
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlloyed molybdenum enables efficient alcohol hydrodeoxygenation with supported bimetallic catalysts†
Christian
Ehinger‡
a,
Stephan
Pollitt‡
b,
Jordan
De Jesus Silva
a,
Xiaoyu
Zhou
a,
Kazutaka
Sakamoto
a,
Maarten
Nachtegaal
ab,
Olga
Safonova
 *b and
Christophe
Copéret
*a
*b and
Christophe
Copéret
*a
aD-CHAB, ETH Zürich, Vladimir–Prelog-Weg 2, 8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch
bCenter for Energy and Environmental Sciences, PSI, Forschungsstrasse 111, 5232 Villigen, Switzerland. E-mail: olga.safonova@psi.ch
First published on 5th March 2025
Abstract
Bimetallic heterogeneous catalysts combining group 9 metals (Rh, Ir) or group 10 metals (Ni, Pd, Pt) with Mo on a silica-based support have been synthesized via surface organometallic chemistry and assessed in their catalytic activity for the hydrodeoxygenation (HDO) of alcohols with particular emphasis on the structural evolution of the catalysts and the role of Mo. The investigation was conducted with an air-free approach to isolate any sample alterations exclusively to those caused by the reaction. Structural analysis was performed using a combination of (S)TEM, IR, and XAS. It was found that Ir–Mo/SiO2, Rh–Mo/SiO2, and Pt–Mo/SiO2 display high activity for primary, secondary, and tertiary alcohol deoxygenation, while Pd–Mo/SiO2 selectively catalyses tertiary alcohol deoxygenation. Other combinations as well as the corresponding monometallic materials do not display the same activity. X-ray absorption spectroscopy confirmed metallic states for M (M = Ni, Rh, Pd, Ir, or Pt), while Mo K-edge XANES showed varying amounts of Mo(0), Mo(IV) and Mo(VI) depending on the metal counterpart in fresh materials, and indicated complete conversion of Mo(VI) to lower oxidation states (IV and 0) during the reaction. For Rh, Pd, Ir, and Pt, alloy formation (M–Mo) was identified via M–Mo paths in EXAFS and supported by CO-IR spectroscopy. In contrast to Ir, Rh, and Pt, where some Mo(0) is present at the nanoparticle surface, Pd–Mo forms an alloy but likely retains Mo in the nanoparticle core, as suggested by CO-IR spectroscopy and CO-chemisorption. Reactivity studies suggest that tertiary alcohols primarily undergo dehydration–hydrogenation, evidenced by olefin formation with MoOx/SiO2, as well as Ir/SiO2 and Ir–Mo/SiO2 under inert conditions. In contrast, primary and secondary alcohols follow a different mechanism, correlated with the presence of metallic Mo species on the nanoparticle surface, highlighting their role in C–O bond activation. These findings provide new insights into the structure–activity relationships of Mo-based bimetallic catalysts, underscoring the influence of Mo in different oxidation states and strong substrate dependence on mechanistic pathways.
Introduction
The synthesis of renewable fuels and platform chemicals from oxygen-rich biomass, such as lignin, requires the selective removal of oxygen-containing functional groups with green hydrogen.1–4 This pivotal process, referred to as hydrodeoxygenation (HDO) and used in the petrochemical industry, has been the subject of intensive research in the past decades.5–7 Efficiently achieving this transformation would facilitate the large-scale production of bio-based fuels and chemicals, reducing the dependency on fossil resources and contributing to a sustainable circular economy and a closed carbon cycle.8–11Breaking C–O bonds is associated with a significant energy barrier due to their high kinetic and thermodynamic stability, requiring the use of catalysts.12,13 Heterogeneous catalysts for HDO often comprise late transition metals – to activate dihydrogen – combined with a group V–VII transition metal, in particular Mo, that are thought to facilitate the activation of the C–O bond because of their oxygen affinity and their ability to introduce Brønsted and/or Lewis acidity.14–20 These bifunctional-bimetallic catalysts can lead to enhanced activity compared to the corresponding monometallic materials, suggesting a synergy that is crucial for the desired reactivity.21–25 However, the exact role and interplay of the components in these catalysts are still poorly understood and often remain speculative, mainly due to the complexity of these materials.13,26
In classical synthetic approaches, metallic nanoparticles of late transition metals (M) are commonly prepared via impregnation techniques, often relying on metal chloride (or nitrate) precursors, possibly leading to contamination or uneven particle formation.27,28 While cost and time efficient, these approaches generally afford rather ill-defined materials, with non-uniform distribution of surface sites and particle sizes. This complexity arises from the synthetic process as it involves uncontrolled dissolution and precipitation processes in water. This, in turn, makes it challenging to achieve molecular-level understanding of catalytic processes and to develop informative structure–property relationships. In that context, surface organometallic chemistry (SOMC) has emerged as a synthetic strategy to generate better-defined catalyst structures using tailored (traceless) molecular precursors that are evenly dispersed on the surface by anchoring onto hydroxy groups (grafting), followed by a post treatment to remove the organic ligands; this approach typically generates smaller nanoparticles and a narrower size distribution.29 In addition, these materials provide tailored interfaces and compositions, while avoiding the incorporation of undesired elements.30
In the presented work, we investigate a series of HDO catalysts prepared via SOMC based on the combinations of a transition-metal, known for their hydrogenation properties (M = Rh, Ir, Ni, Pd, and Pt) and Mo, dispersed on a silica support. Among M–Mo/SiO2, we show that the promotional effect of Mo for HDO of alcohols as prototypical oxygenate substrates is highly efficient with Pt, Rh, and particularly so for Ir, while negligible for Ni, and only observed for tertiary alcohols in the case of Pd. Characterization based on the adsorption of CO and pyridine probe molecules, High-Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF-STEM), and X-ray absorption spectroscopy (XAS), performed on the catalysts before and after the HDO reaction and preserved under inert atmosphere, show that Mo is reduced from Mo(VI) to Mo(IV) and Mo(0) in the presence of the primary metal (M) and incorporated in narrowly dispersed 2–3 nm alloyed nanoparticles (for M = Rh, Ir, Pd, Pt), without significant changes in Brønsted or Lewis acidity. Overall, the data point to the importance of a metallic state of Mo and alloy formation, indicating M–Mo phases play a critical role in efficient HDO processes.
