 wt% demonstrates a tenfold increase in efficiency. This superior performance highlights the importance of the attachment chemistry and process of Pd SAs within C3N4,i.e., processes that lead to Pd SAs located at the most active sites can lead to maximized efficiency with minimal Pd usage, avoiding the waste of Pd associated with random Pd SA or NP deposition.
Conclusion
In this work, we successfully integrated Pd SAs onto/into exfoliated g-C3N4via a reactive deposition method, achieving a controllable uniform loading of highly active Pd SAs in a Pd–N configuration on g-C3N4. Notably, using reactive deposition from a Pd(NH3)4Cl2 precursor a low loading of 0.05 wt% Pd SAs on C3N4 with a density of 1.6 × 106 μm−2 can achieve a maximum H2 production rate of 0.24 mmol h−1 mg−1 Pd, significantly higher than that of Pd nanoparticles decorated on g-C3N4 and also ten times higher than that of Pd SAs decorated on g-C3N4 using other reported approaches. Other tested precursors may also deliver SA attachment but lack the high co-catalytic activity. These results underline the importance of the attachment mechanism in creating a SA/substrate coupling with minimized charge transfer resistance and thus maximized co-catalytic activity – in a most effective way, the process is self-homing, i.e., activation takes place where it is most effective.Data availability
The data that support the findings of this study are available within the article and ESI.†Author contributions
Velu Jeyalakshmi conducted the majority of the experimental work, including synthesis, characterization, data analysis, and drafting of the manuscript. Siming Wu contributed significantly to data analysis and revision of the original draft. Shanshan Qin participated in data analysis, while Xin Zhou performed HAADF-STEM analysis. Bidyut Bikash Sarma and Dimitry E. Doronkin conducted XAS and EXAFS analyses and Jan Kolařík carried out AAS analysis. Miroslav Šoóš supervised the experiments and managed funding acquisition. Patrik Schmuki conceived the idea, supervised the experiments, analyzed the data, revised the manuscript, and secured funding. All authors contributed to the preparation of the manuscript and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Johannes Amos Comenius Programme, European Structural and Investment Funds, project ‘CHEMFELLS VI (No. CZ.02.01.01/00/22_010/0008122). The authors would like to acknowledge DFG and the Operational Program Research, Development and Education (European Regional Development Fund Project No. CZ.02.1.01/0.0/0.0/15_003/0000416 of the Ministry of Education, Youth and Sports of the Czech Republic). P. S. particularly thanks the GA CR-EXPRO Project (Grant No. 23-08019X) from the Czech Science Foundation for financial support. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to thank Dr Edmund Welter for his assistance in using the beamline P65. Beam time was allocated for the proposal II-20230692. The authors would also like to acknowledge the support of the Center for Nanoanalysis and Electron Microscopy (CENEM, Friedrich-Alexander-Universität Erlangen-Nürnberg).References
- Y. Li and S. C. E. Tsang, Recent progress and strategies for enhancing photocatalytic water splitting, Mater. Today Sustain., 2020, 9, 100032 Search PubMed.
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Photocatalytic water splitting, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS.
- K. Nakata and A. Fujishima, TiO2 photocatalysis: design and applications, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- P. Roy, S. Berger and P. Schmuki, TiO2 nanotubes: synthesis and applications, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- S. Peiris, H. B. de Silva, K. N. Ranasinghe, S. V. Bandara and I. R. Perera, Recent development and future prospects of TiO2 photocatalysis, J. Chin. Chem. Soc., 2021, 68, 738–769 CrossRef CAS.
- M. D. Hernández-Alonso, F. Fresno, S. Suárez and J. M. Coronado, Development of alternative photocatalysts to TiO 2: challenges and opportunities, Energy Environ. Sci., 2009, 2, 1231–1257 RSC.
- D. Bhanderi, P. Lakhani and C. K. Modi, Graphitic carbon nitride (g-C3N4) as an emerging photocatalyst for sustainable environmental applications: a comprehensive review, RSC sustain., 2024, 2, 265–287 RSC.
- Q. Wang, Y. Li, F. Huang, S. Song, G. Ai, X. Xin, B. Zhao, Y. Zheng and Z. Zhang, Recent advances in g-C3N4-based materials and their application in energy and environmental sustainability, Molecules, 2023, 28, 432 CrossRef CAS PubMed.
