
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInnovative synergistic control of electric fields and Zn2+ dynamics for revolutionizing zinc metal battery stability†
Shengqiang
Zhang
a,
Chengxin
Liu
b,
Yangyang
Wang
b,
Ao
Xu
b,
Chunxia
Chen
b and
Xiaojie
Liu
 *b
*b
aCollege of Chemistry & Chemical Engineering, Yan’an University, Yan’an, Shaanxi 716000, P. R. China
bKey Laboratory of Synthetic and Natural Functional Molecule of the Ministry of Education, College of Chemistry & Materials Science, Northwest University, Xi'an 710127, P. R. China. E-mail: xiaojie.liu@nwu.edu.cn
First published on 3rd March 2025
Abstract
Aqueous zinc-ion batteries (ZIBs) are emerging as promising next-generation energy storage systems due to their inherent safety, environmental sustainability, and cost-effectiveness. However, their widespread application is hindered by challenges such as dendritic Zn growth, hydrogen evolution, and corrosion-induced passivation, which compromise performance and scalability. To overcome these obstacles, we developed a novel dual-interface modified zinc anode by integrating a zinc fluoride (ZnF2)-silicon (Si) interface using fluorine-doped silicon nanoparticles encapsulated within hollow mesoporous carbon nanospheres (F-Si@HMCS). The in situ formation of a ZnF2 layer provides high electrochemical stability, effectively suppressing dendrite formation, mitigating zinc corrosion, and reducing side reactions with the electrolyte. The silica layer further facilitates uniform Zn electrodeposition by forming Si–O–Zn bonds, which regulate electric field distribution and lower nucleation energy barriers. Additionally, the hollow mesoporous carbon structure facilitates efficient ion transport and acts as a buffer against volume changes during cycling. Consequently, the F-Si@HMCS@Zn electrode exhibits a long lifespan of over 2500 h at 5 mA cm−2 with a capacity of 0.5 mA h cm−2 in a symmetrical cell test. When coupled with α-MnO2 cathodes, the resulting ZIBs exhibit outstanding stable cycle life over 2000 cycles at 2 A g−1.
Introduction
Aqueous batteries are emerging as a safer, more cost-effective, and environmentally friendly alternative to nonaqueous batteries, which face safety risks and the challenges of toxic organic electrolytes. Aqueous electrolytes offer several benefits, including enhanced safety, reduced cost, environmental sustainability, and high ionic conductivity. Unlike nonaqueous batteries, aqueous batteries are more adaptable and easier to handle under manufacturing conditions, positioning them as promising candidates for energy storage solutions.1 Notably, rechargeable aqueous zinc-ion batteries (AZIBs) are regarded as one of the most promising candidates for next-generation energy storage systems (EESs) due to their inherent safety, affordability, and high theoretical capacity (820 mA h g−1 or 5854 mA h cm−2).2–4 However, large-scale application of AZIBs faces several critical challenges. Firstly, the thermodynamic instability between zinc and water leads to significant interfacial side reactions during the zinc stripping/plating process, such as the hydrogen evolution reaction (HER)5,6 and zinc corrosion reaction (ZCR).7 Additionally, the uneven surface resulting from the “tip effect” accelerates the growth of zinc dendrites. The continued growth of these dendrites can puncture the separator, ultimately causing a short circuit in the battery.8,9 Furthermore, the excessive accumulation of zinc dendrites forms dead zinc particles, reducing the utilization of the Zn anode, significantly lowering coulombic efficiency, and accelerating battery failure.10 To overcome these challenges, researchers have focused on modifying the zinc anode through various strategies aimed at suppressing dendrite formation, improving electrochemical stability, and enhancing overall performance.11–15 Among these approaches, interface engineering has proven to be an effective strategy. By introducing interfacial protective layers of the Zn anode, it is possible to regulate the electrochemical behavior, reduce side reactions, and improve the uniformity of zinc deposition.16,17An ideal interfacial protective layer should possess high ionic conductivity and electronic insulation to isolate the Zn anode from direct contact with the electrolyte.18 According to previous reports, carbon materials such as CNTs, graphene, MXene and hollow carbon nanospheres have garnered significant attention due to their ability to provide uniform electric field distribution on the Zn surface and promote fast Zn2+ diffusion kinetics.19–23 Especially, the hollow mesoporous carbon nanospheres (HMCS) possess several advantageous properties, including low density, porous shells, accessible interior space, high surface area, and large pore volume, compared to microporous or nonporous materials.24 The high surface area and porous structure enhance the electronic conductivity of the Zn anode, thereby improving battery efficiency, particularly during high-rate cycling. However, carbonaceous materials remain suboptimal as hosts for Zn due to their insufficient interaction with Zn and high catalytic activity toward the HER.25 Therefore, integrating zincophilic sites into carbonaceous materials is essential to inhibit the formation of Zn dendrites effectively. Recently, various studies have focused on regulating the electrochemical reduction of Zn2+ by integrating zincophilic sites (e.g., Si, Sn, Cu, In, and Sb) into conductive hosts.26–30 This approach helps to homogenize the electric field distribution and provide more nucleation sites, thereby improving the uniformity of zinc deposition. However, it cannot fully eliminate interfacial side reactions due to the direct contact between the post-deposited Zn and the electrolyte. Besides, inorganic zincophilic sites, including CaCO3, ZnO, and ZnF2, have also been explored. In particular, ZnF2 has been widely used as the interface protection layer due to its high Zn2+ conductivity and mechanical strength, which enhances the Zn2+ ion flux and effectively inhibits the growth of dendrites.31,32 For instance, Yang et al. designed a 3D interconnected ZnF2 matrix on Zn foil (Zn@ZnF2), serving as a multifunctional protective layer that redistributes Zn2+ ion flux and reduces desolvation activation energy.33 Nevertheless, these ZnF2 protective layers are typically constructed ex situ, making them susceptible to cracking and detachment from the Zn surface during volume changes in cycling.34 In addition, ex situ ZnF2 layers tend to allow zinc to deposit on their surface, failing to provide continuous protection against side reactions with the electrolyte.35 In contrast, the in situ construction of ZnF2-based interfacial protective layers presents a more practical strategy to address these issues. However, achieving significant improvement in AZIB performance via single interface regulation of ZnF2 remains challenging. To overcome this limitation, double interface regulation is emerging as a more effective strategy. Silicon-based materials have gained attention in solar cells, lithium-ion batteries, and semiconductors due to their high bandgap energy and specific surface area, but there has been little research on AZIBs.36 Moreover, silicon-based materials exhibit hydrophobic properties, which can inhibit the redox reactions of H2O at 0–2 V, thereby enhancing CE and cycle stability.37 Therefore, the development of innovative strategies for the seamless integration of ZnF2 and Si-based dual-interface layers on the zinc anode is imperative to mitigate interfacial side reactions and suppress zinc dendrite growth.