Results and discussion
Synthesis and characterization
All bimetallic materials are prepared in two steps. The first step involves the dispersion of 0.3 wt% Mo on SiO2 (Degussa–Evonik, Aerosil-200), which corresponds to ca. 0.1 Mo nm−2via incipient-wetness impregnation of ammonium heptamolybdate ((NH4)6Mo7O24),31,32 followed by calcination (500 °C under a flow of dry synthetic air). The low Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M ratio (ca. 1:8, vide infra) was chosen because small amounts of Mo have been shown to significantly enhance deoxygenation activity,17 and it also ensures that Mo predominantly contributes to the active phase while minimizing spectroscopic interference from inactive species. The second metal (ca. 1 M atom per nm2) is next introduced using SOMC (Fig. 1a). We select amidinate-based precursors, because they provide a structurally unified platform for all five investigated metals M (Ni, Pd, Pt, Rh, and Ir) and afford supported nanoparticles with relatively narrow size distribution upon treatment under H2.33–35 In short, after grafting the amidinate-based precursors on isolated surface OH groups, as evidenced by IR spectroscopy (Fig. 1c, middle spectrum), the obtained materials are treated under a flow of H2 at 500 °C. The resulting IR spectrum (Fig. 1c, top spectrum) confirms the removal of all organic ligand fragments while the isolated OH groups are restored. The respective monometallic materials are prepared from the same precursors, using SiO2–700 (SiO2 subjected to a temperature treatment at 700 °C at 10−6 mbar) in place of the Mo-doped support.
:M ratio (ca. 1:8, vide infra) was chosen because small amounts of Mo have been shown to significantly enhance deoxygenation activity,17 and it also ensures that Mo predominantly contributes to the active phase while minimizing spectroscopic interference from inactive species. The second metal (ca. 1 M atom per nm2) is next introduced using SOMC (Fig. 1a). We select amidinate-based precursors, because they provide a structurally unified platform for all five investigated metals M (Ni, Pd, Pt, Rh, and Ir) and afford supported nanoparticles with relatively narrow size distribution upon treatment under H2.33–35 In short, after grafting the amidinate-based precursors on isolated surface OH groups, as evidenced by IR spectroscopy (Fig. 1c, middle spectrum), the obtained materials are treated under a flow of H2 at 500 °C. The resulting IR spectrum (Fig. 1c, top spectrum) confirms the removal of all organic ligand fragments while the isolated OH groups are restored. The respective monometallic materials are prepared from the same precursors, using SiO2–700 (SiO2 subjected to a temperature treatment at 700 °C at 10−6 mbar) in place of the Mo-doped support.
 | ||
| Fig. 1 (a) General synthesis route of bimetallic materials. (b) HAADF-STEM micrographs of bimetallic M–Mo/SiO2 materials. (c) IR spectra of MoO3/SiO2 (black spectrum), Ir(COD)(DIA)/MoO3/SiO2 (blue spectrum), Ir–Mo/SiO2 (green spectrum). | ||
HAADF-STEM micrographs of all as-prepared materials are recorded and analysed without prior exposure to air (Table 1, Fig. 1b, and S20–S24†). All materials contain narrowly dispersed nanoparticles of 2–3 nm. With the exception of PtMo, all bimetallic materials exhibit smaller and more narrowly dispersed nanoparticles compared to their monometallic counterparts.36 For NiMo, the sub-nanometre particles are not resolved. For PdMo, the particles are considerably better dispersed, than in the monometallic case; this effect had been previously observed for alloyed bimetallic Pd–Ga/SiO2 derived from the same precursor.33,37 Both IrMo and RhMo also display nanoparticles that are ca. 1 nm smaller than the respective monometallic materials. The strong influence of Mo is particularly remarkable considering that the M:Mo ratio is ca. 8:1, suggesting a strong interaction between the two metals.
| Material | μ (nm) | σ (nm) | N | M (wt%) |
|---|---|---|---|---|
| Ni/SiO2 | 1.9 | 0.6 | 205 | 1.36 |
| Ni–Mo/SiO2 | <1 | — | — | 1.33 |
| Pd/SiO2 | 3.1 | 1.6 | 210 | 2.59 |
| Pd–Mo/SiO2 | 2.5 | 0.5 | 205 | 2.41 |
| Pt/SiO2 | 1.8 | 0.5 | 213 | 3.98 |
| Pt–Mo/SiO2 | 1.8 | 0.6 | 211 | 3.56 |
| Rh/SiO2 | 3.3 | 0.7 | 134 | 2.38 |
| Rh–Mo/SiO2 | 2.4 | 0.6 | 207 | 2.44 |
| Ir/SiO2 | 2.9 | 0.9 | 230 | 4.32 |
| Ir–Mo/SiO2 | 2.0 | 0.6 | 222 | 4.26 |
HDO activity
Next, the synthesized materials are evaluated for the HDO of primary, secondary, and tertiary alcohols. For 1-nonanol (at a catalyst loading of 0.75 mol% M, 130 °C, 20 bar of H2 and decalin as a solvent) the corresponding alkane, n-nonane, is formed in good to high yield within 24 h for the bimetallic materials containing Rh, Ir, and Pt (Fig. 2a). On the other hand, none of the monometallic materials, including MoOx/SiO2 (1% n-nonane, Fig. S32†), enable efficient HDO under the same conditions. Notably, n-octane (ca. 7%) and traces of n-heptane besides n-nonane are also observed with Rh–Mo/SiO2, indicating that C–C cleavage also occurs as a side reaction with Rh. In sharp contrast, bimetallic Ni–Mo/SiO2 and Pd–Mo/SiO2 do not catalyse the HDO reaction of primary alcohols under the investigated conditions. In the case of 2-nonanol, Rh, Pt, and Ir, in combination with Mo, are again highly active materials affording the deoxygenated products in good yield (Fig. 2b). Pd–Mo/SiO2 enables low conversion, yielding only ca. 6% n-nonane. Only traces of product (∼1%) are observed for Ni–Mo/SiO2. In the case of 2-nonanol, no C–C cracking-derived side products are observed even for Rh–Mo/SiO2. In contrast to 1-nonanol, some of the monometallic materials display minor productivity in HDO of 2-nonanol: ca. 2% yield with Rh/SiO2 and ca. 6% yield for Ir/SiO2 and Pt/SiO2, and ca. 1% for MoOx/SiO2. Note that the conversion could not be determined due to the overlap of the 2-nonanol GC signal with one isomer of decalin. Finally, the materials are also evaluated for the deoxygenation of a tertiary alcohol, namely 2,6-dimethylheptan-2-ol (Fig. 2c). All of the monometallic materials display deoxygenation activity, in particular Rh/SiO2 and Ir/SiO2 afford the product, 2,6-dimethylheptane, in 18% and 44% yield, respectively. The monometallic materials based on Ni, Pd, and Pt show lower yields of 1%, 3%, and 5%, respectively. Yet, addition of Mo again increases the HDO activity, resulting in high yields of HDO product (∼80%) for RhMo, IrMo, PtMo, and PdMo. In contrast to primary and secondary alcohols, the PdMo material gives good yield of deoxygenated product with the tertiary alcohol. However, while Mo slightly increases the reactivity, NiMo remains poorly active with a HDO product yield of ca. 5%. Notably, monometallic MoOx/SiO2 affords 3% of 2,6-dimethylheptane, and 12% of the corresponding olefins (2,6-dimethylhept-1-ene and 2,6-dimethylhept-2-ene in a ca. 1:1 ratio) formed via dehydration (Fig. S32†), potentially indicative that a different mechanism can be at play for the deoxygenation of tertiary alcohols, namely via a sequential dehydration–hydrogenation pathway involving olefins as reaction intermediates.
 | ||
| Fig. 2 Conversion and HDO product yields for mono- and bimetallic materials in the deoxygenation of (a) 1-nonanol, (b) 2-nonanol, and (c) 2,6-dimethylheptan-2-ol. Solvent = decahydronaphthalene (decalin). The conversion of 2-nonanol could not be determined due to the overlap of the 2-nonanol GC signal with one isomer of decalin. | ||
Considering that Pd–Mo/SiO2 is inactive for 1-nonanol and 2-nonanol, we highlight the possibility of selectively deoxygenating tertiary alcohols over primary and secondary alcohols. Subjecting a mixture of 1-nonanol and 2,6-dimethylheptan-2-ol to HDO conditions with Pd–Mo/SiO2 leads to the selective formation of 2,6-dimethylheptane in 64% yield (76% conversion), while no n-nonane is observed (Fig. S34†). The activity of the Pd–Mo/SiO2 catalyst seems to be partially inhibited by the presence of 1-nonanol, however, the deoxygenation is highly selective. Note that, in the analogous experiment with Ir–Mo/SiO2, both alcohols in the mixture are efficiently deoxygenated affording yields of 88% 2,6-dimethylheptane and 98% n-nonane (Fig. S34†), highlighting the specificity of Pd–Mo/SiO2 in the HDO of tertiary alcohols, suggesting that a dehydration–hydrogenation pathway is dominant with Pd rather than a more direct HDO process.
Since Ir–Mo/SiO2 displays the most promising properties as a general deoxygenation catalyst for aliphatic alcohols, we further investigate the reactivity of this catalyst. First, the reaction profile is examined by monitoring the reaction products at various reaction times (1, 3, and 8 h), which confirms that 24 h reaction time is necessary for achieving full conversion (Fig. S36†). We also check the possibility of recycling the catalyst, which reveals a constant decrease of activity, giving 86%, 60%, and 15% yields after the 1st, 2nd, and 5th cycle, respectively (Fig. S37†). While the catalyst productivity is reduced to 70% after one cycle, with an overall conversion corresponding to ca. 6 h reaction time for the fresh catalyst, the material remains catalytically active, enabling meaningful characterization of the spent catalyst. This allows us to study the structural changes that occur during the reaction, providing insights into the active phase under operating conditions. The treatment of the spent material with H2 or calcination followed by H2 cannot fully restore the high activity observed in the first cycle. Elemental analysis after five cycles reveals a partial loss of metals, with approximately 35% reduction in Mo content and 15% reduction in Ir. While this loss likely contributes to some of the observed decline in activity, it does not account for the entirety of the decrease, suggesting that irreversible structural changes may have occurred under reaction conditions.
Investigation of structure–activity relationship
The significant difference in reactivity between monometallic and bimetallic materials suggests a key influence of Mo, especially for the HDO of primary and secondary alcohols, leading to a synergistic enhancement of catalytic performance. One possible origin of enhanced activity could be due to the decreased particle size of Rh-, Ir- (and Pd-) based bimetallic materials upon the addition of Mo. However, the particle size of Pt–Mo/SiO2 is within error of monometallic Pt/SiO2, with a strong increase in activity, while Ni–Mo/SiO2 exhibits a particle size decrease, with no concomitant increase in activity, suggesting that particle size alone cannot explain the marked promotional effect of Mo.Surface acidity
Another possible effect leading to enhanced HDO activity is an increase in Brønsted or Lewis acidity upon adding Mo to the silica surface. This acidity could facilitate alcohol activation through protonation, promoting C–O activation via acid-catalyzed dehydration followed by hydrogenation of the resulting olefin.13,15 To probe the potentially enhanced Brønsted and/or Lewis acidity induced by Mo, and its involvement in the HDO of alcohols, we next evaluate the reactivity of 2,6-dimethylheptan-2-ol and 1-nonanol under inert atmosphere (5 bar Ar, 130 °C, 20 h). For the tertiary alcohol, 2,6-dimethylheptan-2-ol, the Ir–Mo/SiO2 catalyst affords a significant yield of olefins (48% total), while monometallic Ir/SiO2 yielded only 9%, suggesting a moderate increase in acidity in the bimetallic material, promoting acid-catalysed dehydration. Conversely, 1-nonanol showed no olefin formation under the same conditions with Ir–Mo/SiO2, indicating that acid-catalyzed dehydration is not a significant pathway for primary alcohols. To further investigate the formation of acid sites we turn to 15N-pyridine adsorption on the most active catalyst (Ir–Mo/SiO2) and analyse the pyridine-exposed material by 15N magic angle spinning solid-state (MAS-SS) NMR spectroscopy (Fig. S31†).38 Notably, only H-bonded pyridine interacting with surface silanol groups is observed, with no evidence of pyridinium formation or pyridine bound to Lewis acidic surface sites, indicating no significant enhancement of Brønsted or Lewis acidity in bimetallic materials. These results align with observations from monometallic MoOx/SiO2, where only tertiary alcohols are converted to olefins. Together, these findings imply that the primary promoting role of Mo in (efficient) deoxygenation of primary and secondary alcohols is not predominantly linked to acidity but likely involves a different mechanistic pathway.Probing electronic effects of Mo with CO adsorption
To investigate whether the presence of Mo can affect the electronic structure of late transition metal nanoparticles, we turn to IR spectroscopy of adsorbed CO as a probe molecule, without exposing the as-prepared materials to air. Looking first at the Ir-based systems, the IR stretching frequency of adsorbed terminal CO is observed at 2079 cm−1 for Ir/SiO2vs. 2072 cm−1 in the bimetallic material, Ir–Mo/SiO2 (Fig. 3). The slight red-shift suggests more electron-rich Ir centres, consistent with alloying (vide infra) and/or the decrease in particle size. Furthermore, this band decreases in intensity, indicative of the presence of fewer accessible Ir (surface) sites, which is seemingly contradictory to the smaller particle size observed by STEM. This could indicate, that part of the surface is blocked by the presence of Mo. Notably, such observation has been reported for a comparable Rh–Mo/SiO2 system, where Mo was specifically introduced on the nanoparticle surface via atomic layer deposition to study the effect of Mo in syngas conversion.39 To further investigate this possibility, we turn to CO-chemisorption (Fig. S27 and S28†).40,41 Considering the different surface to bulk ratios for the nanoparticle sizes measured by HAADF-STEM imaging (Table 1), the CO-chemisorption experiments reveal that for Ir–Mo/SiO2, the CO uptake per surface atom is reduced by over 20%. This is consistent with Mo blocking the adsorption of CO on Ir. A similar trend of CO-IR signatures between mono- vs. bimetallic materials is also observed for Rh- and, most strongly, for Pt-based materials, consistent with alloy formation that decrease the number of CO adsorption sites due to the presence of surface Mo and/or the restructuring of the nanoparticle shape and size by incorporation of Mo (Fig. S11–S19†). | ||
| Fig. 3 IR spectra of adsorbed CO on Ir/SiO2 (grey spectrum) and Ir–Mo/SiO2 (blue spectrum) after dosing ca. 15 mbar of CO, followed by evacuation at 10−5 mbar for 10 minutes. The spectra are baseline corrected and normalized by the intensity of the siloxane band at 1870 cm−1. | ||
Notably, Pd is the only system where the normalized intensity of both terminal and bridging CO increases moving from monometallic to the bimetallic material, without a significant shift of the bands. To further investigate this, we again turn to CO-chemisorption (Fig. S29 and S30†). It is known that the Pd:CO stoichiometry is highly dependent on particle size (among other variables), ranging from 2:1 for large particles to 1:1 for small clusters, as the preferential binding mode transitions from bridging to linear.42,43 Again comparing the nanoparticle surface normalized CO uptake, the value obtained for Pd–Mo/SiO2 is 60% higher than for monometallic Pd/SiO2. This is consistent with the smaller particle size but not indicative of any interference of Mo with CO adsorption on the nanoparticle surface, in contrast to Ir–Mo/SiO2. Among the metals investigated by CO adsorption (Rh, Ir, Pd, Pt), Pd has the lowest surface energy for fcc low index surfaces, hence the lowest driving force for exposing Mo on the nanoparticle surface.44,45 This might explain the inactivity in the HDO of primary and secondary alcohols, potentially highlighting the role of Mo in the mechanism for these substrates.
Monitoring the state of Mo and M via XAS
XAS characterization is next performed to gather further information on the structure and state of Mo and M in these catalysts, before (fresh) and after HDO of 1-nonanol (spent, after 20 h reaction time). In order to minimize sample alteration by air and moisture, all samples are handled and transferred without air-exposure prior to and during characterization.For both fresh and spent Ir–Mo/SiO2, analysis of their X-ray absorption near-edge structure (XANES) spectra at the Ir L3-edge confirm the metallic nature of Ir as analogously observed for the monometallic Ir/SiO2.35 However, the spectroscopic signature in Mo K-edge XANES is more complex: Mo already appears to be present in several oxidation states in the fresh Ir–Mo/SiO2 catalyst. Linear combination fit (LCF) analysis identifies Mo(VI), Mo(IV), and metallic Mo(0) as main components, with the respective reference spectra reproducing the observed spectrum (Fig. 4a and 5a; see ESI† for details). Note that due to the presence of several (at least 3) Mo (oxidation) states, Mo K–edge extended X-ray absorption fine structure (EXAFS) spectra of the fresh Ir–Mo/SiO2 catalyst cannot reliably be fitted. Regarding the spent Ir–Mo/SiO2 catalyst, all remaining Mo(VI) species are converted yielding an increased amount of both Mo(IV) and Mo(0) according to Mo K-edge XANES (Fig. S41†) and LCF analysis (Fig. 5b). Notably, the respective EXAFS spectrum can be fitted with two scattering paths corresponding to Mo–O and Mo–Ir (Fig. 4b and Table 2 column 1, Fig. S59–S61†), indicating the formation of an Ir–Mo alloyed phase. Alternative fits using a Mo–Mo path instead of Mo–Ir resulted in higher R-factors (Table S23†) suggesting a lower probability of Mo–Mo bond formation. In addition, the fitted Mo–Mo bond for Ir–Mo/SiO2 was significantly elongated (ca. 2.91 ± 0.05 Å) in comparison to fitted Mo–Mo bonds in Mo foil (2.700 ± 0.004 Å), and to fitted Ir–Ir (2.697 ± 0.003 Å) and Mo–Ir (2.70 ± 0.05 Å) in the same sample, further supporting the hypothesis of Mo bonding to Ir over forming segregated Mo(0) domains.
 | ||
| Fig. 4 (a) LCF of fresh Ir–Mo/SiO2 with Mo(VI), Mo(IV), and Mo(0) references of Mo K-edge XANES spectra. (b) Mo K-edge EXAFS spectrum and fit of spent Ir–Mo/SiO2 after HDO of 1-nonanol. | ||
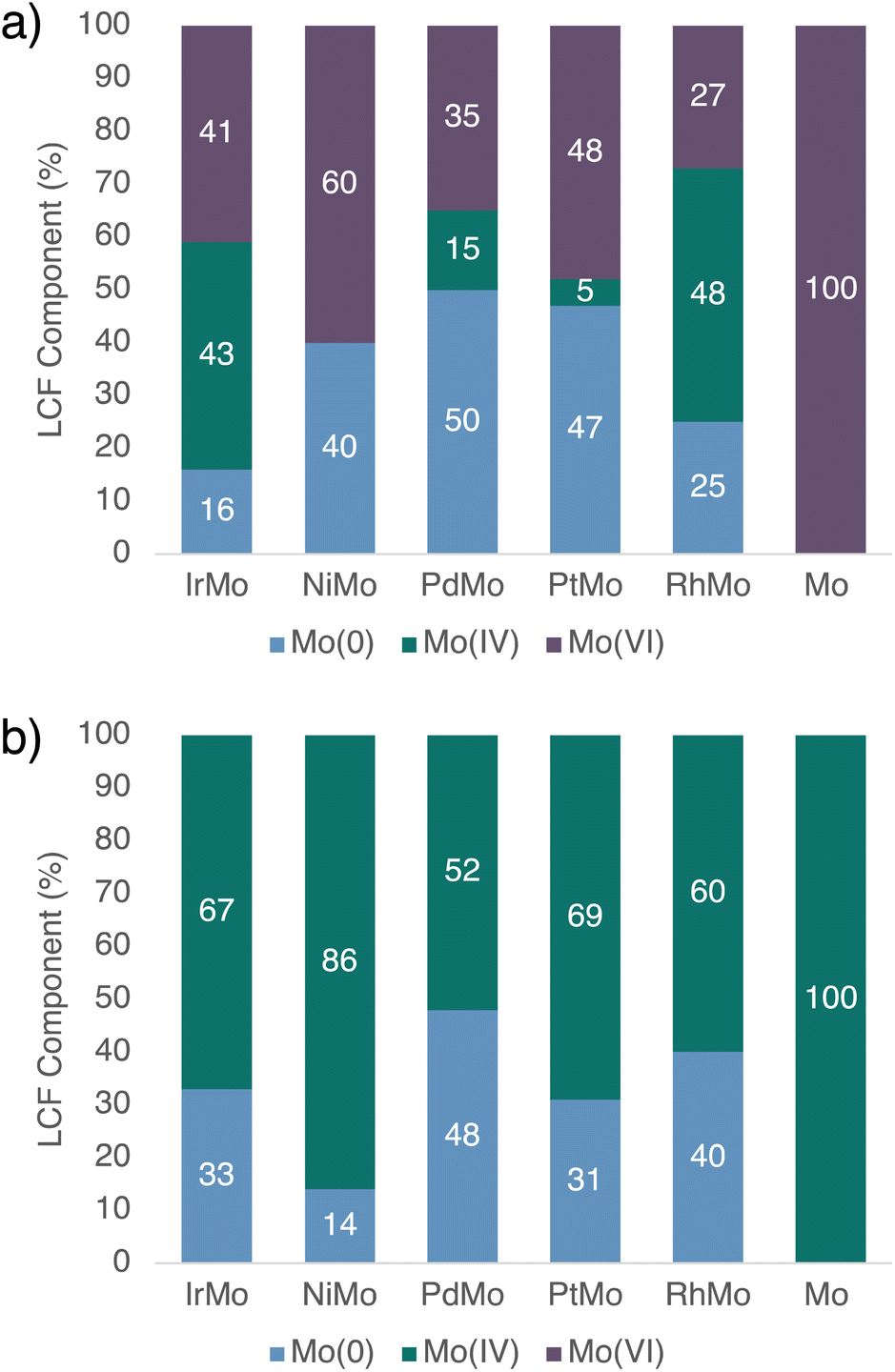 | ||
| Fig. 5 Relative fractions of the Mo(0), Mo(IV) and Mo(VI) components in the catalysts determined by LCF of Mo K-edge XANES spectra of (a) as-prepared materials (fresh) and (b) after HDO reaction with 1-nonanol as substrate (spent). | ||
| Scattering path/Sample | IrMo | PtMo | RhMo | PdMo | NiMo |
|---|---|---|---|---|---|
| M–M fresh | 9.7 ± 0.7 | 10.3 ± 0.4 | 10.2 ± 0.8 | 8.3 ± 0.7 | 8.6 ± 0.9 |
| M–M spent | 9.7 ± 0.6 | 9.7 ± 0.4 | 9.7 ± 0.5 | 10.9 ± 0.5 | 8.3 ± 0.6 |
| Mo–O spent | 3.3 ± 2.0 | 3.5 ± 1.3 | 3.2 ± 1.5 | 3.3 ± 0.8 | 3.5 ± 0.6 |
| Mo–M spent | 2.5 ± 1.4 | 2.8 ± 0.9 | 3.7 ± 0.5 | 4.1 ± 0.4 | 0 |
From the perspective of Ir L3-edge EXAFS, no respective Ir–Mo path can be fitted for both the fresh and spent Ir–Mo/SiO2 catalyst, and only an Ir–Ir path consistent with metallic Ir is found. This is likely due to the low concentration of Mo with respect to Ir (1:8). Assuming only two types of Mo species are present (metallic and oxidic), consistent with the XANES spectra, Mo(0) and Mo(IV) are coordinated by Ir and O respectively, where the metallic Mo exhibits a Mo–Ir coordination number of 7.5 (=2.5/33%, from Table 2 and Fig. 5, respectively), which is within error of the respective coordination number for Ir–Ir (9.7), in accordance with Mo being incorporated in the nanoparticles. This is in line with the observations from CO chemisorption and parallels what has been reported for the Rh–Mo/SiO2 syngas conversion catalyst discussed above.39 On the other hand, the coordination number of Mo–O in Mo(IV) is found to be 4.9 (=3.3/67%, from Table 2 and Fig. 5, respectively), well in the range of nanostructured MoO2 with a coordination of 6 in the bulk structure. In this simplified picture, we assume that the oxidized Mo is distributed on the support, while Mo(0) is located in the metallic nanoparticles. Note, that the presence of an intermediate case of partially oxidized Mo on the nanoparticle surface, as proposed to form under syngas conversion conditions with Rh–Mo/SiO2, cannot be excluded. However, consideration of these species would result in the same conclusion of Mo–Ir undercoordination in the nanoparticle, hence a preferential localization of Mo on the nanoparticle surface.39