- J. Pei, H. Li, S. Zhuang, D. Zhang and D. Yu, Recent Advances in g-C3N4 Photocatalysts: A Review of Reaction Parameters, Structure Design and Exfoliation Methods, Catalysts, 2023, 13, 1402 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Advancing the electrochemistry of the hydrogen-evolution reaction through combining experiment and theory, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Heterogeneous single-atom photocatalysts: fundamentals and applications, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- Y. Xia, M. Sayed, L. Zhang, B. Cheng and J. Yu, Single-atom heterogeneous photocatalysts, Chem Catal., 2021, 1, 1173–1214 CrossRef CAS.
- U. Kerketta, A. B. Tesler and P. Schmuki, Single-atom Co-catalysts employed in titanium dioxide photocatalysis, Catalysts, 2022, 12, 1223 CrossRef CAS.
- Y. Akinaga, T. Kawawaki, H. Kameko, Y. Yamazaki, K. Yamazaki, Y. Nakayasu, K. Kato, Y. Tanaka, A. T. Hanindriyo and M. Takagi, Metal Single-Atom Cocatalyst on Carbon Nitride for the Photocatalytic Hydrogen Evolution Reaction: Effects of Metal Species, Adv. Funct. Mater., 2023, 33, 2303321 CrossRef CAS.
- X. Jia, J. Zhao, W. Zhang, X. Fu, J. Long, Q. Gu and Z. Gao, Single-Atomic Pd Embedded 2D g-C3N4 Homogeneous Catalyst Analogues for Efficient LMCT Induced Full-Visible-Light Photocatalytic Suzuki Coupling, ChemistrySelect, 2022, 7, e202202973 CrossRef CAS.
- F. Hu, L. Leng, M. Zhang, W. Chen, Y. Yu, J. Wang, J. H. Horton and Z. Li, Direct synthesis of atomically dispersed palladium atoms supported on graphitic carbon nitride for efficient selective hydrogenation reactions, ACS Appl. Mater. Interfaces, 2020, 12, 54146–54154 CrossRef CAS PubMed.
- Z. Chen, S. Mitchell, E. Vorobyeva, R. K. Leary, R. Hauert, T. Furnival, Q. M. Ramasse, J. M. Thomas, P. A. Midgley and D. Dontsova, Stabilization of single metal atoms on graphitic carbon nitride, Adv. Funct. Mater., 2017, 27, 1605785 CrossRef.
- R. Xu, B. Xu, X. You, D. Shao, G. Gao, F. Li, X.-L. Wang and Y.-F. Yao, Preparation of single-atom palladium catalysts with high photocatalytic hydrogen production performance by means of photochemical reactions conducted with frozen precursor solutions, J. Mater. Chem. A, 2023, 11, 11202–11209 RSC.
- M. Ren, X. Zhang, Y. Liu, G. Yang, L. Qin, J. Meng, Y. Guo and Y. Yang, Interlayer palladium-single-atom-coordinated cyano-group-rich graphitic carbon nitride for enhanced photocatalytic hydrogen production performance, ACS Catal., 2022, 12, 5077–5093 CrossRef CAS.
- S. Cao, H. Li, T. Tong, H. C. Chen, A. Yu, J. Yu and H. M. Chen, Single-atom engineering of directional charge transfer channels and active sites for photocatalytic hydrogen evolution, Adv. Funct. Mater., 2018, 28, 1802169 CrossRef.
- P. Suja, J. John, T. Rajan, G. M. Anilkumar, T. Yamaguchi, S. C. Pillai and U. Hareesh, Graphitic carbon nitride (gC 3 N 4) based heterogeneous single atom catalysts: synthesis, characterisation and catalytic applications, J. Mater. Chem. A, 2023, 11, 8599–8646 RSC.
- S. Li, Z. Kan, H. Wang, J. Bai, Y. Liu, S. Liu and Y. Wu, Single-atom photo-catalysts: synthesis, characterization, and applications, Nano Mater. Sci., 2023, 284–304 Search PubMed.
- S. M. Wu and P. Schmuki, Single Atom Cocatalysts in Photocatalysis, Adv. Mater., 2024, 2414889 CrossRef.