Based on the above considerations, we proposed and designed fluorine (F)-doped silicon nanoparticles encapsulated within hollow mesoporous carbon nanospheres (F-Si@HMCS) as double interface layers to enable uniform nucleation and dendrite-free zinc metal anodes (ZMAs). The in situ formation of a ZnF2 layer provides excellent electrochemical stability and suppresses dendrite growth, while the silica layer further facilitates uniform zinc electrodeposition by forming Si–O–Zn bonds that regulate the electric field distribution. The hollow mesoporous carbon structure not only aids in ion transport but also provides mechanical buffering, reducing the stress caused by volume expansion during cycling (Fig. 1a). The combined effects of these modifications significantly improve the cycling stability and performance of the zinc anode, endowing ZIBs with superior cycle life, enhanced rate capability, and high capacity retention. The modified zinc anode demonstrates a remarkable lifespan of over 2500 hours with stable performance in symmetrical cell tests, and when paired with α-MnO2 cathodes, the ZIBs exhibit outstanding cycle stability over 2000 cycles at high current densities. Our work presents a promising approach for enhancing the performance of aqueous ZIBs, paving the way for their practical application in energy storage systems, electric vehicles, and other large-scale applications.
 | ||
| Fig. 1 (a) The synthesis process of F-Si@HMCS and schematic illustrations of Zn metal plating behaviors on bare Zn and F-Si@HMCS@Zn electrodes. (b and c) SEM images; (d) TEM image; (e) HRTEM image; (f) the corresponding energy-dispersive X-ray mapping of F-Si@HMCS. (g) XRD pattern; (h) Raman spectrum of F-Si@HMCS. XPS spectrum of F-Si@HMCS: (i) C 1s. (j) FTIR spectrum of F-Si@HMCS. | ||
Results and discussion
The F-Si@HMCS composite was synthesized using a conventional method (Fig. 1a), as detailed in the experimental section. Silicon powder was partially oxidized to form Si@SiO2 (Fig. S1†). Resorcinol (R) and formaldehyde (F) were then used for RF polycondensation, coating the Si@SiO2 surface to create a shell. This intermediate, Si@SiO2@RF, was carbonized in an argon atmosphere to produce Si@SiO2@C. Afterwards, the RF resin was carbonized and SiO2 was completely etched by HF to obtained Si@HMCS. Finally, fluorination was performed via NH4F pyrolysis, where NH4F decomposes at 300 °C, releasing fluorine atoms to form F-Si@HMCS, enabling efficient fluorine incorporation and enhancing the material's properties. The structure and morphology of F-Si@HMCS were characterized by SEM and TEM. SEM images (Fig. 1b and c) reveal a core–shell structure with silicon nanoparticles as the core and carbon as the shell, confirmed by TEM (Fig. 1d). HRTEM (Fig. 1e) shows a lattice spacing of 0.31 nm, corresponding to the (111) plane of silicon. EDS mapping indicates a uniform distribution of C, F, O, and Si, while comparative TEM and EDS images (Fig. S2†) show that fluorine doping does not alter morphology. XRD confirmed the crystalline structure, with peaks at 28.3°, 47.3°, and 56.2°, matching the (111), (220), and (311) planes of silicon (JCPDS No.27-1402) (Fig. 1g).38 Raman spectra (Fig. 1h) showed two peaks at 506 and 920.5 cm−1 related to silicon and Si–O–Si, respectively,39 and an ID/IG ratio of 0.95 for F-Si@HMCS compared to 0.79 for Si@HMCS (Fig. S3†), indicating higher defect density in F-Si@HMCS. XPS analysis revealed bonding states in F-Si@HMCS. The C 1s spectrum (Fig. 1i) showed peaks for C–C, C–O, and C–F bonds. The Si 2p spectrum displays peaks at 100.5 and 101.6 eV, attributed to Si0 and Si4+, respectively.40 The Si4+ signal, associated with SiO2, results from surface oxidation by atmospheric oxygen and water in the air, further verified by Fourier-transform infrared (FTIR) spectroscopy. FTIR spectra (Fig. 1j) exhibit characteristic peaks at 1068.6 and 980.6 cm−1, corresponding to Si–O–Si and Si–OH bonds, respectively. The SiO2 generated by the oxidation of the silicon surface contributes to the formation of a uniform network of Zn2+ transport channels. Additionally, the SiO2 provides extra mechanical strength, which helps resist the growth of Zn dendrites, ultimately preventing the puncture of the separator.41 Nitrogen adsorption–desorption isotherms analyzed by the BJH method showed specific surface areas of 173 and 164 m2 g−1 for F-Si@HMCS and Si@HMCS, respectively (Fig. S4a and b†). Both materials exhibited mesoporous structures (Fig. S4c and d†), enabling efficient electrolyte ion transport.Electrochemical characteristics
The electrochemical performance of the Zn anode is significantly enhanced by the F-Si@HMCS protective layer, which inhibits Zn directional growth, reduces side reactions, and accelerates ion transport. Coulombic efficiency (CE), a critical indicator of zinc-ion battery (AZIB) performance, reflects energy utilization and stability during charge–discharge cycles. To investigate Zn2+ behavior during deposition/stripping, asymmetric cells (bare Zn//Cu, Si@HMCS@Zn//Cu, and F-Si@HMCS@Zn//Cu) were tested. As shown in Fig. 2a, F-Si@HMCS@Zn//Cu achieved an ultra-high average CE of ≈99.9% over 800 cycles at 2 mA cm−2, outperforming bare Zn//Cu and Si@HMCS@Zn//Cu, demonstrating that fluorine ions effectively stabilize the deposition/stripping process by reducing interfacial side reactions. Further analysis of plating/stripping voltage curves (Fig. 2b–d) revealed that the polarization potential of F-Si@HMCS@Zn//Cu stabilized at 16 mV after 250 cycles, compared to 26 mV for bare Zn//Cu and 20 mV for Si@HMCS@Zn//Cu. Obviously, the observed phenomenon is that the polarization voltage of the bare Zn//Cu battery is smaller than that of the Si@HMCS@Zn//Cu battery in the first 50 cycles. Unfortunately, the voltage of the bare Zn//Cu battery fluctuates sharply after only 150 cycles, probably owing to irreversible by-product formation and causing the short-circuit of the battery. This phenomenon is consistent with the results reported in previous literature42–46 This demonstrates the ability of the F-Si@HMCS layer to suppress Zn directional growth, minimize side reactions, and enhance ion transport. Additionally, nucleation overpotential measurements at 5 mA cm−2 (Fig. 2e–g) showed that F-Si@HMCS@Zn exhibited a lower nucleation overpotential compared to bare Zn and Si@HMCS@Zn, indicating enhanced Zn2+ desolvation and nucleation. The hollow mesoporous structure of the F-Si@HMCS layer further prevents direct contact between the electrolyte and electrode, promoting uniform Zn2+ deposition. The rate performance (Fig. 2h) demonstrated that the F-Si@HMCS@Zn symmetric cell maintained a lower hysteresis and stable voltage plateau across current densities ranging from 1 to 5 mA cm−2. When the current density increased to 5 mA cm−2, the polarization of bare Zn rose to 287 mV due to sluggish ion transfer, while F-Si@HMCS@Zn exhibited only 124 mV. Long-term plating/stripping cycling at 5 mA cm−2 with a fixed capacity of 0.5 mA h cm−2 (Fig. 2i) further confirmed the superior stability of F-Si@HMCS@Zn, which showed minimal voltage fluctuations, unlike bare Zn and Si@HMCS@Zn, which lost voltage characteristics after 83.8 and 126.5 hours, respectively. These improvements are attributed to the interaction between Si nanoparticles with hydroxyl groups and Zn2+ ions, forming a Si–O–Zn cross-linked structure that regulates interfacial charge distribution and reduces the nucleation barrier of Zn deposition. However, the weaker driving force of the Si@HMCS layer limits Zn2+ ion diffusion. F doping enables the in situ formation of ZnF2, which enhances interfacial bonding and Zn2+ ion flux, effectively inhibiting zinc dendrite growth and promoting electrochemical performance. | ||
| Fig. 2 (a) Coulombic efficiency of the bare Zn electrode, Si@HMCS@Zn electrode, and F-Si@HMCS@Zn electrode for stripping/deposition on copper foil at a current density of 2 mA cm−2. Charge–discharge curves of (b) bare Zn//Cu; (c) Si@HMCS@Zn//Cu asymmetric cell; (d) F-Si@HMCS@Zn//Cu asymmetric cells under constant current cycling at different cycle numbers. Stripping/deposition curves of (e) bare Zn, (f) Si@HMCS@Zn, and (g) F-Si@HMCS@Zn symmetric cells. (h) Rate performance of bare Zn, Si@HMCS@Zn, and F-Si@HMCS@Zn symmetric cells within the current range of 0.5 to 5 mA cm−2. (i) Long-term cycling performance of bare Zn, Si@HMCS@Zn, and F-Si@HMCS@Zn symmetric cells at a current density of 5 mA cm−2. | ||
The transference number of Zn2+ (tZn2+) was measured using electrochemical impedance spectroscopy (EIS) and calculated via the Bruce–Vincent method to evaluate Zn2+ diffusion under the F-Si@HMCS protective layer. The calculation formula is:47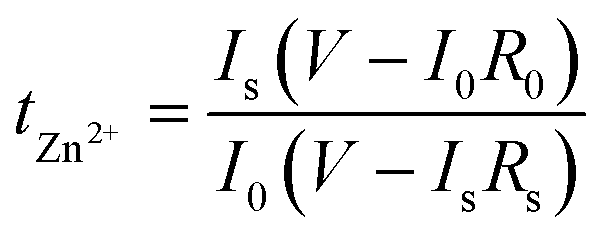 , where I0, R0, Is, Rs, and V represent the initial current, charge transfer resistance before polarization, steady-state current, charge transfer resistance after polarization, and the applied potential (15 mV), respectively. As shown in Fig. 3a–c, tZn2+ for the F-Si@HMCS@Zn symmetric cell reached 0.84, significantly higher than the 0.63 for Si@HMCS@Zn and 0.48 for bare Zn, demonstrating that the F-Si@HMCS layer greatly enhances Zn2+ transport and facilitates uniform Zn plating/stripping. EIS curves before and after polarization (Fig. 3d–f) indicate increased impedance for all cells due to side reactions and surface accumulation under applied potential, resulting in higher internal resistance. Further analysis using the Distribution of Relaxation Times (DRT) method provided detailed insights into charge transfer impedance (Rct), as shown in Fig. 3g–i. The DRT method employs regularization to process EIS data, calculating the distribution function g (τ) for characteristic times (τ) and subsequently, based on g(τ) analysis, revealing the internal dynamics of the battery. After polarization, the Rct of the F-Si@HMCS@Zn symmetric cell was 142.9 Ω, compared to 228 Ω for Si@HMCS@Zn and 937 Ω for bare Zn. These results confirm that the F-Si@HMCS modification layer significantly mitigates side reactions between the Zn anode and electrolyte, accelerates Zn2+ transport, and ensures stable electrochemical performance.
, where I0, R0, Is, Rs, and V represent the initial current, charge transfer resistance before polarization, steady-state current, charge transfer resistance after polarization, and the applied potential (15 mV), respectively. As shown in Fig. 3a–c, tZn2+ for the F-Si@HMCS@Zn symmetric cell reached 0.84, significantly higher than the 0.63 for Si@HMCS@Zn and 0.48 for bare Zn, demonstrating that the F-Si@HMCS layer greatly enhances Zn2+ transport and facilitates uniform Zn plating/stripping. EIS curves before and after polarization (Fig. 3d–f) indicate increased impedance for all cells due to side reactions and surface accumulation under applied potential, resulting in higher internal resistance. Further analysis using the Distribution of Relaxation Times (DRT) method provided detailed insights into charge transfer impedance (Rct), as shown in Fig. 3g–i. The DRT method employs regularization to process EIS data, calculating the distribution function g (τ) for characteristic times (τ) and subsequently, based on g(τ) analysis, revealing the internal dynamics of the battery. After polarization, the Rct of the F-Si@HMCS@Zn symmetric cell was 142.9 Ω, compared to 228 Ω for Si@HMCS@Zn and 937 Ω for bare Zn. These results confirm that the F-Si@HMCS modification layer significantly mitigates side reactions between the Zn anode and electrolyte, accelerates Zn2+ transport, and ensures stable electrochemical performance.
 | ||
| Fig. 3 (a–c) Current-time plot of assembled symmetric cells of bare Zn, Si@HMCS@Zn, and F-Si@HMCSC@Zn after constant potential polarization at 15 mV. (d–i) EIS curves and DRT analysis before and after polarization for (d and g) F-Si@HMCS@Zn, (e and h) Si@HMCS@Zn, and (f and i) bare Zn. | ||
The corrosion behavior of zinc anodes was investigated by immersing bare Zn and F-Si@HMCS@Zn electrodes in 2 M ZnSO4 solution for 10 days. As shown in Fig. 4a and b, the surface of bare Zn became rough after immersion, indicating severe interfacial side reactions with ZnSO4. In contrast, the F-Si@HMCS@Zn electrode retained its morphology, demonstrating excellent resistance to interfacial reactions. XRD analysis (Fig. 4c and d) further supports these observations: the bare Zn electrode exhibited characteristic peaks at 8.3° and 16.3°, corresponding to Zn4SO4(OH)6·5H2O (JCPDS No. 39-0688),48 a typical corrosion byproduct. However, no such peaks were observed for the F-Si@HMCS@Zn electrode, confirming its superior corrosion resistance. To quantify the anti-corrosion performance, linear polarization measurements were conducted (Fig. 4e). The F-Si@HMCS@Zn electrode exhibited the lowest corrosion current density (Icorr, 1.71 mA cm−2) compared to Si@HMCS@Zn (1.91 mA cm−2) and bare Zn (2.21 mA cm−2), highlighting the enhanced corrosion resistance provided by the F-Si@HMCS layer. Additionally, linear sweep voltammetry (LSV) in 1 M Na2SO4 solution (Fig. 4f) revealed that the F-Si@HMCS@Zn electrode had the highest overpotential, indicating the hydrophobic effect of fluorine in the protective layer. This hydrophobicity inhibits water molecule decomposition on the electrode surface, preventing the formation of basic zinc sulfate caused by local pH changes, and thereby extending battery lifespan. These results demonstrate that the F-Si@HMCS layer effectively mitigates corrosion and hydrogen evolution, ensuring prolonged stability of the zinc anode. Controlling Zn electrodeposition morphology is crucial for mitigating dendrite growth and corrosion during repeated cycling. The morphologies of bare Zn and F-Si@HMCS@Zn electrodes were analyzed using ex situ SEM after 10, 25, and 50 electroplating/stripping cycles at a current density of 1 mA cm−2 and a cutoff capacity of 1 mA h cm−2. After just 10 cycles, bare Zn showed significant moss-like deposits and the formation of “dead zinc” products (Fig. 4g), resulting from uncontrolled dendrite growth and severe electrolyte corrosion. These issues arise from high localized current densities, uneven Zn2+ diffusion, and subsequent exacerbation of dendrite growth. In contrast, the F-Si@HMCS@Zn electrode maintained a dense, dendrite-free surface with no significant structural changes even after 50 cycles (Fig. 4h). The uniform Zn2+ transport channel enabled by the F-Si@HMCS layer ensures orderly deposition. Additionally, XRD patterns (Fig. 4i and j) of cycled F-Si@HMCS@Zn show no detectable Zn4SO4(OH)6·5H2O peaks, confirming the excellent anti-passivation performance of the protective layer. XPS further revealed the role of the F-Si@HMCS layer in stabilizing Zn deposition. After 50 cycles, the O 1s spectrum (Fig. 4k) showed a peak at 529.1 eV, indicating the formation of Si–O–Zn bonds.49 The F 1s spectrum (Fig. 4f) displayed a ZnF2 peak, demonstrating in situ formation of ZnF2 during cycling. The Si 2p spectrum (Fig. 4m) further confirmed the presence of Si–O–Zn cross-linked structures, formed via interactions between S–OH groups and Zn2+ ions during plating. These results were further supported by XRD and FTIR measurements. As shown in Fig. S5a,† a new diffraction peak located at 28.6° was detected in the F-Si@HMCS@Zn electrode, corresponding to the (111) planes of tetragonal ZnF2 (JCPDS No. 21-1480). This confirms the successful formation of the ZnF2 composite. Additionally, FTIR spectra of the F-Si@HMCS@Zn electrode were collected to verify the existence of Si–O–Zn bonds during the deposition process. In Fig. S5b,† the F-Si@HMCS@Zn electrode exhibits a characteristic peak at 963.8 cm−1, representing the Si–O–Zn bond.50 These findings highlight the critical role of the F-Si@HMCS layer in ensuring stable, efficient, and dendrite-free zinc deposition, significantly enhancing the electrochemical performance of Zn anodes.
 | ||
| Fig. 4 SEM images of (a) bare Zn and (b) F-Si@HMCS@Zn electrodes after immersion in 2 M ZnSO4 electrolyte for 10 days. (c and d) XRD patterns of different electrodes after immersion. (e) Corrosion; (f) HER curves of different electrodes. SEM images of (g) bare Zn and (h) F-Si@HMCS@Zn electrodes in 2 M ZnSO4 + 0.1 M MnSO4 electrolyte after different cycles at 1 mA cm−2 with a capacity of 1 mA h cm−2. (i and j) The corresponding XRD analysis of the bare Zn and F-Si@HMCS@Zn anodes after different cycles. (k) High-resolution O 1s XPS spectra. (l) High-resolution O 1s XPS spectra. (m) High-resolution Zn 2p XPS spectra. | ||
To visually monitor the dynamic deposition behavior of Zn, in situ optical microscopy was employed. For the bare Zn electrode, non-uniform granular Zn deposits appeared within 10 minutes of deposition (Fig. 5a). With continued electrodeposition, a significant accumulation of granular zinc dendrites was observed, leading to an increasingly rough surface. This phenomenon is attributed to Zn2+ desolvation in the electrolyte, where active water molecules near the electrode undergo hydrogen evolution, raising the local pH. This results in surface corrosion and the formation of basic zinc sulfate, which passivates the anode and promotes uneven Zn deposition and dendrite growth. Over time, these dendrites risk puncturing the separator, potentially causing short circuits. In contrast, the F-Si@HMCS@Zn electrode exhibited uniform and smooth Zn deposition throughout the process, with no dendrite formation (Fig. 5b). Numerical simulations of the electric field intensity further revealed significant differences between the electrodes. The bare Zn anode displayed an uneven electric field distribution, leading to irregular protrusions and intensified electric fields at the tips, which promote bulky dendrite growth (Fig. 5c). Conversely, the F-Si@HMCS@Zn electrode, with its electronic shielding effect, redistributed and homogenized interface charges and Zn2+ flux, effectively suppressing irregular deposition and ensuring smooth, dendrite-free Zn plating (Fig. 5d).51 The Rct of F-Si@HMCS@Zn symmetric batteries was also evaluated at various temperatures (30–70 °C) using EIS (Fig. 5e1). DRT analysis of EIS curves (Fig. 5e2 and e3) and in situ EIS spectra (Fig. 5f and g) provided further insights into interfacial dynamics.52 The superior deposition kinetics of F-Si@HMCS@Zn are primarily attributed to its hollow mesoporous structure, which enhances ion transport and ensures stable, uniform Zn deposition, as verified by the electrode overpotential. In the symmetrical cell (Fig. S6a†), HMCS@Zn exhibits a lower nucleation overpotential (41.1 mV) compared to bare Zn (95.2 mV). The lower nucleation overpotential reflects a reduced nucleation energy barrier for Zn deposition, indicating that the HMCS coating facilitates rapid Zn plating. The symmetrical battery test results indicate that 2 h oxidation time of silicon to silica is optimal (Fig. S6b†).
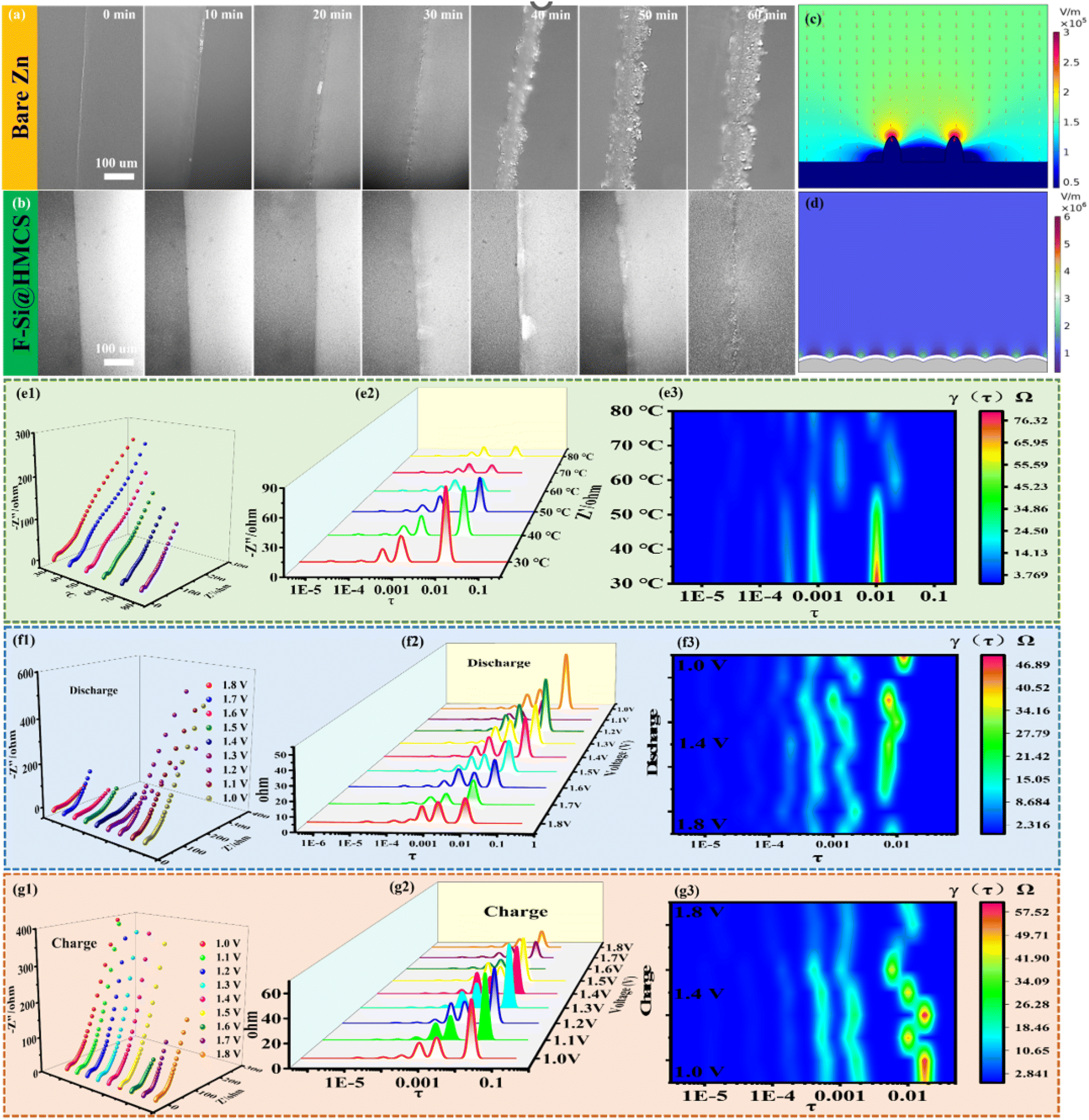 | ||
| Fig. 5 In situ optical observation of different materials at 5 mA cm−2 current density: (a) bare Zn; (b) F-Si@HMCS@Zn electrode. Simulations electric field distributions. (c) on bare Zn and (d) on F-Si@HMCS@Zn electrode. The EIS curves of F-Si@HMCS@Zn symmetrical cells: (e1) at different temperatures and (e2 and e3) a corresponding analysis of the distribution of DRT upon galvanostatic; (f1) discharge process and (f2 and f3) a corresponding analysis of the distribution of DRT upon galvanostatic; (g1) charge process and (g2 and g3) a corresponding analysis of the distribution of DRT upon galvanostatic. | ||
To evaluate the performance of Zn-ion full batteries, F-Si@HMCS@Zn was used as the anode and α-MnO2 as the cathode. Cyclic voltammetry (CV) curves (Fig. 6a) revealed two pairs of well-defined redox peaks in all three cells, corresponding to the co-embedding of Zn2+ and H+ in the cathode.53 The similar CV shapes across bare Zn//MnO2, Si@HMCS@Zn//MnO2, and F-Si@HMCS@Zn//MnO2 suggest that surface modifications of the Zn anode do not significantly alter the electrochemical reactions. However, F-Si@HMCS@Zn//MnO2 exhibited higher reduction and lower oxidation voltages, indicating improved reaction kinetics due to the F-Si@HMCS layer. The charge–discharge curves at 2 A g−1 (Fig. 6b) demonstrate that F-Si@HMCS@Zn//MnO2 achieves the highest reversible capacity among the cells. Additional charge–discharge studies (Fig. 6c and S7†) at 1 A g−1 revealed consistent charge–discharge platforms aligned with the CV peaks. Rate performance testing (Fig. 6d and S8†) further highlighted the superior performance of F-Si@HMCS@Zn//MnO2, achieving specific capacities of 230.9 to 74.3 mA h g−1 as current densities increased from 0.1 to 5.0 A g−1, far exceeding bare Zn//MnO2, which retained only 6.3 mA h g−1 at 5.0 A g−1. These results underscore the role of F in reducing the Zn2+ nucleation overpotential and enhancing interfacial ion transfer kinetics. Long-term cycling tests (Fig. 6e and f) further demonstrated the exceptional stability of F-Si@HMCS@Zn//MnO2. At 1 A g−1, the reversible capacity remained at 182.4 mA h g−1 after 800 cycles, compared to 36.8 and 139.6 mA h g−1 for bare Zn//MnO2 and Si@HMCS@Zn//MnO2, respectively. At 2 A g−1, F-Si@HMCS@Zn//MnO2 maintained 77.4 mA h g−1 with a coulombic efficiency of 99.85% after 2000 cycles, while the capacities of bare Zn//MnO2 and Si@HMCS@Zn//MnO2 rapidly declined to 35.3 and 54.9 mA h g−1, respectively. This highlights the F-Si@HMCS layer's ability to promote stable Zn deposition kinetics and enhance battery performance. The exceptional performance of F-Si@HMCS@Zn is attributed to the in situ formation of ZnF during charging. The strong Zn–F bond enriches active sites, accelerates Zn2+ diffusion, and reduces desolvation energy barriers.54 Furthermore, the mesoporous structure of Si@HMCS facilitates Zn2+ transport and ensures uniform deposition.55 Silicon nanoparticles in the core–shell structure also provide zincophilic sites, enhancing Zn2+ interaction and ionic dynamics.56 Together, these features suppress hydrogen evolution, prevent basic zinc sulfate passivation, and maintain low polarization, ensuring prolonged battery stability and high performance.