The monometallic Mo material, MoO3/SiO2, in the absence of Ir, is also reduced under HDO conditions (with 1-nonanol), but only from Mo(VI) to Mo(IV), while no metallic Mo(0) is observed, corroborating the strong interaction between Ir and Mo in Ir–Mo/SiO2 and the necessity of a second metal to achieve a reduction to Mo(0). Overall, these combined results confirm the reduction of Mo by Ir and the alloying of Mo into the Ir nanoparticles, consistent with what is observed by CO IR spectroscopy (vide supra).
Analysis of the XANES and EXAFS spectra of the corresponding Rh and Pt materials show similar trends, and the respective coordination numbers for M–M, M–Mo, and Mo–O after HDO (spent) are in the same range (Table 2 columns 2–4). In these systems, M remains metallic. Regarding the Mo K-edge, Mo(IV) and Mo(0) are present in the fresh materials along with Mo(VI), which is transformed to Mo(IV) and Mo(0) in the spent materials (Fig. 5, S40 and S41†). While EXAFS spectra at the Mo K-edge of fresh materials can again not be fitted reliably due to the coexistence of several species (vide supra), the EXAFS spectra of spent materials can be fitted with Mo–M paths, confirming the presence of M–Mo alloyed phases (Tables S6, S10, S14 and S18† and associated figures). Paralleling the observation with Ir–Mo/SiO2, the inclusion of Mo–Mo paths instead of Mo–M again results in higher R-factors (Table S23†) suggesting alloy formation to be the more likely scenario.
At 50% Mo(0), Pd–Mo/SiO2 exhibits the highest proportion of metallic Mo among the bimetallic materials, and in contrast to the other metals, the Mo(0) content does not evolve significantly during the reaction, paralleling the inactivity of this material in the HDO of 1-nonanol. The Pd–Pd coordination number after the reaction is higher than for the other metals and increases significantly during the reaction, while for the other bimetallic systems it remains constant within error. HAADF-STEM imaging of the spent material reveals the formation of a few large nanoparticles under reaction conditions in addition to smaller nanoparticles, consistent with sintering. EDX mapping shows a spatial correlation of Mo and Pd in larger nanoparticles, in line with alloy formation (Fig. S26†). In contrast to the other metals, this observed sintering behaviour parallels the inactivity in HDO with primary and secondary alcohols as well as the absence of (high concentrations of) Mo on the nanoparticle surface observed by CO-chemisorption, and it is likely that these phenomena are connected. On the other hand, the deoxygenation of tertiary alcohols can occur via a dehydration–hydrogenation pathway which is likely promoted by the tandem reactivity of oxidized Mo located on the support for dehydrogenation coupled to hydrogenation with (alloyed) Pd nanoparticles.
In the case of Ni-based materials, Ni K-edge XANES indicates a small but detectable amount of oxidized Ni(II) slightly variable (0–12%) across all samples (Fig. S42 and Table S1†). Mo K-edge XANES spectra show that in the fresh sample, there is a significant proportion of metallic Mo(0), while there is no Mo(IV) present. However, for the spent catalyst, all of Mo(VI) and most of Mo(0) are converted to Mo(IV). Moreover, no Mo–Ni path was detected in the Fourier transform of the Mo K-edge (or Ni K-edge) EXAFS of the spent Ni–Mo/SiO2 catalyst. In contrast to the other metals, while Mo(0) is present in the fresh Ni–Mo/SiO2 material, presumably in an alloyed state or the interface, it does not withstand the HDO conditions. This absence of an alloy phase parallels the absence of (or very low) HDO reactivity.
In summary, it can be concluded that across all the investigated bimetallic catalysts based on the motif M–Mo/SiO2, for which HDO activity is observed (M = Ir, Rh, Pt, and Pd for tertiary alcohols), Mo is reduced to a metallic state in significant extent and incorporated in the nanoparticles of M, forming an alloy. In contrast, no significant alloying is observed for Ni exposed to HDO conditions.
In contrast to primary and secondary alcohols (1-nonanol and 2-nonanol), the tertiary alcohol (2,6-dimethylheptan-2-ol) undergoes dehydration to olefins with MoOx/SiO2 under reaction conditions. Similarly, olefinic products are observed for the tertiary alcohol with both monometallic Ir/SiO2 and, to a greater extent, with Ir–Mo/SiO2 under inert atmosphere, while they are absent for 1-nonanol with Ir–Mo/SiO2 under the same conditions. This indicates that Mo increases surface acidity, which selectively impacts tertiary alcohols, and suggests a dehydration–hydrogenation mechanism is operative for these substrates (Fig. 6a). This explains the high activity of Pd-, Rh-, Ir-, and Pt-based bimetallic materials towards tertiary alcohols, and the modest activity of monometallic materials likely following the same mechanism. Ni–Mo/SiO2 shows slightly enhanced activity compared to Ni/SiO2 with tertiary alcohols while overall not exhibiting great compatibility with alcohols under the investigated conditions. For primary and secondary alcohols, a different mechanism is likely dominant and appears to be linked to the presence of reduced Mo species on the nanoparticle surface of Ir-, Rh-, and Pt-based bimetallic materials.
 | ||
| Fig. 6 (a) Proposed main mechanistic pathway for the deoxygenation of tertiary alcohols involving a dehydration–hydrogenation tandem reaction. (b) Proposed involvement of reduced Mo species on alloyed nanoparticles in HDO, that abstract oxygen functionalities and are re-reduced by neighboring metal-hydrides. | ||
In light of the high Mo–O bond strength, one can propose that Mo sites on the surface of M–Mo alloyed nanoparticles help to activate the C–O bond by strongly binding oxygen functionalities, while the late transition metal activates H2 and helps to (re-)reduce both Mo and the hydrocarbyl part (Fig. 6b).15,23 This parallels what is observed with bimetallic reverse-water–gas-shift (RWGS) catalysts.39,46,47 We propose that the difficulty of maintaining reduced Mo on the surface contributes to the low activity of Ni–Mo, which undergoes dealloying under reaction conditions. Regarding Pd, CO chemisorption suggests Mo not to be present on the nanoparticle surface, which is also reflected by the negligible variation in Mo(0) content before and after the reaction with 1-nonanol. This suggests that reduced Mo is not accessible for alcohol substrates. However, oxidized Mo sites and/or silanols on the support can partake in acid-catalysed dehydration of tertiary alcohols, which is the presumed origin of the excellent chemoselectivity towards HDO of these substrates.
Conclusion
In this study, we have systematically investigated the effect of Mo on alcohol deoxygenation for bimetallic materials based on M–Mo/SiO2 (M = Ni, Pd, Pt, Rh, and Ir). The SOMC preparation protocol, combined with an air-free approach for characterization before and after catalysis, allows for a direct comparison of activity and structure between the five investigated metals. The promotional effect of Mo was particularly strong in combination with Ir, but also significant with Rh and Pt. For Pd–Mo/SiO2, efficient HDO activity is observed only with tertiary alcohols, while almost no conversion to product was seen for the secondary and primary alcohols, opening the possibility to explore the selective deoxygenation of diols. For Ni-based catalysts, no significant deoxygenation activity is observed, which parallels the lack of formation of a stable alloy with Mo under the investigated reaction conditions.Coming to the role of Mo, with tertiary alcohols, the slight increase of Brønsted (and/or Lewis) acidity (not detected by pyridine as a probe molecule) appears to be sufficient to drive deoxygenation via a dehydration–hydrogenation pathway. However, for primary and secondary alcohols, a different mechanism is likely dominant. HDO activity for these substrates is linked to the formation of a Mo surface alloy, evidenced by XAS, CO-IR spectroscopy, and CO-chemisorption, suggesting reduced Mo sites on alloyed nanoparticles to be key. Presumably, Mo activates the C–O bond by coordination to oxygen followed by a redox process, while the other transition metal (Pt, Rh, or Ir) helps to activate H2, which is used to keep Mo in a reduced state and enable catalytic turnover. Keeping Mo reduced and accessible on the nanoparticle surface is likely to be the key balance for high activity with primary and secondary alcohols. While Ni does not form a (stable) alloy, Pd–Mo/SiO2 exhibits a high proportion of alloyed Mo, however, likely due to the low surface energy of Pd, Mo is concentrated in the bulk of nanoparticles. Possibly, this could be a mechanism for deactivation observed with Ir–Mo/SiO2, where the concentration of metallic Mo increases during the reaction.
Data availability
The data supporting this article have been included as part of the ESI (PDF).†Author contributions
C. E. contributed to project design, synthesized materials, conducted routine characterization and catalytic tests, performed data analysis, conceptualized results, and co-wrote the original manuscript draft. S. P. contributed to project design, conducted X-ray absorption spectroscopy (XAS) experiments and data processing, performed data analysis, conceptualized results, and co-wrote the original manuscript draft. J. D. J. S. enabled the experimental workflow for catalytic reactions and contributed to data interpretation. X. Z. performed transmission electron microscopy (TEM) measurements. K. S. conducted chemisorption measurements and data analysis. M. N. provided input on XAS experiments, conceptualized results, and secured funding. O. S. and C. C. contributed to project design, conceptualized results, co-wrote the original manuscript draft, and secured funding. All authors contributed to writing and editing the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
C. C. and C. E. acknowledge the Swiss National Science Foundation (grant 200020B_192050) and the Scholarship Fund of the Swiss Chemical Industry (SSCI). C. C., O. S., M. N., S. P., and X. Z. acknowledge the National Centre of Competence in Research (NCCR) Catalysis (grant 180544). J. D. J. S. was supported by the National Research Fund, Luxembourg (AFR Individual PhD Grant 12516655). The authors thank Barnabé Berger, Mattia Ciuoli, and Max Candrian for fruitful discussions and help with material synthesis. The authors thank Merlin Seidel and Enzo Brack for help with 15N-pyridine adsorption experiments, and STEM-imaging, respectively. The authors also thank the Swiss Light Source for beamtime (SuperXAS beamline X10DA) (Proposal No. 20222179) and the beamline staff for assistance during measurements. ScopeM is gratefully acknowledged for their support and assistance in this work through project no. 2460.References
- M. M. Ambursa, J. C. Juan, Y. Yahaya, Y. H. Taufiq-Yap, Y.-C. Lin and H. V. Lee, A review on catalytic hydrodeoxygenation of lignin to transportation fuels by using nickel-based catalysts, Renewable Sustainable Energy Rev., 2021, 138, 110667 CrossRef CAS.
- F. Kourougianni, A. Arsalis, A. V. Olympios, G. Yiasoumas, C. Konstantinou, P. Papanastasiou and G. E. Georghiou, A comprehensive review of green hydrogen energy systems, Renewable Energy, 2024, 231, 120911 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering, Chem. Rev., 2006, 106(9), 4044 CrossRef CAS PubMed.
- M. Dusselier, M. Mascal and B. F. Sels, Top Chemical Opportunities from Carbohydrate Biomass: A Chemists View of the Biorefinery, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. Nicholas, K. M., Springer International Publishing, 2014, p. 1, DOI:10.1007/128_2014_544.
- W. Jin, L. Pastor-Pérez, D. Shen, A. Sepúlveda-Escribano, S. Gu and T. Ramirez Reina, Catalytic Upgrading of Biomass Model Compounds: Novel Approaches and Lessons Learnt from Traditional Hydrodeoxygenation – a Review, ChemCatChem, 2019, 11(3), 924 CrossRef CAS.
- L. Qu, X. Jiang, Z. Zhang, X.-g. Zhang, G.-y. Song, H.-l. Wang, Y.-p. Yuan and Y.-l. Chang, A review of hydrodeoxygenation of bio-oil: model compounds, catalysts, and equipment, Green Chem., 2021, 23(23), 9348 RSC.
- J. G. Tillou, C. J. Ezeorah, J. J. Kuchta, S. C. D. Dissanayake Mudiyanselage, J. D. Sitter and A. K. Vannucci, A review on recent trends in selective hydrodeoxygenation of lignin derived molecules, RSC Sustainability, 2023, 1(7), 1608 RSC.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Highly Selective Catalytic Conversion of Phenolic Bio-Oil to Alkanes, Angew. Chem., Int. Ed., 2009, 48(22), 3987 CrossRef CAS PubMed.
- S. Sethupathy, G. Murillo Morales, L. Gao, H. Wang, B. Yang, J. Jiang, J. Sun and D. Zhu, Lignin valorization: Status, challenges and opportunities, Bioresour. Technol., 2022, 347, 126696 CrossRef CAS PubMed.
- C. Yang, W. Wang, D. Wang, M. Gong, Y. Xin, L. Xiao, O. V. Kikhtyanin, D. Kubicka and W. Wu, The promotion effects of MoOx species in the highly effective NiMo/MgAl2O4 catalysts for the hydrodeoxygenation of methyl palmitate, J. Environ. Chem. Eng., 2022, 10(3), 107761 CrossRef CAS.
- A. Deneyer, T. Renders, J. Van Aelst, S. Van den Bosch, D. Gabriëls and B. F. Sels, Alkane production from biomass: chemo-, bio- and integrated catalytic approaches, Curr. Opin. Chem. Biol., 2015, 29, 40 CrossRef CAS PubMed.
- Bond Lengths And Strengths, in Lange's Handbook of Chemistry, ed. Speight, J. G., McGraw-Hill Education, 17th edn, 2017 Search PubMed.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts, Green Chem., 2019, 21(14), 3715 RSC.
- N. Arun, R. V. Sharma and A. K. Dalai, Green diesel synthesis by hydrodeoxygenation of bio-based feedstocks: Strategies for catalyst design and development, Renewable Sustainable Energy Rev., 2015, 48, 240 CrossRef CAS.
- A. M. Robinson, J. E. Hensley and J. W. Medlin, Bifunctional Catalysts for Upgrading of Biomass-Derived Oxygenates: A Review, ACS Catal., 2016, 6(8), 5026 CrossRef CAS.
- S. Haida, S. Löbner, H. Lund, S. Bartling, C. Kreyenschulte, H. Atia, A. M. Abdel-Mageed, C. Kubis and A. Brückner, Investigations into the influence of nickel loading on MoO3-modified catalysts for the gas-phase hydrodeoxygenation of anisole, Catal. Sci. Technol., 2024, 14(8), 2201 RSC.
- S. Liu, W. Zheng, J. Fu, K. Alexopoulos, B. Saha and D. G. Vlachos, Molybdenum Oxide-Modified Iridium Catalysts for Selective Production of Renewable Oils for Jet and Diesel Fuels and Lubricants, ACS Catal., 2019, 9(9), 7679 CrossRef CAS.
- K. Yuan, Y. Yamazaki, X. Jin and K. Nozaki, Multifunctional WO3–ZrO2-Supported Platinum Catalyst for Remarkably Efficient Hydrogenolysis of Esters to Alkanes, J. Am. Chem. Soc., 2023, 145(6), 3454 CrossRef CAS PubMed.
- L. Jin, W. Li, Q. Liu, L. Ma, C. Hu, A. T. Ogunbiyi, M. Wu and Q. Zhang, High performance of Mo-promoted Ir/SiO2 catalysts combined with HZSM-5 toward the conversion of cellulose to C5/C6 alkanes, Bioresour. Technol., 2020, 297, 122492 CrossRef CAS PubMed.
- S. Liu, T. Simonetti, W. Zheng and B. Saha, Selective Hydrodeoxygenation of Vegetable Oils and Waste Cooking Oils to Green Diesel Using a Silica-Supported Ir–ReO Bimetallic Catalyst, ChemSusChem, 2018, 11(9), 1446 CrossRef CAS PubMed.
- Y. Zhang, G. Fan, L. Yang, L. Zheng and F. Li, Cooperative Effects between Ni-Mo Alloy Sites and Defective Structures over Hierarchical Ni-Mo Bimetallic Catalysts Enable the Enhanced Hydrodeoxygenation Activity, ACS Sustainable Chem. Eng., 2021, 9(34), 11604 CrossRef CAS.
- B. Liu, Y. Nakagawa, M. Yabushita and K. Tomishige, Highly Efficient Iridium–Iron–Molybdenum Catalysts Condensed on Boron Nitride for Biomass-Derived Diols Hydrogenolysis to Secondary Monoalcohols, J. Am. Chem. Soc., 2024, 146(14), 9984 CrossRef CAS PubMed.
- A. Robinson, G. A. Ferguson, J. R. Gallagher, S. Cheah, G. T. Beckham, J. A. Schaidle, J. E. Hensley and J. W. Medlin, Enhanced Hydrodeoxygenation of m-Cresol over Bimetallic Pt–Mo Catalysts through an Oxophilic Metal-Induced Tautomerization Pathway, ACS Catal., 2016, 6(7), 4356 CrossRef CAS.
- J. Wang, Y. Chen, C. Liu, Y. Lu, X. Lin, D. Hou, C. Luo, D. Wang, Z. Zheng and Y. Zheng, Highly stable Mo-based bimetallic catalysts for selective deoxygenation of oleic acid to fuel-like hydrocarbons, J. Environ. Chem. Eng., 2023, 11(1), 109104 CrossRef CAS.