- Y. Wang, S. Qin, N. Denisov, H. Kim, Z. Bad'ura, B. B. Sarma and P. Schmuki, Reactive Deposition Versus Strong Electrostatic Adsorption (SEA): A Key to Highly Active Single Atom Co-Catalysts in Photocatalytic H2 Generation, Adv. Mater., 2023, 35, 2211814 CrossRef CAS PubMed.
- S. Qin, J. Will, H. Kim, N. Denisov, S. Carl, E. Spiecker and P. Schmuki, Single atoms in photocatalysis: low loading is good enough, ACS Energy Lett., 2023, 8, 1209–1214 CrossRef CAS.
- G. Cha, A. Mazare, I. Hwang, N. Denisov, J. Will, T. Yokosawa, Z. Badura, G. Zoppellaro, A. B. Tesler and E. Spiecker, A facile “dark”-deposition approach for Pt single-atom trapping on facetted anatase TiO2 nanoflakes and use in photocatalytic H2 generation, Electrochim. Acta, 2022, 412, 140129 CrossRef CAS.
- Z. Wu, I. Hwang, G. Cha, S. Qin, O. Tomanec, Z. Badura, S. Kment, R. Zboril and P. Schmuki, Optimized Pt single atom harvesting on TiO2 nanotubes—Towards a most efficient photocatalyst, Small, 2022, 18, 2104892 CrossRef CAS PubMed.
- A. Torres-Pinto, M. J. Sampaio, C. G. Silva, J. L. Faria and A. M. Silva, Metal-free carbon nitride photocatalysis with in situ hydrogen peroxide generation for the degradation of aromatic compounds, Appl. Catal., B, 2019, 252, 128–137 CrossRef CAS.
- C. Marchal, T. Cottineau, M. G. Méndez-Medrano, C. Colbeau-Justin, V. Caps and V. Keller, Au/TiO2–gC3N4 nanocomposites for enhanced photocatalytic H2 production from water under visible light irradiation with very low quantities of sacrificial agents, Adv. Energy Mater., 2018, 8, 1702142 CrossRef.
- M. Karimi-Nazarabad, H. Ahmadzadeh and E. K. Goharshadi, Porous perovskite-lanthanum cobaltite as an efficient cocatalyst in photoelectrocatalytic water oxidation by bismuth doped g-C3N4, Sol. Energy, 2021, 227, 426–437 CrossRef CAS.
- F. Fina, S. K. Callear, G. M. Carins and J. T. Irvine, Structural investigation of graphitic carbon nitride via XRD and neutron diffraction, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS.
- N. Wang, J. Wang, J. Hu, X. Lu, J. Sun, F. Shi, Z.-H. Liu, Z. Lei and R. Jiang, Design of palladium-doped g-C3N4 for enhanced photocatalytic activity toward hydrogen evolution reaction, ACS Appl. Energy Mater., 2018, 1, 2866–2873 CrossRef CAS.
- L. Li, X. Dai, M. Lu, C. Guo, S. M. Wabaidur, X.-L. Wu, Z. Lou, Y. Zhong and Y. Hu, Electron-enriched single-Pd-sites on g-C3N4 nanosheets achieved by in-situ anchoring twinned Pd nanoparticles for efficient CO2 photoreduction, Adv. Powder Mater., 2024, 3, 100170 CrossRef.
- S. Mondal, L. Sahoo, M. Banoo, Y. Vaishnav, C. Prabhakaran Vinod and U. K. Gautam, Enhancing the Catalytic Activity of Pd Nanocrystals towards Suzuki Cross-Coupling by g-C3N4 Photosensitization, ChemNanoMat, 2024, 10, e202300451 CrossRef CAS.
- S.-M. Wu, L. Wu, N. Denisov, Z. Badura, G. Zoppellaro, X.-Y. Yang and P. Schmuki, Pt Single Atoms on TiO2 Can Catalyze Water Oxidation in Photoelectrochemical Experiments, J. Am. Chem. Soc., 2024, 16363–16368 CrossRef CAS PubMed.