 | ||
| Fig. 6 (a) CV curves of bare Zn//MnO2, Si@HMCS@Zn//MnO2, and F-Si@HMCS@Zn//MnO2 at a scan rate of 0.1 mV s−1 (b) Charge–discharge curves of bare Zn//MnO2, Si@HMCS@Zn//MnO2, and F-Si@HMCS@Zn//MnO2. (c) Charge–discharge curves of F-Si@HMCS@Zn//MnO2 after 10, 50, and 100 cycles at 2 A g−1. (d) Rate performance; cycling performance of bare Zn//MnO2, Si@HMCS@Zn//MnO2, and F-Si@HMCS@Zn//MnO2 cells at current densities of (e) 1 A g−1 and (f) 2 A g−1. (g) CV curve of the F-Si@HMCS@Zn//MnO2 battery at different scan rates. (h) Fit plot of log(i) vs. log(v) in peaks 1, 2, 3 and 4. (j) The EIS curves of Bare Zn//MnO2 and F-Si@HMCS@Zn//MnO2 batteries before cycling and after 21 cycles. (i) Pseudocapacitive contribution plots at different scan rates for F–Si@HMCS@Zn//MnO2. | ||
The effect of interfacial modification on Zn2+ diffusion kinetics was further investigated using cyclic voltammetry (CV). The peak current (ip) is described by the equation:57ip = 2.69 × 105n3/2AD1/2cv1/2, where ip, F, A, D, c, v, R,and T are peak current (A), Faraday's constant (96485 C mol−1), working electrode, the diffusion coefficient, potential scanning rate, concentration, ideal gas constant, and absolute temperature, respectively. The formula shows that ip is proportional to D−1/2, thus the diffusion coefficient of Zn2+ can be compared by comparing the peak current ip. As shown in Fig. 6g, peak currents increase with higher sweep rates for all cells. Among the three batteries (Fig. S9†), F-Si@HMCS@Zn//MnO2 demonstrated the highest peak current, while bare Zn//MnO2 had the lowest, indicating that the F-Si@HMCS@Zn significantly enhances Zn2+ diffusion rates. Additionally, the redox peak shapes remained consistent across scan rates, demonstrating stable electrochemical behavior. However, the oxidation peak of the coin cell with Zn//MnO2 and Si@HMCS@Zn//MnO2 under the scan rate of 1 mV s−1 is at 1.66 V, and is shifted by nearly 0.2 V compared to the case of 0.1 mV s−1. This behavior can be attributed to the coating of the zinc anode with the F-Si@HMCS improving the overall electrochemical kinetics by enhancing the conductivity and providing a more stable reaction site. This phenomenon is consistent with the literature.58 The relationship between peak current (i) and scan rate (v) follows i = abv, where a and b are constants; when the b value is 0.5, the electrochemical reaction process is mainly affected by the diffusion mechanism, and when the b value is 1, the electrochemical reaction process is mainly controlled by the surface chemical reaction.59 As shown in Fig. 6h, the b-values for F-Si@HMCS@Zn (0.51, 0.64, 0.73, 0.48) are higher than those for Si@HMCS@Zn (Fig. S10a) and bare Zn (Fig. S10b†) batteries, emphasizing the role of F-Si@HMCS in facilitating surface-controlled processes. As shown in Fig. 6h, the b values of peaks 1, 2, 3, and 4 are 0.51, 0.64, 0.73 and 0.48, which are larger than those of Si@HMCS@Zn//MnO2 and bare Zn//MnO2 batteries, suggesting the F-Si@HMCS@Zn facilitates the surface-controlled process. The contribution ratio of diffusion control and surface pseudocapacitance was further evaluated using: iV = k1v1/2 + k2v. The pseudocapacitance contributions for F-Si@HMCS@Zn increased with scan rate, reaching 89% at higher rates (Fig. 6i), significantly surpassing those of Si@HMCS@Zn (Fig. S11a†) and bare Zn (Fig. S11b†) batteries. This highlights the ability of the F-Si@HMCS layer to enhance Zn2+ diffusion and promote pseudocapacitive behavior. EIS spectra (Fig. 6j) revealed that the interfacial charge transfer impedance of F-Si@HMCS@Zn is consistently lower than that of bare Zn//MnO2 in both fresh and cycled states, confirming superior kinetic performance and interfacial dynamics for F-Si@HMCS@Zn.
Conclusion
This study introduces an innovative F-Si@HMCS coating for the Zn anode, effectively isolating the zinc surface from the electrolyte to mitigate corrosion and suppress hydrogen evolution. The strong zincophilic interaction between fluorine and Zn significantly lowers the diffusion barrier for Zn2+ ions, enhancing ion transfer numbers and promoting robust Zn–F bonding. By redistributing ionic flux at the interface and directing uniform Zn2+ deposition, this interaction improves both the efficiency and stability of the deposition process. As a result, the F-Si@HMCS@Zn asymmetric cell achieves an exceptional coulombic efficiency of 99.9% and maintains a low voltage gap of 16 mV over 800 cycles. Furthermore, the F-Si@HMCS@Zn//MnO2 full battery demonstrates remarkable durability, retaining a reversible specific capacity of 77.4 mA h g−1 after 2000 cycles at a current density of 2 A g−1. This work offers valuable insights into advanced coating strategies for enhancing Zn2+ deposition and extending the lifespan of Zn anodes in AZIBs.Data availability
The data that support this study are available in the corresponding ESI.†Author contributions
Shengqiang Zhang: writing – original draft, software, methodology, formal analysis, data curation, conceptualization. Chengxin Liu: software, methodology, formal analysis. Yangyang Wang: software. Ao Xu: methodology, formal analysis. Chunxia Chen: software. Jinbo Bai: software. Xiaojie Liu: funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22075227) and the Science Foundation of Shaanxi Province (No. 2022GD-TSLD-18).References
- M. Xia, J. Zhou and B. Lu, Adv. Energy Mater., 2024, 2404032, DOI:10.1002/aenm.202404032.
- J. Zhou, F. Wu, Y. Mei, Y. Hao, L. Li, M. Xie and R. Chen, Adv. Mater., 2022, 34, 2200782 CrossRef CAS PubMed.
- P. Xue, C. Guo, L. Li, H. Li, D. Luo, L. Tan and Z. Chen, Adv. Mater., 2022, 34, 2110047 CrossRef CAS PubMed.
- F. Xie, H. Li, X. Wang, X. Zhi, D. Chao, K. Davey and S. Qiao, Adv. Energy Mater., 2021, 11, 2003419 CrossRef CAS.
- M. Zhou, Y. Chen, G. Fang and S. Liang, Energy Storage Mater., 2022, 45, 618–646 CrossRef.
- A. Naveed, T. Rasheed, B. Raza, J. Chen, J. Yang, N. Yanna and J. Wang, Energy Storage Mater., 2022, 44, 206–230 CrossRef.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS PubMed.
- S. Higashi, S. Lee, J. Lee, K. Takechi and Y. Cui, Nat. Commun., 2016, 7, 11801 CrossRef PubMed.
- P. Zou, Y. Wang, S. Chiang, X. Wang, F. Kang and C. Yang, Nat. Commun., 2018, 9, 464 CrossRef PubMed.
- M. Chamoun, B. Hertzberg, T. Gupta, D. Davies, S. Bhadra, B. Van Tassell, C. Erdonmez and D. Steingart, NPG Asia Mater., 2015, 7, e178 CrossRef.
- M. K. Peng, X. Tang, K. Xiao, T. Hu, K. Yuan and Y. Chen, Angew. Chem., Int. Ed., 2023, e2302701, DOI:10.1002/anie.202302701.
- D. Xie, Y. Sang, D. Wang, W. Diao, F. Tao, C. Liu, J. Wang, H. Sun, J. Zhang and X. Wu, Angew. Chem., Int. Ed., 2023, 62, e202216934 CrossRef CAS PubMed.
- C. Xi, Y. Xiao, C. Yang, M. Li, L. Li, Y. Chao, L. Li, C. He and Y. Yu, J. Mater. Chem. A, 2023, 11, 6522–6529 RSC.
- Y. Li, H. Yao, X. Liu, X. Yang and D. Yuan, Nano Res., 2023, 16, 9179–9194 CrossRef CAS.
- S. Wu, Z. Hu, P. He, L. Ren, J. Huang and J. Luo, eScience, 2023, 3, 100120 CrossRef.
- J. Yang, R. Zhao, Y. Wang, Z. Hu, Y. Wang, A. Zhang, C. Wu and Y. Bai, Adv. Funct. Mater., 2023, 33, 2213510 CrossRef CAS.
- H. He, H. Qin, J. Wu, X. Chen, R. Huang, F. Shen, Z. Wu, G. Chen, S. Yin and J. Liu, Energy Storage Mater., 2021, 43, 317–336 CrossRef.
- K. Xie, K. Ren, Q. Wang, Y. Lin, F. Ma, C. Sun, Y. Li, X. Zhao and C. Lai, eScience, 2023, 3, 100153 CrossRef.
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1903675 CrossRef PubMed.
- Z. Gong, Z. Li, P. Wang, K. Jiang, Z. Bai, K. Zhu, J. Yan, K. Ye, G. Wang, D. Cao and G. Chen, Energy Mater. Adv., 2023, 2023, 0035 CrossRef.
- C. L. Wang, Y. Gao, L. Sun, Y. Zhao, D. Yin, H. Wang, J. Cao, Y. Cheng and L. M. Wang, Nano Res., 2022, 15, 8076–8082 CrossRef CAS.
- G. Zhang, X. Zhang, H. Liu, J. Li, Y. Chen and H. Duan, Adv. Energy Mater., 2021, 11, 2003927 CrossRef CAS.
- Y. An, Y. Tian, S. Xiong, J. Feng and Y. Qian, ACS Nano., 2021, 15, 11828–11842 CrossRef CAS PubMed.
- H. Zhang, O. Noonan, X. Huang, Y. Yang, C. Xu, L. Zhou and C. Yu, ACS Nano, 2016, 10, 4579–4586 CrossRef CAS PubMed.
- P. Sun, Z. Cao, Y. Zeng, W. Xie, N. Li, D. Luan, S. Yang, L. Yu and X. Lou, Angew. Chem., Int. Ed., 2022, 61, e202115649 CrossRef CAS PubMed.
- K. Lee, E. Kim, J. Kim, K. Kim, Y. Lee, M. Lee, K. Ryu, S. Shin, J. Choi, S. Kwon, H. Lee, J. Kim, B. Kim, B. Kim and S. Lee, Adv. Energy Mater., 2024, 14, 2303803 CrossRef CAS.
- Y. Zeng, P. Sun, Z. Pei, Q. Jin, X. Zhang, L. Yu and X. Lou, Adv. Mater., 2022, 34, 2200342 CrossRef CAS PubMed.
- H. Yu, Y. Zeng, N. Li, D. Luan, L. Yu and X. Lou, Sci. Adv., 2022, 8, eabm5766 CrossRef CAS PubMed.
- F. Ling, L. Wang, F. Liu, M. Ma, S. Zhang, X. Rui, Y. Shao, Y. Yang, S. He, H. Pan, X. Wu, Y. Yao and Y. Yu, Adv. Mater., 2023, 35, 2208764 CrossRef CAS PubMed.
- L. Wang, W. Huang, W. Guo, Z. Guo, C. Chang, L. Gao and X. Pu, Adv. Funct. Mater., 2022, 32, 2108533 CrossRef CAS.
- L. Lei, F. Chen, Y. Wu, J. Shen, X. Wu, S. Wu and S. Yuan, Sci. China:Chem., 2022, 65, 2205–2213 CrossRef CAS.
- X. Xie, S. Liang, J. Gao, S. Guo, J. Guo, C. Wang, G. Xu, X. Wu, G. Chen and J. Zhou, Energy Environ. Sci., 2020, 13, 503–510 RSC.
- Y. Yang, C. Liu, Z. Lv, H. Yang, Y. Zhang, M. Ye, L. Chen, J. Zhao and C. Li, Adv. Mater., 2021, 33, 2007388 CrossRef CAS PubMed.
- Y. L. Mu, T. Zhou, D. Li, W. Liu, P. Jiang, L. Chen, H. Zhou and G. Ge, Chem. Eng. J., 2022, 430, 132839 CrossRef CAS.
- W. Du, E. Ang, Y. Yang, Y. Zhang, M. Ye and C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- D. Ouyang, X. Wang, Q. Liu, G. Wang, X. Zhang, J. Chen, H. Zhou, Y. Yao, Y. Zhang, Y. Liu and X. Zhu, Silicon, 2023, 15, 6541–6549 CrossRef CAS.
- X. Pu, H. Wang, D. Zhao, H. Yang, X. Ai, S. Cao, Z. Chen and Y. Cao, Small, 2019, 15, 1805427 CrossRef PubMed.
- Z. Hu, L. Zhao, T. Jiang, J. Liu, A. Rashid, P. Sun, G. Wang, C. Yan and L. Zhang, Adv. Funct. Mater., 2019, 29, 1906548 CrossRef CAS.
- T. Meng, B. Li, Q. Wang, J. Hao, B. Huang, F. Gu, H. Xu, P. Liu and Y. Tong, ACS Nano, 2020, 14, 7066–7076 CrossRef CAS PubMed.
- S. Karuppiah, C. Keller, P. Kumar, P. H. Jouneau, D. Aldakov, J. Ducros, G. Lapertot, P. Chenevier and C. Haon, ACS Nano, 2020, 14, 12006–12015 CrossRef CAS PubMed.
- Y. Yang, T. Chen, M. Zhu, G. Gao, Y. Wang, Q. Nie, Y. Jiang, T. Xiong, W. Lee and J. Xue, ACS Appl. Mater. Interfaces, 2022, 14, 37759–37770 CrossRef CAS PubMed.
- W. Chen, Y. Tan, C. Guo, X. Zhang, X. He, W. Kuang, H. Weng, H. Du, D. Huang, Y. Huang, J. Xu and H. He, J. Colloid Interface Sci., 2024, 669, 104–116 CrossRef CAS PubMed.
- S. Wang, Q. Ran, R. Yao, H. Shi, Z. Wen, M. Zhao, X. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef CAS PubMed.
- Y. Feng, Y. Wang, L. Sun, K. Zhang, J. Liang, M. Zhu, Z. Tie and Z. Jin, Small, 2023, 19, 2302650 CrossRef CAS PubMed.
- Q. Yang, F. Mo, Z. Liu, L. Ma, X. Li, D. Fang, S. Chen, S. Zhang and C. Zhi, Adv. Mater., 2019, 31, 1901521 CrossRef PubMed.
- H. Wang, A. Zhou, X. Hu, Z. Song, B. Zhang, S. Gao, Y. Huang, Y. Cui, Y. Cui, L. Li, F. Wu and R. Chen, Adv. Mater., 2024, 36, 2407145 CrossRef CAS PubMed.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- Q. Cao, Y. Gao, J. Pu, X. Zhao, Y. Wang, J. Chen and C. Guan, Nat. Commun., 2023, 14 DOI:10.1038/s41467-023-36386-3.
- M. Xia, H. Fu, K. Lin, A. Rao, L. Cha, H. Liu, J. Zhou, C. Wang and B. Lu, Energy Environ. Sci., 2024, 17, 1255–1265 RSC.
- G. Kong, J. Lu, S. Zhang, C. Che and H. Wu, Surf. Coat. Technol., 2010, 205, 545–550 CrossRef CAS.
- M. Zhu, H. Wang, H. Wang, C. Li, D. Chen, K. Wang, Z. Bai, S. Chen, Y. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2024, 63, e2316904 Search PubMed.
- Y. Dai, C. Zhang, W. Zhang, L. Cui, C. Ye, X. Hong, J. Li, R. Chen, W. Zong, X. Gao, J. Zhu, P. Jiang, Q. An, D. Brett, I. Parkin, G. He and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202301192 CrossRef CAS PubMed.
- X. Chen, W. Li, D. Reed, X. Li and X. Liu, Energy Rev., 2023, 6, 33 CAS.
- S. Yang, H. Du, Y. Li, X. Wu, B. Xiao, Z. He, Q. Zhang and X. Wu, Green Energy Environ., 2023, 8, 1531–1552 CrossRef CAS.
- H. Peng, W. Ge, X. Ma, X. Jiang, K. Zhang and J. Yang, Chemsuschem, 2024, 17, 2400076 Search PubMed.
- M. Kwon, J. Lee, S. Ko, G. Lim, S. Yu, J. Hong and M. Lee, Energy Environ. Sci., 2022, 15, 2889–2899 RSC.
- Z. Wu, X. Chen, X. Pan, X. Li, J. Yao, B. Jiang, L. Lv, H. Wang, B. Zhang and H. Wan, ACS Appl. Energy Mater., 2024, 7, 834–844 CrossRef CAS.
- X. Gao, C. Zhang, Y. Dai, S. Zhao, X. Hu, F. Zhao, W. Zhang, R. Chen, W. Zong, Z. Du, H. Dong, Y. Liu, H. He, J. Li, I. Parkin, G. He and C. Carmalt, Small Struct., 2023, 4, 2200316 CrossRef CAS.
- A. Yu, W. Zhang, N. Joshi and Y. Yang, Recent advances in anode design for mild aqueous Zn-ion batteries, Energy Storage Mater., 2024, 64, 103075 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08518c |
| This journal is © The Royal Society of Chemistry 2025 |