- Z. Wang, B. Pholjaroen, M. Li, W. Dong, N. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, Chemoselective hydrogenolysis of tetrahydrofurfuryl alcohol to 1,5-pentanediol over Ir-MoOx/SiO2 catalyst, J. Energy Chem., 2014, 23(4), 427 CrossRef.
- A. N. Kay Lup, F. Abnisa, W. M. A. W. Daud and M. K. Aroua, A review on reaction mechanisms of metal-catalyzed deoxygenation process in bio-oil model compounds, Appl. Catal., A, 2017, 541, 87 CrossRef CAS.
- G. J. Hutchings and J. C. Védrine, Heterogeneous Catalyst Preparation, in Basic Principles in Applied Catalysis, ed. Baerns, M., Springer Berlin Heidelberg, 2004, p. 215, DOI:10.1007/978-3-662-05981-4_6.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, the Gap between Industrial and Well-Defined Supported Catalysts, Angew. Chem., Int. Ed., 2018, 57(22), 6398 CrossRef PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Surface Organometallic and Coordination Chemistry toward Single-Site Heterogeneous Catalysts: Strategies, Methods, Structures, and Activities, Chem. Rev., 2016, 116(2), 323 CrossRef PubMed.
- C. Copéret, Single-Sites and Nanoparticles at Tailored Interfaces Prepared via Surface Organometallic Chemistry from Thermolytic Molecular Precursors, Acc. Chem. Res., 2019, 52(6), 1697 CrossRef PubMed.
- E. L. Lee and I. E. Wachs, In Situ Spectroscopic Investigation of the Molecular and Electronic Structures of SiO2 Supported Surface Metal Oxides, J. Phys. Chem. C, 2007, 111(39), 14410 CrossRef CAS.
- Z. J. Berkson, R. Zhu, C. Ehinger, L. Lätsch, S. P. Schmid, D. Nater, S. Pollitt, O. V. Safonova, S. Björgvinsdóttir, A. B. Barnes, Y. Román-Leshkov and G. A. Price, et al., Active Site Descriptors from 95Mo NMR Signatures of Silica-Supported Mo-Based Olefin Metathesis Catalysts, J. Am. Chem. Soc., 2023, 145(23), 12651 CrossRef CAS PubMed.
- C. Ehinger, X. Zhou, M. Candrian, S. R. Docherty, S. Pollitt and C. Copéret, Group 10 Metal Allyl Amidinates: A Family of Readily Accessible and Stable Molecular Precursors to Generate Supported Nanoparticles, JACS Au, 2023, 3(8), 2314 CrossRef CAS PubMed.
- W. Zhou, S. R. Docherty, C. Ehinger, X. Zhou and C. Copéret, The promotional role of Mn in CO2 hydrogenation over Rh-based catalysts from a surface organometallic chemistry approach, Chem. Sci., 2023, 14(20), 5379 RSC.
- W. Zhou, C. Hansen, W. Cao, E. Brack, S. R. Docherty, C. Ehinger, Y. Wang, C. Wang and C. Copéret, Gallium: A Universal Promoter Switching CO2 Methanation Catalysts to Produce Methanol, JACS Au, 2025, 5(1), 217 CrossRef CAS PubMed.
- M. H. Youn, J. G. Seo, P. Kim and I. K. Song, Role and effect of molybdenum on the performance of Ni-Mo/γ-Al2O3 catalysts in the hydrogen production by auto-thermal reforming of ethanol, J. Mol. Catal. A: Chem., 2007, 261(2), 276 CrossRef CAS.
- S. R. Docherty, N. Phongprueksathat, E. Lam, G. Noh, O. V. Safonova, A. Urakawa and C. Copéret, Silica-Supported PdGa Nanoparticles: Metal Synergy for Highly Active and Selective CO2-to-CH3OH Hydrogenation, JACS Au, 2021, 1(4), 450 CrossRef CAS PubMed.
- I. B. Moroz, K. Larmier, W.-C. Liao and C. Copéret, Discerning γ-Alumina Surface Sites with Nitrogen-15 Dynamic Nuclear Polarization Surface Enhanced NMR Spectroscopy of Adsorbed Pyridine, J. Phys. Chem. C, 2018, 122(20), 10871 CrossRef CAS.
- A. S. Asundi, A. S. Hoffman, P. Bothra, A. Boubnov, F. D. Vila, N. Yang, J. A. Singh, L. Zeng, J. A. Raiford, F. Abild-Pedersen, S. R. Bare and S. F. Bent, Understanding Structure–Property Relationships of MoO3-Promoted Rh Catalysts for Syngas Conversion to Alcohols, J. Am. Chem. Soc., 2019, 141(50), 19655 CrossRef CAS PubMed.
- X. Zhang, Y. Lu, L. Kovarik, P. Dasari, D. Nagaki and A. M. Karim, Structure sensitivity of n-butane hydrogenolysis on supported Ir catalysts, J. Catal., 2021, 394, 376 CrossRef CAS.
- G. Bergeret and P. Gallezot, Particle Size and Dispersion Measurements, in Handbook of Heterogeneous Catalysis, p. 738, DOI:10.1002/9783527610044.hetcat0038.
- P. Canton, G. Fagherazzi, M. Battagliarin, F. Menegazzo, F. Pinna and N. Pernicone, Pd/CO Average Chemisorption Stoichiometry in Highly Dispersed Supported Pd/γ-Al2O3 Catalysts, Langmuir, 2002, 18(17), 6530 CrossRef CAS.
- A. Horváth, A. Beck, A. Sárkány, Z. Koppány, A. Szűcs, I. Dékány, Z. E. Horváth and L. Guczi, Effect of different treatments on Aerosil silica-supported Pd nanoparticles produced by ‘controlled colloidal synthesis, Solid State Ionics, 2001, 141–142, 147 CrossRef.
- W. R. Tyson and W. A. Miller, Surface free energies of solid metals: Estimation from liquid surface tension measurements, Surf. Sci., 1977, 62(1), 267 CrossRef CAS.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, The surface energy of metals, Surf. Sci., 1998, 411(1), 186 CrossRef CAS.
- C. Sener, T. S. Wesley, A. C. Alba-Rubio, M. D. Kumbhalkar, S. H. Hakim, F. H. Ribeiro, J. T. Miller and J. A. Dumesic, PtMo Bimetallic Catalysts Synthesized by Controlled Surface Reactions for Water Gas Shift, ACS Catal., 2016, 6(2), 1334 CrossRef CAS.
- C. Hansen, W. Zhou, E. Brack, Y. Wang, C. Wang, J. Paterson, J. Southouse and C. Copéret, Decoding the Promotional Effect of Iron in Bimetallic Pt–Fe-nanoparticles on the Low Temperature Reverse Water–Gas Shift Reaction, J. Am. Chem. Soc., 2024, 146(40), 27555 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08532a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |