
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zirconium-mediated carbon–fluorine bond functionalisation through cyclohexyne “umpolung”†‡
Sara
Bonfante
 ab,
Theo F. N.
Tanner
b,
Christian
Lorber
*a,
Jason M.
Lynam
*b,
Antoine
Simonneau
*a and
John M.
Slattery
*b
ab,
Theo F. N.
Tanner
b,
Christian
Lorber
*a,
Jason M.
Lynam
*b,
Antoine
Simonneau
*a and
John M.
Slattery
*b
aLCC-CNRS, Université de Toulouse, CNRS, UPS205route de Narbonne, BP44099, F-31077 Toulouse cedex 4, France
bDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK
First published on 16th January 2025
Abstract
Polarity reversal, or “umpolung”, is a widely acknowledged strategy to allow organic functional groups amenable to react in alternative ways to the usual preference set by their electronic features. In this article, we demonstrate that cyclohexyne umpolung, realized through complexation to zirconocene, makes the small strained cycloalkyne amenable to C–F bond functionalisation. Such strong bond activation chemistry is unprecedented in “free” aryne and strained alkyne chemistry. Our study also reveals that the reactivity of the Zr–cyclohexyne complex is highly sensitive to the degree of fluorination of the heteroarene. In addition, parasitic reactions of the ancillary ligand PMe3 were observed when pentafluoropyridine was the substrate.
Introduction
Arynes and strained small cycloalkynes are fleeting intermediates that have experienced renewed interest over the past 15 years.1 Easier methods for their in situ generation have allowed the development of several useful synthetic protocols. In particular, cycloadditions or multicomponent reactions as well as formal insertions into polar X–Y bonds have been achieved, whereas their combination with late transition-metal-based methodologies have further expanded the portfolio of reactions achievable for these fascinating species.1e,g Their intrinsic electrophilicity, driving their reactivity, is the consequence of poor overlap of the p-orbitals involved in the second π bond, resulting in significant lowering of the LUMO compared to linear alkynes.2 Arynes and strained alkynes can be stabilized by coordination to a low-valent transition metal center,3 from which a high degree of back-bonding may impart the strained fragment a nucleophilic behaviour (Scheme 1). This is a manifestation of the ability of transition metals to reverse the innate reactivity of organic ligands.4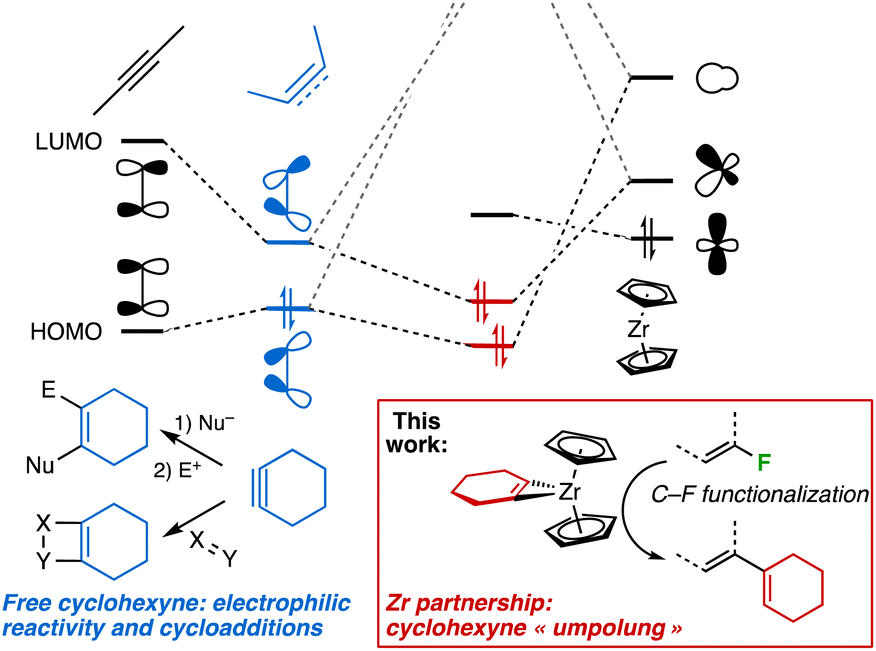 | ||
| Scheme 1 As a result of strong back-bonding to the LUMO of cyclohexyne, polarity reversal (“umpolung”) ensues from Zr complexation and makes it amenable to C–F bond activation. | ||
Unlike transition-metal alkylidenes, alkylidynes or imides, which can promote the cleavage of C–H5 and C–F5c,f,6 bonds, there are only very limited examples in which aryne/strained alkyne complexes undergo bond activation and functionalisation processes on exogenous substrates.7 Conceptually, this would demonstrate that their “umpolung”8 may lead to bond activation processes that have remained elusive for “free” arynes and small cycloalkynes. We now report a successful application of this concept for C–F bond functionalisation with a metal-coordinated strained alkyne. Such a transformation is attractive given the ubiquity of C–F bonds in pharmaceuticals or agrochemicals,9 as a late-stage derivatization strategy, or to elaborate fluorinated synthons from simple and widely available per- or poly-fluorinated molecules.10
An important body of work exists concerning the functionalisation of polyfluoro-pyridines and -azines employing transition metals in either stoichiometric or catalytic amounts. Approaches based on the use of late transition metals10a dominate this chemistry and generally rely on oxidative addition of the C–F bond followed by transmetalation and allow the introduction of aryl,11 vinyl,12 alkyl,13 acyl14 as well as silyl12b or boryl15 groups. Although group 4 and 5 transition-metal complexes have been shown to be competent for CAr–F bond activation5c,f,6,16 and their catalytic hydrodefluorination,17 there is in contrast only a handful of early transition metal (ETM) species reported to mediate C–F bond functionalisation of polyfluoropyridines. In situ generated imido5c or η2-cyclopropene16i zirconocene complexes as well as a titanium alkylidyne5f,6 react with C–F bonds of fluoropyridines to form C–N or C–C bonds through C–F-addition over the metal–ligand bond. The undeniable advantage of these methods is their perfect regioselectivity for the 2-position of the aryl compound, especially challenging when the 4-position also carries a fluorine atom.16i,18,19 Of note, the groups of Rosenthal and Beckhaus have explored the reactivity of group 4 metallocene–alkyne complexes with fluoropyridines, revealing the preference of the systems for 1,2-C–H addition or C–F bond oxidative addition, but no coupling of the alkyne with the fluorinated heterocycle was observed.16d,e This work demonstrates that a cyclohexyne–zirconocene complex is effectively capable of C–F bond functionalisation of fluoropyridines. This is achievable thanks to the ring strain of the small cyclic alkyne ligand as well as its “umpolung” in the vicinity or zirconium, and results in net alkenylation of the heterocycle.
Results and discussion
The Zr complex [Cp2Zr(PMe3)(c-C2(CH2)4)] 1 (Scheme 2) was first prepared by Buchwald and co-workers. It was selected for this study as it reacts with various unsaturated organic molecules by insertion into the Zr–C bond.20 We commenced our study by treating 1 with 3 equiv. of 2,6-difluoropyridine (Py-F2) or 2,4,6-trifluoropyridine (Py-F3) in C6D6. In both cases, the targeted C–F functionalisation took place, unlike the previously reported reactivity of group 4 metallocene–alkyne complexes with polyfluoropyridines that leads to C–F metalation.16d,e We thus obtained compounds [Cp2ZrF(κ2C,N–C11H11−nNF1+n)] (n = 0 or 1) 2 and 3, respectively (Scheme 2), which were identified on the basis of their 1H and 19F NMR spectra. Liberation of PMe3 was evidenced by its resonance at −62.0 ppm in the 31P NMR spectrum. The addition product 3 was characterised by a singlet at δ (19F) = −35.4 ppm, corresponding to the zirconium fluoride, in the 19F NMR spectrum. Moreover, a doublet at δ (19F) = −60.7 ppm (4JFF = 24.0 Hz) and a doublet of doublets of doublets at δ (19F) = −94.8 ppm (4JFF = 24.0, 3JFH = 9.5 Hz, 3JFH = 7.5 Hz) were associated with the ortho- and para-fluorine of the pyridine ligand, respectively. Both 2 and 3 are highly soluble and sensitive compounds, making their purification and isolation from the reaction mixture difficult. However, an optimal isolated yield of 71% for 3 was obtained when the reaction was carried out in cyclohexane instead of benzene. | ||
| Scheme 2 Reaction of 1 with Py-Fn (n = 2, 3) leads to C–F alkenylation. aNMR yield. bIsolated yield. | ||
After treatment of the reaction mixture containing 3 with I2, gas chromatography-mass spectrometry (GC-MS) analysis resulted in the detection of 2-(2-iodocyclohexen-1-yl)-4,6-difluoropyridine. This confirmed that products 2 and 3 were the result of 1,3-addition of the C–F bond over the metal–cyclohexyne linkage (Scheme 2). To the restricted portfolio of Zr-mediated C–F bond functionalizations, allowing introduction of H,16c,17 alkyl,16i aryl16a or amino5c groups, this reaction adds an efficient and regioselective way to insert an alkene moiety. The strain imparted to the alkyne is instrumental in this outcome since zirconocene complexes of unstrained alkynes preferentially lead to oxidative addition of aromatic C–F bonds,16d underpinning the special reactivity of the cyclohexyne ligand.
The molecular structure of 3 was determined by single crystal X-ray diffraction (Fig. 1) and provided definitive evidence for a C–F bond functionalisation at the ortho position of the fluoropyridine. The Zr atom lies in a distorted trigonal bipyramid environment, with apical sites occupied by the nitrogen of the pyridine ligand and the fluoride. The former belongs to a 1-zircona-2-aza-cyclopentadiene motif that connects it to the alkenyl carbon found in the equatorial plan, forcing the F–Zr–N angle to deviate significantly from linearity [F1–Zr1–N1 148.46(6)°] and imposing a tilt angle for the Cp–Zr–Cp moiety of 135.8°. Similar deviations from the ideal trigonal bipyramid geometry are found in other crystallographically characterized five-coordinated, Cp-supported 1-zircona-2-aza-cyclopentadienes.21 Compared to structurally related species (Table S3‡),21,22 the angles measured within the five-membered ring as well as the bond lengths of the conjugated part show no significant irregularity. However, the Zr–C bond is rather short and the Zr–N one, rather long. In the family of compounds selected for comparison, the former varies slightly in length, while the Zr–N distance fluctuates depending on the L or X character of the nitrogen ligand and the N-substituent. In the present case, we explain the long Zr–N bond by the presence of the fluoride ligand in a mutually trans position, coupled to the electron-poor properties of the difluoropyridine ring. The elongation of the Zr–N bond mechanically impacts the length of the Zr–C one due to the rigidity of the 1-zircona-2-aza-cyclopentadiene core. An additional observation is that the N1–C11–F3 angle is quite acute [114.7(2)°], presumably because of a long-range interaction of F3 with the electropositive Zr atom (Zr1–F3 3.469(2) Å < ΣrW).
 | ||
| Fig. 1 Molecular structure of 3 in the solid state. Ellipsoids drawn at the 25% probability level. Selected bond lengths (Å) and angles (°): Zr1–F1 2.008(1), Zr1–C1 2.321(2), Zr1–N1 2.479(2), N1–C7 1.372(3), C1–C6 1.354(3), C6–C7 1.456(3), F1–Zr1–C1 80.26(6), C1–Zr1–N1 68.35(6), F1–Zr1–N1 148.46(6), N1–C11–F3 114.7(2). | ||
We next turned our attention towards the reaction between 1 and 2,3,5,6-tetrafluoropyridine (Py-F4). When 1 was reacted with an equimolar amount of Py-F4 at 60 °C in C6D6, the main product was found to be the C–H activation product [Cp2Zr(1-C6H9)(4-C5F4N)] 5 (Scheme 3), similar to what was observed in the reaction of the Cp2Zr(η2-Me3SiC2SiMe3) complex with Py-F4.16d This species was characterised by two multiplets in the 19F NMR spectrum at δ = −97.7 and −114.8 ppm associated with the ortho- and meta-F of the pyridine ligand, respectively. Small amounts of 1-cyclohexenylzirconocene fluoride (6, 9% after 48 h) also formed according to NMR analysis, that were found to increase with time as 5 decreased (NMR yield of 5 was 53% after 27 h but dropped to 45% after 48 h). We believe 5 arises from 1,2 C–H addition over the Zr–(η2-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) bond in 1, a hypothesis that was further supported by the absence of deuterium incorporation from C6D6 into the cyclohexenyl ligand. Further evidence for C–H activation was obtained from NMR analysis of the reaction mixture after treatment with I2, which indicated the presence of 4-iodo-2,3,5,6-tetrafluoropyridine. Of note, the reaction mixture became more complex, with a range of unidentified products, when excess Py-F4 was employed.
C) bond in 1, a hypothesis that was further supported by the absence of deuterium incorporation from C6D6 into the cyclohexenyl ligand. Further evidence for C–H activation was obtained from NMR analysis of the reaction mixture after treatment with I2, which indicated the presence of 4-iodo-2,3,5,6-tetrafluoropyridine. Of note, the reaction mixture became more complex, with a range of unidentified products, when excess Py-F4 was employed.
 | ||
| Scheme 3 Reaction of 1 with Py-F4 leads to C–H activation. aNMR yields. | ||
Finally, we investigated the reactivity of 1 towards pentafluoropyridine (Py-F5). After four hours at room temperature in C6D6, 1 was fully converted into 1-cyclohexenylzirconocene fluoride ([Cp2ZrF(1-C6H9), 6, 62% NMR yield) (Scheme 4). An unidentified dark red solid that precipitated at the end of the reaction, amounting roughly 25–30% of the reactant mass and proving insoluble in usual organic solvents, including highly polar ones, as well as in water. The elemental analysis of two different samples showed the presence of nitrogen (ca. 3.5%), suggesting it may incorporate pyridinic moieties. While the carbon content was consistent (ca. 35%), the hydrogen varied from a sample to the other (2.7 to 4.9%). Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) revealed that the Zr and P contents fluctuate from 0.5–1.5% to 3–11%, respectively (Table S2‡).
 | ||
| Scheme 4 Species identified from the reaction of 1 with Py-F5. aNMR yields. | ||
These conflicting analyses point to an uncontrolled precipitation of this material. Infrared spectroscopy (ATR method) shows two common intense bands at 957–980 and 487–496 cm−1 within non-superimposable spectra (Fig. S12‡). The first band could be the result of P–F stretches that generally result in strong absorption in the 700–1000 cm−1 region for fluorophosphoranes and around 900 cm−1 for fluorophosphoniums.23 We also identified in the reaction mixture the hydrodefluorination product 2,3,5,6-tetrafluoropyridine (Py-F4, 9% NMR yield), trimethyldifluorophosphorane (F2PMe3, 7, 1% NMR yield) and a third product presumably resulting from the reaction of pentafluoropyridine with a phosphorus species, the precise identification of which remained elusive. The formation of 6, Py-F4 and 7 was ascertained by multinuclear NMR spectroscopy. To further evidence the presence of 6, 1 equivalent of I2 was added into the reaction mixture and the formation of 1-iodocyclohexene, Cp2ZrFI, Cp2ZrF2 and Cp2ZrI2 was observed by 1H NMR analysis. Moreover, the addition of hydrochloric acid to the crude mixture led to the liberation of cyclohexene (see ESI‡ for detailed NMR analysis). Different reaction conditions were investigated: changing solvent, concentration, reagent ratio and lowering of the temperature to 0 °C had only little influence on the reaction outcome. At −40 °C, the system was unreactive.
Even though the mechanism leading to 6 is still unclear, it is proposed that the vinylic proton in 6 comes from the phosphine, since when the solvent used for the reaction was deuterated, no deuterium incorporation was found. The presence of the formal hydrodefluorination product Py-F4 recalls recent works demonstrating the ability of pnictogen compounds to achieve this transformation either stoichiometrically or catalytically.24 In particular, our groups jointly reported that simple trialkylphosphines are able to catalytically hydrodefluorinate Py-F5 to Py-F4,24a and mechanistic investigations revealed that the catalytic cycle involves P(III)/P(V) redox cycling and initiates with nucleophilic attack of the phosphorus atom onto the electron-deficient heterocycle24a,b,f,25via a Meisenheimer-like transition state. It is thus likely that the ancillary ligand PMe3 plays a role in the formation of Py-F4. Indeed, PMe3 reacts rapidly in C6D6 at room temperature with one equivalent of Py-F5 to afford a mixture in which Py-F4 and difluorophosphoranes F2PMe3 and 8 were identified as the major products (47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :34:19 ratio, respectively, >90% NMR yield according to 19F NMR integration, Scheme 5). 8 is likely the result of SNAr of Py-F5ortho-fluoride by the fluorophosphorus ylide Me2FP
:34:19 ratio, respectively, >90% NMR yield according to 19F NMR integration, Scheme 5). 8 is likely the result of SNAr of Py-F5ortho-fluoride by the fluorophosphorus ylide Me2FP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (ref. 26) for which the way and means of its in situ formation are yet to be elucidated. When this mixture was treated with one equivalent of 1, the latter was immediately converted to 6 rather selectively, along with some insoluble materials. Addition of two more equivalent of 1 were necessary to completely consume F2PMe3 and 8, leaving 6 and unreacted Py-F4 as the main identifiable products of the reaction (see ESI‡ for details). The significant P content and low Zr one in the red solid, as well as a strong absorption in the P–F region indicates it presumably stems from another uncontrolled parasitic reaction between PMe3 and Py-F5, but its absence in the case of the reaction thereof without 1 suggests Zr plays a role in its formation.
CH2 (ref. 26) for which the way and means of its in situ formation are yet to be elucidated. When this mixture was treated with one equivalent of 1, the latter was immediately converted to 6 rather selectively, along with some insoluble materials. Addition of two more equivalent of 1 were necessary to completely consume F2PMe3 and 8, leaving 6 and unreacted Py-F4 as the main identifiable products of the reaction (see ESI‡ for details). The significant P content and low Zr one in the red solid, as well as a strong absorption in the P–F region indicates it presumably stems from another uncontrolled parasitic reaction between PMe3 and Py-F5, but its absence in the case of the reaction thereof without 1 suggests Zr plays a role in its formation.
 | ||
| Scheme 5 The reaction of PMe3 with Py-F5 affords Py-F4 and difluorophosphoranes F2PMe3 and 8. The difluorophosphoranes react with 1 to give 6. | ||
Several hypotheses may be given to explain the differences in reactivity of the four polyfluoropyridines towards 1. The absence of a formal 1,2-C–F addition product after treatment with Py-F5 can reasonably be attributed to the fast reaction of the latter with dissociated PMe3. The low Lewis basicity of Py-F5 may also inhibit binding to the phosphine-free cyclohexyne complex, which may be an important prerequisite for the C–F bond activation event. Interestingly, during the synthesis of 1, an unsaturated [Cp2Zr(η2-c-C6H8)] cyclohexyne complex is supposed to form, to which PMe3 is then complexed to furnish 1. Addition of Py-F5 to the reaction mixture before addition of PMe3 resulted in no reaction, suggesting the Zr centre is unable to coordinate Py-F5 and trigger C–F bond activation. Additionally, the structure of 3 (Fig. 1) reveals a 1,3-allylic (1,3A) strain between the cycles' backbones (carbons C5 to C8), along the ring junction. This destabilizing interaction would be exacerbated if the heteroaromatic ring was fully fluorinated and may exist before the C–C bond formation event, imparting a kinetic penalty for a C–F bond activation pathway. The observation of the C–H activation product in the reaction with Py-F4 may be the result of two factors: on the one hand, the high level of fluorination of the heterocycle makes it, akin to Py-F5, a poor Lewis base that is not likely to strongly bind the Zr center. Besides, a potential C–F/CH2 repulsion, resulting in the previously mentioned 1,3A strain in the cyclometalated product (Fig. 1), is likely to arise from Py-F4 coordination. On the other hand, the metalation of the acidic C–H bond provides a metal-4-pyridyl compound with highly stabilizing two-fold ortho fluorine substitution that considerably strengthen the Zr–C bond.27 Delineation of the factors that govern C–F vs. C–H bond activation in fluoroaromatics has been the object of several experimental and theoretical studies,28 revealing marked differences between metals. Focusing on Zr, our observation seems to be in line with the one of the Rosenthal group with [Cp2Zr(η2-Me3SiCCSiMe3)].16d The latter shows preference for C–H over C–F bond activation when reacted with 2,3,5,6- or 2,4,5,6-tetrafluoropyridine. In the case of Py-F3 and Py-F2, higher Lewis basicity may allow the heteroarene to better interact with the Zr atom after PMe3 dissociation and thus bringing its ortho C–F bonds closer to the reactive cyclohexyne ligand—the stage is set for a regioselective C–F bond activation to take place.
A series of calculations using density functional theory (DFT) were undertaken to investigate this proposal. The interaction between the putative 16-electron complex [Cp2Zr(c-C2(CH2)4)] and a range of different fluorine-substituted pyridine ligands, as well as parent pyridine itself, was investigated. This was envisaged to give structure A (Fig. 2). The resulting calculations demonstrated that in all cases PMe3 binds more strongly to the zirconium than all the pyridine ligands investigated, in the order PMe3 ≫ Py-H > Py-F1 > Py-F2 ∼ Py-F3 ∼ Py-F4 ∼ Py-F5. In the case of A-F1, two isomers may be considered, with the fluorine orientated either towards or away from the cyclohexyne ligand. The former (A-F1-1) is 17 kJ mol−1 higher in energy than the latter (A-F1-2) which may represent a repulsive interaction between the fluorine lone pairs and the cyclohexyne ligand. Such an interaction could be playing a role in the polyfluorinated analogues too. An examination of the bond metrics within the optimised structures supported the arguments that the more fluorinated ligands might be expected to show weaker Zr⋯N interactions. As shown in Table S4,‡ there is notable lengthening of this distance with an increasing degree of fluorination.
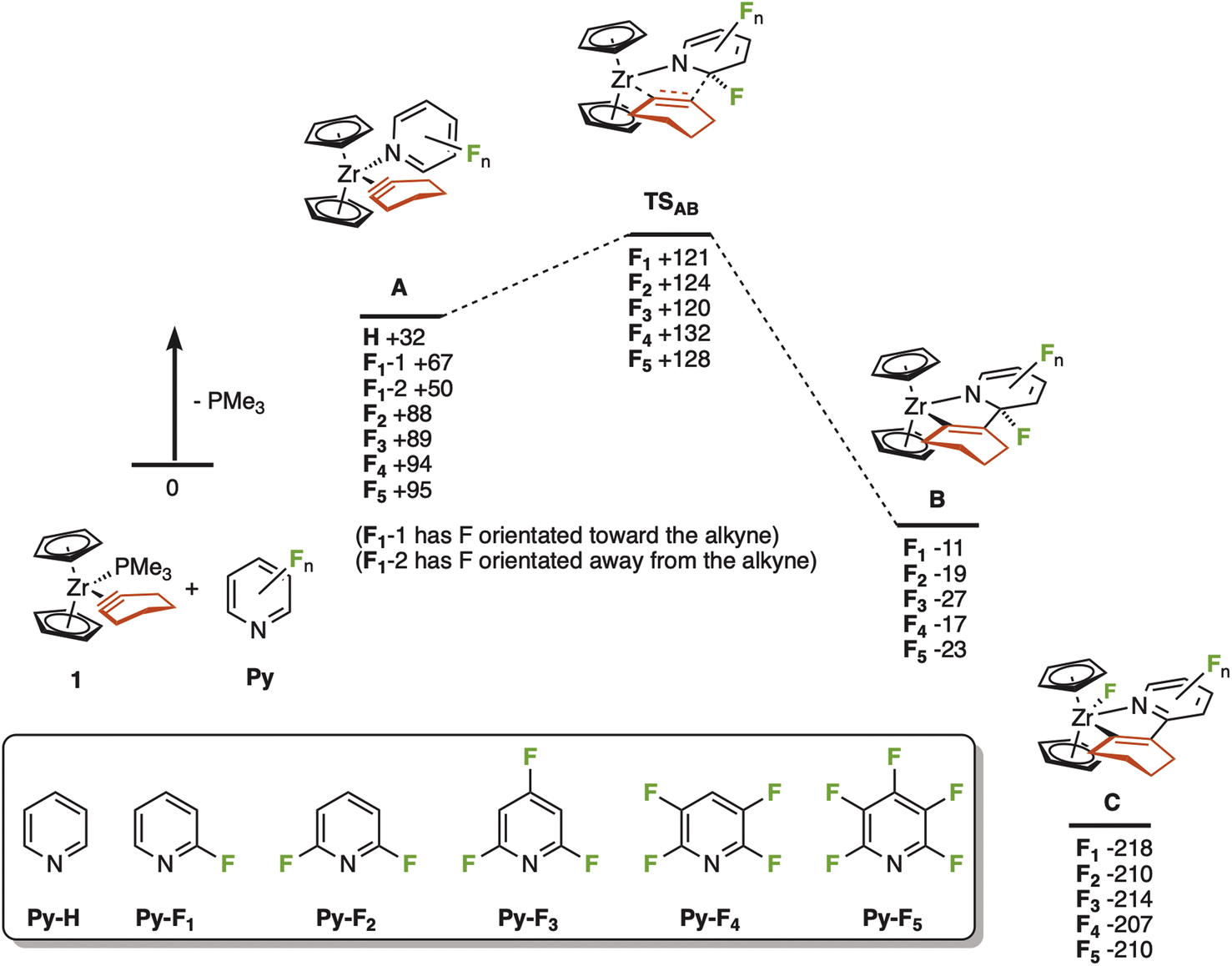 | ||
| Fig. 2 Potential energy surface for the reaction between 1 and substituted pyridines. Energies are Gibbs energies at 298 K in kJ mol−1 at the D3(BJ)-pbe0/def2-TZVPP//bp86/sv(p) level of theory with COSMO solvent correction in cyclohexane relative to 1 and the corresponding uncoordinated pyridine ligand. Insert shows the labelling code for the substituent patterns used. | ||
A transition state corresponding to C–C bond formation between the cyclohexyne and pyridine was located (TSAB, Fig. 2a) for all the fluorinated analogues. There was little overall effect of the degree of fluorination on the barrier to this step with respect to 1 and the free pyridines, but it should be noted that when compared to complex A, the barrier was lower in the cases of F2–F5 (31–38 kJ mol−1) than F1 (54 kJ mol−1). This transition state connects A with a Zr(IV) complex, B, which is probably best viewed as containing a bidentate ligand with coordinating alkenyl and amido groups, the pyridine ligand having been dearomatized.6c,16i
An examination of the calculated bond metrics in the complexes A (Table S4‡) supports the notation that this C–C bond formation is best considered as a case of “umpolung” reactivity. The calculated alkyne C–C bond lengths range from 1.334–1.377 Å, much closer to the corresponding CC bond length for cyclohexene (1.351 Å) than the CC length in cyclohexyne (1.237 Å) calculated at the same level of theory—it is worth noting that the cyclohexyne C–C bond in complex 1 has been measured at 1.295(25) Å,20 lying in between these two extrema. Indeed, in the case of the putative 16-electron complex [Cp2Zr(c-C2(CH2)4)] the C–C bond is 1.356 Å. These data indicate that there is extensive π-backbonding between the Zr and cyclohexyne in this system and is modulated by the electron-richness of the ligand. In the pyridine series, it is probably best considered as a Zr(IV) complex with a coordinated metallacyclopropene. Transition state TSAB can therefore be viewed as an intramolecular organometallic nucleophilic attack onto the pyridine, which is templated by a weak Zr⋯N interaction, consistent with an “umpolung” view of alkyne reactivity.
In all cases, the experimentally observed product arising from the reaction between Py-F2 and Py-F3 (experimental complexes 2 and 3, corresponding to states C-F2 and C-F3) was significantly thermodynamically favourable, presumably representing the formation of a strong Zr–F bond.
The results from this computational study therefore demonstrate that a plausible pathway for the intramolecular “umpolung” reaction is viable in all cases. This provides some evidence to support the argument that in the cases of Py-F4 and Py-F5, the predominance of competing reactions overrides the C–F functionalisation pathway, rather than any intrinsic barrier.
Other polyfluoroarenes were evaluated for C–F bond functionalisation using 1: hexafluorobenzene (C6F6) proved unreactive, in line with N → Zr interaction being a pre-requisite for the C–F bond activation to occur. The reaction between 1 and 2-fluoropyridine (Py-F1) led to full consumption of the former and produced a mixture of Zr fluorides as attested by 19F NMR. Our attempts with 2,4-difluoropyridine (Py-F2′) were not reproducible, but led in any case to intractable mixtures of fluorinated and organometallic compounds. 19F NMR monitoring of these reaction did reveal 6 formed in significant amounts though.
Conclusions
This work demonstrates for the first time that polarity reversal, or “umpolung”, of a small cycloalkyne is a reliable strategy to make fleeting strained intermediates amenable to strong bond activation processes they are normally not competent for. Thus, a metal–cycloalkyne complex can engage in C–F bond functionalisation of fluoropyridines, contrasting with the chemistry of acyclic alkyne complexes of group 4 metallocenes.16d,e We are convinced that this reactivity should be reachable for other metal–aryne or -cycloalkyne complexes,3 such as Ni or Pd ones, showing reactivity that is reminiscent of strained alkyne “umpolung”.4b–e The degree of fluorination of the substrate drastically changes the outcome; kinetic and thermodynamic reasons may be invoked to explain why the desired C–F functionalisation was not observed for Py-F4 and Py-F5. Our results may extend to Zr–aryne complexes giving a likely direction for our future work.Data availability
Raw NMR and IR data are freely available from the Recherche Data Gouv database (https://doi.org/10.57745/VGHSZN). Results of data treatment (NMR, crystallographic details, DFT) as well as atomic coordinates for computed species have been compiled in a .pdf file freely available as part of the ESI.‡ X-ray diffraction data have been deposited on the Cambridge Crystallographic Data Centre (CCDC) under the deposition number 2339708.Author contributions
S. B. ran the experiments, analysed the data and compiled the ESI. J. M. L. performed DFT calculations, analysed the theoretical results and wrote the computational part of the manuscript. T. F. N. T. recorded single-crystal X-ray diffraction data and solved the structures. C. L., J. M. L., A. S. and J. M. S. obtained funding for the project, managed it and took part in the analysis of the data. A. S. wrote the first draft of the manuscript. The manuscript and ESI were then further revised through contributions of all authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 860322. We are grateful to the CNRS (Centre National de la Recherche Scientifique) and the University of York for providing access to facilities. JML is supported by an Industry Fellowship from the Royal Society (INF\R1\221057). We are grateful to Professor Robin Perutz and Professor Michel Etienne for very helpful and insightful discussions.Notes and references
- (a) K. Kamikawa, Nat. Rev. Chem., 2023, 7, 496 CrossRef PubMed; (b) S. M. Anthony, L. G. Wonilowicz, M. S. McVeigh and N. K. Garg, JACS Au, 2021, 1, 897 CrossRef CAS PubMed; (c) J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892 CrossRef CAS; (d) M. Zanda, R. Bucci, N. L. Sloan and L. Topping, Eur. J. Org. Chem., 2020, 5278 CrossRef CAS; (e) R. A. Dhokale and S. B. Mhaske, Synthesis, 2018, 50, 1 CrossRef CAS; (f) H. Takikawa, A. Nishii, T. Sakai and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030 RSC; (g) M. Feng and X. Jiang, Synthesis, 2017, 49, 4414 CAS; (h) A. E. Goetz and N. K. Garg, J. Org. Chem., 2014, 79, 846 CrossRef CAS; (i) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (j) A. M. Dyke, A. J. Hester and G. C. Lloyd-Jones, Synthesis, 2006, 4093 CAS.
- N. G. Rondan, L. N. Domelsmith, K. N. Houk, A. T. Bowne and R. H. Levin, Tetrahedron Lett., 1979, 20, 3237 CrossRef.
- (a) M. A. Bennett, Aust. J. Chem., 2010, 63, 1066 CrossRef CAS; (b) W. M. Jones and J. Klosin, in Adv. Organomet. Chem., ed. F. G. A. Stone and R. West, Academic Press, 1998, pp. 147–221 Search PubMed; (c) S. L. Buchwald and R. B. Nielsen, Chem. Rev., 1988, 88, 1047 CrossRef CAS; (d) M. A. Bennett and H. P. Schwemlein, Angew Chem., Int. Ed. Engl., 1989, 28, 1296 CrossRef.
- A review on ligand “umpolung”: (a) R. H. Morris, Chem. Soc. Rev., 2024, 53, 2808 RSC , for applications of this strategy to the reaction of metal–aryne complexes with organic (pseudo)halides, see: ; (b) Y. Sumida, T. Sumida, D. Hashizume and T. Hosoya, Org. Lett., 2016, 18, 5600 CrossRef CAS PubMed; (c) M. Hatakeyama, Y. Sakamoto, K. Ogata, Y. Sumida, T. Sumida, T. Hosoya and S. Nakamura, Phys. Chem. Chem. Phys., 2017, 19, 26926 RSC; (d) B. N. Denman, E. E. Plasek and C. C. Roberts, Organometallics, 2023, 42, 859 CrossRef CAS; (e) A. Umanzor, N. A. Garcia and C. C. Roberts, ACS Org. Inorg. Au, 2024, 4, 97 CrossRef CAS.
- Selected examples: (a) H. van der Heijden and B. Hessen, J. Chem. Soc. Chem. Commun., 1995, 145 RSC; (b) E. Tran and P. Legzdins, J. Am. Chem. Soc., 1997, 119, 5071 CrossRef CAS; (c) H. M. Hoyt, F. E. Michael and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 1018–1019 CrossRef CAS PubMed; (d) B. C. Bailey, H. Fan, E. W. Baum, J. C. Huffman, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2005, 127, 16016 CrossRef CAS PubMed; (e) B. C. Bailey, H. Fan, J. C. Huffman, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2007, 129, 8781 CrossRef CAS PubMed; (f) B. C. Bailey, J. C. Huffman and D. J. Mindiola, J. Am. Chem. Soc., 2007, 129, 5302 CrossRef CAS; (g) J. G. Andino, U. J. Kilgore, M. Pink, A. Ozarowski, J. Krzystek, J. Telser, M.-H. Baik and D. J. Mindiola, Chem. Sci., 2010, 1, 351 RSC; (h) S. Zhang, M. Tamm and K. Nomura, Organometallics, 2011, 30, 2712 CrossRef CAS . A review on C–H activation by metal-imido bonds: ; (i) P. T. Wolczanski, Organometallics, 2018, 37, 505 CrossRef CAS.
- (a) A. R. Fout, J. Scott, D. L. Miller, B. C. Bailey, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 331 CrossRef CAS; (b) J. G. Andino, H. Fan, A. R. Fout, B. C. Bailey, M.-H. Baik and D. J. Mindiola, J. Organomet. Chem., 2011, 696, 4138 CrossRef CAS; (c) H. Fan, A. R. Fout, B. C. Bailey, M. Pink, M.-H. Baik and D. J. Mindiola, Dalton Trans., 2013, 42, 4163 RSC.
- To the best of our knowledge, this restricts to intra- or inter-molecular CAr–H bond activation by zirconium-benzyne complexes: (a) G. Erker, J. Organomet. Chem., 1977, 134, 189 CrossRef CAS; (b) G. Erker and T. Mühlenbernd, J. Organomet. Chem., 1987, 319, 201 CrossRef CAS; (c) M. Suleiman, F. Taullaj and U. Fekl, Int. J. Quantum Chem., 2021, 121, e26638 CrossRef CAS.
- D. Seebach, Angew Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef.
- (a) C. Zhang, K. Yan, C. Fu, H. Peng, C. J. Hawker and A. K. Whittaker, Chem. Rev., 2022, 122, 167 CrossRef CAS; (b) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed; (c) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633 CrossRef CAS PubMed; (d) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem.–Eur. J., 2019, 25, 11797 CrossRef CAS; (e) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS; (f) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (g) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (h) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- (a) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS; (b) J. Weaver and S. Senaweera, Tetrahedron, 2014, 70, 7413 CrossRef CAS; (c) R. P. Hughes, Eur. J. Inorg. Chem., 2009, 2009, 4591–4606 CrossRef.
- (a) A. Steffen, M. I. Sladek, T. Braun, B. Neumann and H.-G. Stammler, Organometallics, 2005, 24, 4057 CrossRef CAS; (b) D. Breyer, T. Braun and P. Kläring, Organometallics, 2012, 31, 1417 CrossRef CAS; (c) O. He, B. Cula and T. Braun, Helv. Chim. Acta, 2023, 106, e202200156 CrossRef CAS.
- (a) T. Braun, R. N. Perutz and M. I. Sladek, Chem. Commun., 2001, 2254 RSC; (b) T. Braun, J. Izundu, A. Steffen, B. Neumann and H.-G. Stammler, Dalton Trans., 2006, 5118 RSC; (c) D. Breyer, J. Berger, T. Braun and S. Mebs, J. Fluorine Chem., 2012, 143, 263 CrossRef CAS.
- (a) A. D. Sun, K. Leung, A. D. Restivo, N. A. LaBerge, H. Takasaki and J. A. Love, Chem. Eur. J., 2014, 20, 3162–3168 CrossRef CAS; (b) Y. A. Ho, M. Leiendecker, X. Liu, C. Wang, N. Alandini and M. Rueping, Org. Lett., 2018, 20, 5644–5647 CrossRef CAS PubMed; (c) X. Gu, K. Liu, L. Yang, C. Xie, M. Li and J. (Joelle) Wang, Chem. Sci., 2022, 13, 12498–12502 RSC.
- T. Braun, S. Parsons, R. N. Perutz and M. Voith, Organometallics, 1999, 18, 1710 CrossRef CAS.
- (a) R. J. Lindup, T. B. Marder, R. N. Perutz and A. C. Whitwood, Chem. Commun., 2007, 3664 RSC; (b) M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947 CrossRef CAS PubMed; (c) A. L. Raza, J. A. Panetier, M. Teltewskoi, S. A. Macgregor and T. Braun, Organometallics, 2013, 32, 3795 CrossRef CAS.
- Selected examples: (a) B. L. Edelbach, B. M. Kraft and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 10327–10331 CrossRef CAS; (b) B. M. Kraft, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 10973–10979 CrossRef CAS; (c) B. M. Kraft and W. D. Jones, J. Organomet. Chem., 2002, 658, 132–140 CrossRef CAS; (d) U. Jäger-Fiedler, P. Arndt, W. Baumann, A. Spannenberg, V. V. Burlakov and U. Rosenthal, Eur. J. Inorg. Chem., 2005, 2005, 2842–2849 CrossRef; (e) I. M. Piglosiewicz, S. Kraft, R. Beckhaus, D. Haase and W. Saak, Eur. J. Inorg. Chem., 2005, 938–945 CrossRef CAS; (f) R. D. Rieth, W. W. Brennessel and W. D. Jones, Eur. J. Inorg. Chem., 2007, 2839–2847 CrossRef CAS; (g) G. Podolan, D. Lentz and H.-U. Reissig, Angew. Chem., Int. Ed., 2013, 52, 9491–9494 CrossRef CAS PubMed; (h) M. Nechayev, T. L. Gianetti, R. G. Bergman and J. Arnold, Dalton Trans., 2015, 44, 19494–19500 RSC; (i) N. Romero, Q. Dufrois, N. Crespo, A. Pujol, L. Vendier and M. Etienne, Organometallics, 2020, 39, 2245–2256 CrossRef CAS , for a review detailing stoichiometric and catalytic C–F bond activation by lanthanides and early transition metals, see: ; (j) M. Klahn and U. Rosenthal, Organometallics, 2012, 31, 1235–1244 CrossRef CAS.
- (a) U. Jäger-Fiedler, M. Klahn, P. Arndt, W. Baumann, A. Spannenberg, V. V. Burlakov and U. Rosenthal, J. Mol. Catal. A:Chem., 2007, 261, 184–189 CrossRef; (b) S. Yow, S. J. Gates, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2012, 51, 12559–12563 CrossRef CAS PubMed; (c) T. L. Gianetti, R. G. Bergman and J. Arnold, Chem. Sci., 2014, 5, 2517–2524 RSC.
- A review discussing the reactivity of polyfluoro-aromatic and heteroaromatic compounds: G. M. Brooke, J. Fluorine Chem., 1997, 86, 1 CrossRef.
- For recent examples illustrating this difficulty, see: (a) M. Ohashi, R. Doi and S. Ogoshi, Chem.–Eur. J., 2014, 20, 2040 CrossRef CAS PubMed; (b) L. Finck and M. Oestreich, Chem.–Eur. J., 2021, 27, 11061 CrossRef CAS PubMed.
- S. L. Buchwald, R. T. Lum and J. C. Dewan, J. Am. Chem. Soc., 1986, 108, 7441 CrossRef CAS.
- (a) P. Binger, F. Sandmeyer and C. Krueger, Organometallics, 1995, 14, 2969 CrossRef CAS; (b) P. Arndt, C. Lefeber, R. Kempe and U. Rosenthal, Chem. Ber., 1996, 129, 207 CrossRef CAS; (c) J. Zhao, S. Zhang, W.-X. Zhang and Z. Xi, Organometallics, 2011, 30, 3464 CrossRef CAS; (d) J. Zhao, S. Zhang, W.-X. Zhang and Z. Xi, Organometallics, 2012, 31, 8370 CrossRef CAS; (e) G. Bender, G. Kehr, C. G. Daniliuc, B. Wibbeling and G. Erker, Dalton Trans., 2013, 42, 14673 RSC; (f) V. V. Burlakov, L. Becker, V. S. Bogdanov, M. V. Andreev, P. Arndt, A. Spannenberg, W. Baumann and U. Rosenthal, Eur. J. Inorg. Chem., 2014, 2014, 5304–5310 CrossRef CAS.
- L. Becker, F. Strehler, M. Korb, P. Arndt, A. Spannenberg, W. Baumann, H. Lang and U. Rosenthal, Chem.–Eur. J., 2014, 20, 3061 CrossRef CAS.
- (a) A. J. Downs and R. Schmutzler, Spectrochim. Acta, Part A, 1965, 21, 1927–1939 CrossRef CAS; (b) A. J. Downs and R. Schmutzler, Spectrochim. Acta, Part A, 1967, 23, 681–701 CrossRef CAS; (c) J. A. Gibson, G.-V. Röschenthaler and R. Schmutzler, J. Chem. Soc., Dalton Trans., 1975, 918–924 RSC; (d) F. Seel and H.-J. Bassler, Z. Anorg. Allg. Chem., 1975, 418, 263–272 CrossRef CAS.
- (a) S. Bonfante, C. Lorber, J. M. Lynam, A. Simonneau and J. M. Slattery, J. Am. Chem. Soc., 2024, 146, 2005 CrossRef CAS; (b) K. Chulsky, I. Malahov, D. Bawari and R. Dobrovetsky, J. Am. Chem. Soc., 2023, 145, 3786 CrossRef CAS; (c) J. Zhang, X. Zhao, J.-D. Yang and J.-P. Cheng, J. Org. Chem., 2022, 87, 294 CrossRef CAS PubMed; (d) J. Zhang, J.-D. Yang and J.-P. Cheng, Nat. Commun., 2021, 12, 2835 CrossRef CAS PubMed; (e) Y. Pang, M. Leutzsch, N. Nöthling, F. Katzenburg and J. Cornella, J. Am. Chem. Soc., 2021, 143, 12487 CrossRef CAS PubMed; (f) S. Lim and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 16188 CrossRef CAS PubMed; (g) A. A. Facundo, A. Arévalo, G. Fundora-Galano, M. Flores-Álamo, E. Orgaz and J. J. García, New J. Chem., 2019, 43, 6897 RSC; (h) S. S. Chitnis, F. Krischer and D. W. Stephan, Chem.–Eur. J., 2018, 24, 6543 CrossRef CAS PubMed; (i) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS.
- Examples of SNAr by trivalent phosphorus compound on pentafluoropyridine: (a) L. N. Markovskii, G. G. Furin, Yu. G. Shermolovich and G. G. Yakobson, Zh. Obshch. Khim., 1979, 49, 531 CAS; (b) L. N. Markovskii, G. G. Furin, Y. G. Shermolovich and G. G. Yakobson, Izv. Akad. Nauk SSSR, Ser. Khim., 1981, 867 CAS; (c) N. N. Demik, M. M. Kabachnik, Z. S. Novikova and I. P. Beletskaya, Izv. Akad. Nauk SSSR, Ser. Khim., 1992, 2432 CAS; (d) Yu. A. Veits, N. B. Karlstedt, A. V. Chuchuryukin and I. P. Beletskaya, Russ. J. Org. Chem., 2000, 36, 750–756 CAS; (e) L. I. Goryunov, J. Grobe, V. D. Shteingarts, B. Krebs, A. Lindemann, E.-U. Würthwein and C. Mück-Lichtenfeld, Chem.–Eur. J., 2000, 6, 4612 CrossRef CAS PubMed; (f) R. M. Bellabarba, M. Nieuwenhuyzen and G. C. Saunders, Organometallics, 2003, 22, 1802 CrossRef CAS; (g) I. P. Beletskaya, E. G. Neganova and Y. A. Veits, Russ. J. Org. Chem., 2004, 40, 1782 CrossRef CAS; (h) L. I. Goryunov, J. Grobe and V. D. Shteingarts, Collect. Czech. Chem. Commun., 2008, 73, 1612 CrossRef CAS; (i) B. Van Nguyen and D. J. Burton, J. Fluorine Chem., 2012, 135, 144 CrossRef; (j) S. I. Zhivetyeva, L. I. Goryunov, I. Y. Bagryanskaya, J. Grobe, V. D. Shteingarts and E.-U. Würthwein, J. Fluorine Chem., 2014, 164, 58 CrossRef CAS.
- J. Vicente, M. T. Chicote, J. Fernandez-Baeza, A. Fernandez-Baeza and P. G. Jones, J. Am. Chem. Soc., 1993, 115, 794–796 CrossRef CAS.
- E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2009, 131, 7817 CrossRef CAS PubMed.
- (a) O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed; (b) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Michel Etienne and Prof. Robin Perutz. |
| ‡ Electronic supplementary information (ESI) available: General methods, experimental procedures, NMR spectra, crystallographic methods, details of computational methods, collated energies and xyz coordinates, and CIF file. CCDC 2339708. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08522a |
| This journal is © The Royal Society of Chemistry 2025 |