- P. Liu, Z. Huang, X. Gao, X. Hong, J. Zhu, G. Wang, Y. Wu, J. Zeng and X. Zheng, Synergy between palladium single atoms and nanoparticles via hydrogen spillover for enhancing CO2 photoreduction to CH4, Adv. Mater., 2022, 34, 2200057 CrossRef CAS PubMed.
- P. H. Ho, J.-W. Woo, R. F. Ilmasani, J. Han and L. Olsson, The role of Pd–Pt interactions in the oxidation and sulfur resistance of bimetallic Pd–Pt/γ-Al2O3 diesel oxidation catalysts, Ind. Eng. Chem. Res., 2021, 60, 6596–6612 CrossRef CAS.
- M. Wang, S. Shen, L. Li, Z. Tang and J. Yang, Effects of sacrificial reagents on photocatalytic hydrogen evolution over different photocatalysts, J. Mater. Sci., 2017, 52, 5155–5164 CrossRef CAS.
- V. Kumaravel, M. D. Imam, A. Badreldin, R. K. Chava, J. Y. Do, M. Kang and A. Abdel-Wahab, Photocatalytic hydrogen production: role of sacrificial reagents on the activity of oxide, carbon, and sulfide catalysts, Catalysts, 2019, 9, 276 CrossRef CAS.
- N. Denisov, S. Qin, J. Will, B. N. Vasiljevic, N. V. Skorodumova, I. A. Pašti, B. B. Sarma, B. Osuagwu, T. Yokosawa and J. Voss, Light-Induced Agglomeration of Single-Atom Platinum in Photocatalysis, Adv. Mater., 2023, 35, 2206569 CrossRef CAS PubMed.
- J. E. B. Randles, Kinetics of rapid electrode reactions, Discuss. Faraday Soc., 1947, 1, 11–19 RSC.
- Z. Wang, A. Murphy, A. O'Riordan and I. O'Connell, Equivalent impedance models for electrochemical nanosensor-based integrated system design, Sensors, 2021, 21, 3259 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Ein stabiler “Single-site”-Palladiumkatalysator für Hydrierungen, Angew. Chem., 2015, 127, 11417–11422 CrossRef.
- L. Liu, X. Wu, L. Wang, X. Xu, L. Gan, Z. Si, J. Li, Q. Zhang, Y. Liu, Y. Zhao and R. Ran, Atomic palladium on graphitic carbon nitride as a hydrogen evolution catalyst under visible light irradiation, Commun. Chem., 2019, 2(1), 18, DOI:10.1038/s42004-019-0117-4.
- A. Ryabov, G. Kazankov, A. Yatsimirskii, L. Kuz'mina, O. Y. Burtseva, N. Dvortsova and V. Polyakov, Synthesis by ligand exchange, structural characterization, and aqueous chemistry of ortho-palladated oximes, Inorg. Chem., 1992, 31, 3083–3090 CrossRef CAS.
- P. Maitlis, Metal Complexes: The Organic Chemistry of Palladium, Elsevier, 2012, pp. 1–103 Search PubMed.
- L. Pazderski, 15N NMR coordination shifts in Pd (II), Pt (II), Au (III), Co (III), Rh (III), Ir (III), Pd (IV), and Pt (IV) complexes with pyridine, 2, 2′-bipyridine, 1, 10-phenanthroline, quinoline, isoquinoline, 2, 2′-biquinoline, 2, 2′: 6′, 2′-terpyridine and their alkyl or aryl derivatives, Magn. Reson. Chem., 2008, 46, S3–S15 CrossRef PubMed.
- N. Wang, J. Wang, J. Hu, X. Lu, J. Sun, F. Shi, Z.-H. Liu, Z. Lei and R. Jiang, Design of Palladium-Doped g-C 3 N 4 for Enhanced Photocatalytic Activity toward Hydrogen Evolution Reaction, ACS Appl. Energy Mater., 2018, 1(6), 2866–2873, DOI:10.1021/acsaem.8b00526.
- R. Xu, B. Xu, X. You, D. Shao, G. Gao, F. Li, X.-L. Wang and Y.-F. Yao, Preparation of single-atom palladium catalysts with high photocatalytic hydrogen production performance by means of photochemical reactions conducted with frozen precursor solutions, J. Mater. Chem. A, 2023, 11(21), 11202–11209 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08589b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |