
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzolium-2-dithiocarboxylates as redox active ligands in nickel chemistry†‡
Martin S.
Luff
 a,
Tin M.
Filipovic
a,
Celine S.
Corsei
a,
Kai
Oppel
a,
Ivo
Krummenacher
ab,
Rüdiger
Bertermann
ab,
Maik
Finze
ab,
Holger
Braunschweig
ab and
Udo
Radius
*a
a,
Tin M.
Filipovic
a,
Celine S.
Corsei
a,
Kai
Oppel
a,
Ivo
Krummenacher
ab,
Rüdiger
Bertermann
ab,
Maik
Finze
ab,
Holger
Braunschweig
ab and
Udo
Radius
*a
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: u.radius@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 11th February 2025
Abstract
The coordination chemistry of carbene-CS2 adducts of selected NHCs and cAACs and their redox active nature in nickel complexes is reported. These azolium-2-dithiocarboxylate ligands can be considered as 1,1-isomeric dithiolene analogues bearing a 2π electron reservoir. Depending on the co-ligands attached to nickel, square planar mono- or bis-(carbene-CS2) complexes of the types [Ni(IiPr)2(carbene-CS2)] (2a–g) (carbene = cAACMe, IDipp, IMes, BIMe, BIiPr, IiPr, and IiPrMe) and [Ni(carbene-CS2)2] (3a–c) (carbene = cAACMe, IDipp, and IMes) are accessible by alkene substitution using [Ni(IiPr)2(ƞ2-C2H4)] or [Ni(COD)2] as the starting material (cAACMe = 1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene, IR = 1,3-diorganylimidazolin-2-ylidene, IRMe = 1,3-diorganyl-4,5-dimethylimidazolin-2-ylidene, and BIR = 1,3-diorganylbenzimidazolin-2-ylidene). In the complexes 2a–g and 3a–c, all carbene-CS2 ligands are coordinated in a κ2-S,S′ fashion to nickel(II) and are ligated either in their formally one electron reduced (3a–c) or two electron reduced (2a–g) redox states. The complexes 3a–c reveal intense NIR absorptions, which shift upon metallic reduction to the nickelate salts of the type [Cat]+[Ni(carbene-CS2)2]˙− (4a–bcat). In these nickelates, an additional electron is shared across a ligand-centered SOMO of π-symmetry which is delocalized over both azolium-2-dithiocarboxylate ligands and results in carbene-CS2 moieties with a formal −1.5 charge per ligand, further demonstrating the flexible redox active nature of these azolium-2-dithiocarboxylate ligands.
Introduction
Redox active ligands can adopt different oxidation states upon coordination and can act as electron reservoirs for transition metal and main group elements. Therefore they have become a very important class of ligands over the last few decades.1 Redox active ligands allow access to a plethora of different oxidation or spin states in their complexes.2 The use of such ligands which behave as electron reservoirs with reversible redox activity and which can donate or accept one or more electrons on demand during a catalytic cycle has emerged as a powerful tool in catalysis. Such ‘‘actor ligands’’ can extend the electronic flexibility of the central element and directly participate in crucial bond breaking or bond forming steps, thus acting as metal–ligand bifunctional catalysts. The implementation of such metal–ligand cooperation strategies for the design of functional catalysts has been very successful in recent years. Thus, a broad variety of interesting applications of these ligands have been developed, ranging from stabilization of subvalent main group compounds1g,3 to inexpensive 3d transition metal based complexes which mimic the catalytic activity of efficient noble metal catalysts.1b,4 Furthermore, redox active ligands are essential components in artificial photosynthetic,5 photoredox,6 and electrocatalytic systems7 as well as in heteroatom based diradicaloids.8 Employed in these processes are mainly a broad variety of easily tunable, functionalized nitrogen or oxygen containing π-conjugated systems such as catecholates, 2,2′-bipyridines, ortho-amidophenolates or diazabutadienes.3One of the prominent examples of such redox active ligands is sulfur-based dithiolenes, which have been a subject of interest since the 1960s as components in, for example, prototypical homoleptic square planar or trigonal prismatic transition metal complexes with interesting bonding situations.9 In these types of compounds, the formal oxidation state of the central metal atom may not always be unambiguously determined, thus rendering them redox non-innocent.10 For example, in square planar bis-1,2-dithiolene group 10 complexes, the central metal atom M may formally exist in oxidation states 0 (A), +II (B), or + IV (C) (Scheme 1), as both attached 1,2-dithiolene ligands may exist in three possible redox states: a regular neutral 1,2-dithiolene (A), a radical monoanionic form (B), or a dianionic 1,2-dithiolate ligand (C).
 | ||
| Scheme 1 Square planar group 10 bis-1,2-dithiolene complexes (M = Ni, Pd, Pt) in three possible resonance structures: A, B and C. | ||
This mesomeric flexibility, i.e., the redox active nature, of dithiolenes often leads to unique physical properties.11 For example, group 10 bis-dithiolene complexes, especially those of nickel, have attracted considerable interest for application as switchable, modulable near-infrared (NIR) dyes for medicinal and technical purposes.12 Due to the synthetic availability of the ligands, the majority of known group 10 bis-dithiolene systems are mostly limited to the 1,2-substitutional pattern with just a few examples known for related 1,1-isomeric dithiolate complexes.13
An interesting class of molecules in this respect includes 1,1-dithiolene analogues derived from Lewis-acid–base adducts of N-heterocyclic carbenes (NHCs) or cyclic (alkyl)(amino)carbenes (cAACs) with CS2, which form zwitterionic azolium-2-dithiocarboxylates (vide infra, Schemes 2 and 3).14 Although these compounds have been used as ligands in transition metal chemistry before,14b,k,15 they have so far not been considered for the synthesis of bis-dithiolene nickel complexes. Just recently we reported that NHC- and cAAC-based neutral carbene-CS2 adducts (A), i.e., azolium-2-dithiocarboxylates, undergo reductive complexation with metallic main group reductants to yield magnesium or potassium complexes of radical-anions [carbene-CS2]˙− (B).16 Furthermore, earlier studies have also reported the synthesis of alkali metal clusters of dianions of the type [carbene-CS2]2− (C),17 demonstrating that the π-system of carbene-CS2 adducts may act as a π-electron reservoir for up to two added electrons (Scheme 2). Despite these interesting properties, reports on the redox activity of azolium-2-dithiocarboxylates as ligands are limited to two very recent studies.15s,t
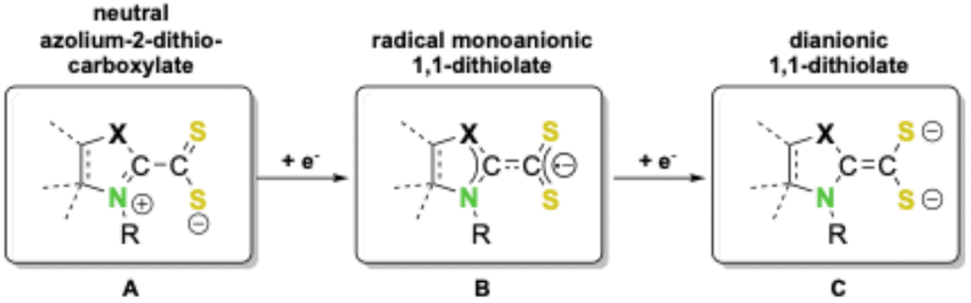 | ||
| Scheme 2 Three redox-states of azolium-2-dithiocarboxylates (X = CMe2, NR). | ||
 | ||
| Scheme 3 Synthesis of azolium-2-dithiocarboxylates 1a–g.14d,g–j,16 | ||
Considering the significance of such redox active complexes in materials science and catalysis we were interested in developing azolium-2-dithiocarboxylate nickel chemistry based on NHCs and cAACs. We herein report the synthesis and characterization of several azolium-2-dithiocarboxylate nickel complexes, thereby clearly demonstrating the redox active nature of this class of ligands.
Results and discussion
We started our study with the synthesis of different carbene-CS2 adducts of a set of electronically and sterically different N-heterocyclic carbenes (NHCs), such as the cAAC-based adduct cAACMe-CS2 (1a; cAACMe = 1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene), several N-aryl substituted NHC-adducts such as IDipp-CS2 (1b) and IMes-CS2 (1c; IR = 1,3-diorganylimidazolin-2-ylidene), π-benzannulated adducts such as BIMe-CS2 (1d) and BIiPr-CS2 (1e: BIR = 1,3-diorganylbenzimidazolin-2-ylidene), and nitrogen and/or backbone alkylated carbene-CS2 adducts such as IiPr-CS2 (1f) and IiPrMe-CS2 (1g; IRMe = 1,3-diorganyl-4,5-dimethylimidazolin-2-ylidene). These NHC-CS2 adducts can be synthesized in large quantities and high yields from the reaction of CS2 and the free NHC (see Scheme 3), as reported previously for most of these azolium-2-dithiocarboxylates.14d,g–j,16 As the synthesis of BIiPr-CS2 (1e) is unknown we present the synthesis and characterization of this compound in the ESI.‡18 Recently, we also presented the detailed redox chemistry and the electronic properties of many of these azolium-2-dithiocarboxylates, which typically feature quasi-reversible redox events. These redox waves correspond to the reduction to either radical monoanions ([NHC-CS2]˙−) or dianions ([carbene-CS2]2−) (for an overview of the cyclic voltammograms recorded for 1a–g see Fig. S1 of the ESI‡).16Due to our long-standing interest in nickel chemistry,19 we were interested in the reductive coordination of these neutral carbene-CS2 ligands with suitable electron-rich nickel(0) precursors.20 Therefore, we chose [Ni(IiPr)2(ƞ2-C2H4)]20a as the starting material, as it provides access to the reactive, coordinatively unsaturated complex {Ni(IiPr)2}.19,20 Solutions of [Ni(IiPr)2(ƞ2-C2H4)] in toluene react immediately with azolium-2-dithiocarboxylates 1a–g accompanied by a color change from orange to yellow (2a–c) or green (2d–g) (see Scheme 4). Depending on the nature of the carbene-CS2 adduct used, the resulting products of these reactions either precipitated (2f–g) over several hours from these solutions or remained entirely (2a–c) or partially (2d–e) in solution. Workup led to the isolation of the complexes [Ni(IiPr)2(carbene-CS2)] (2a–g) in 51–94% yield (Scheme 4). Although NMR scale synthesis of 2a–g indicated quantitative formation of the products, isolation on a preparative scale in high purity afforded lower yields for 2b (51%) and 2c (65%) due to material loss during the purification process, i.e., due to the high sensitivity against oxygen and moisture and the high solubility of these complexes in nonpolar solvents like pentane or hexane.
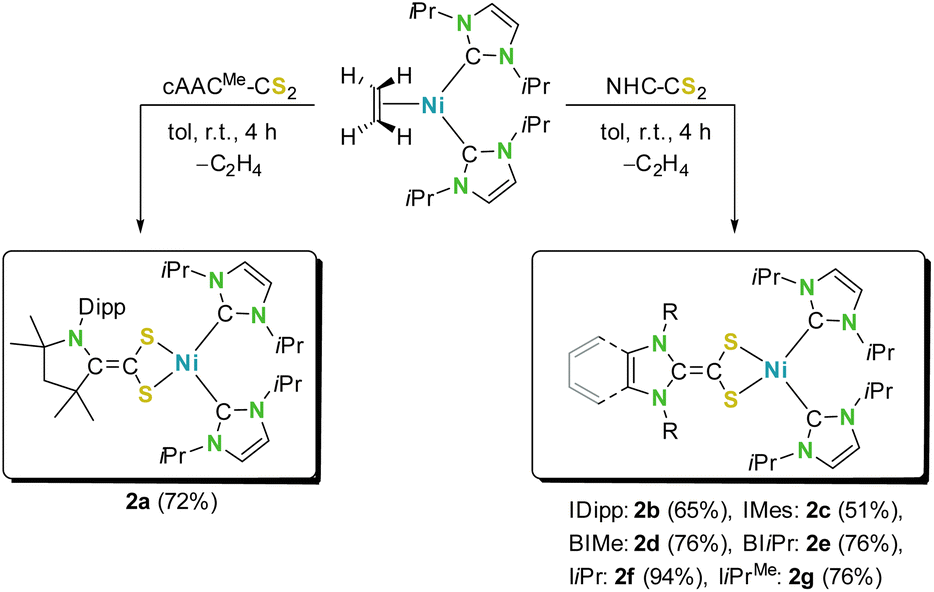 | ||
| Scheme 4 Synthesis of mono-azolium-2-dithiocarboxylate nickel complexes of the type [Ni(IiPr)2(carbene-CS2)] (2). | ||
The products obtained were in general sensitive towards air and moisture and not stable in many common solvents such as MeCN or chlorinated hydrocarbons, except for the cAAC-derivative 2a, which proved to be more stable. The complexes 2a–g were characterized by using elemental analysis, high resolution mass spectrometry (HRMS), NMR, IR and UV/VIS/NIR spectroscopy. However, low solubility and instability even in THF solutions of the complexes [Ni(IiPr)2(IiPr-CS2)] (2f) and [Ni(IiPr)2(IiPrMe-CS2)] (2g) prevented characterization of these compounds in solution. HRMS measurements of highly diluted THF solutions of these compounds led to the detection of protonated molecular ions [M–H]+. Nevertheless, CP/MAS (CP: cross polarization; MAS: magic angle spinning) NMR spectroscopic data obtained from solid samples of 2f–g are in line with solution NMR data of 2a–e and confirm the formation of the complexes [Ni(IiPr)2(NHC-CS2)] (NHC = IiPr and IiPrMe, see below). The molecular structures of 2a–d were confirmed by using single crystal X-ray diffraction (SC-XRD) analysis performed on single crystals obtained either from saturated toluene or from saturated THF solutions.
Solution NMR spectroscopy of complexes 2a–e confirmed the presence of two IiPr NHC ligands coordinated to nickel, showing resonances in the 1H and 13C{1H} NMR spectra of the complexes which were clearly shifted relative to the non-ligated IiPr. These IiPr shifts were in principle independent of the nature of the carbene-CS2 co-ligand attached to nickel. Characteristic CHmethine proton resonances were found in a small range of 6.05–6.27 ppm and the nickel-coordinated carbene carbon atom resonances were detected in a range between 181.2 ppm and 183.9 ppm. For complex 2a, the signals of IiPr were split into two slightly shifted sets of resonances due to the asymmetry imposed by the coordinated cAACMe-CS2 (1a) ligand. The coordinated azolium-2-dithiocarboxylate ligands in complexes 2 should be considered as dianionic 1,1-dithiolates (see Scheme 2) coordinated to Ni(II). This dianionic nature of the [carbene-CS2]2− ligands in 2 is evident from the 1H and 13C{1H} NMR spectroscopic shifts in solution as well as in the solid state (13C and 15N CP/MAS), which are significantly different from those of the non-coordinated, neutral carbene-CS2 molecules. The most significant changes upon coordination were observed in the 13C NMR resonances of the carbenoid (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS2) and the CS2 carbon atoms. While these resonances were found for the neutral carbene-CS2 adducts 1a–g at values in the range of 146.7–187.5 ppm (carbenoid, C1) and 219.7–229.2 ppm (CS2, C2),14b,h,14i,16,17 reductive coordination of the ligand led to shifts down to 130.0–141.5 ppm for the C1 carbon atom of the NHC and to 78.5–113.4 ppm for the C2 carbon atom of the CS2 group. Moreover, these values differ clearly from values obtained for analogous 1,1-ene-dithiolate nickel complexes of the type [NiL2(S2CCR2)] (R = ketyl) bearing electronegative ketyl groups at the C2 carbon atom, which typically give rise to resonances at ca. 120 ppm (R2CCS2, C1) and 210 ppm (CS2, C2).2113C NMR values for comparable carbene-ligated CE2 dianions (E = O, S) have so far not been reported.17,22 Furthermore, in the 15N CP/MAS NMR spectra of 2f–g, two almost identically shifted low field resonances were detected at −173.5 ppm and −175.2 ppm for 2f and at −173.3 ppm and −175.4 ppm for 2g, which must be assigned to the nitrogen atoms of the NHC ligands coordinated to nickel, as the nitrogen atoms of the dianionic ligands [IiPr-CS2]2− and [IiPrMe-CS2]2− in 2f and 2g give rise to resonances at significantly more negative chemical shifts of −267.0 ppm in 2f and −268.6 ppm in 2g. This is in the typical signal range observed for delocalized enamines, in line with the formal Lewis structure of the dianionic redox state C (Scheme 2).23 The nitrogen atoms of the NHC ligands attached to nickel most probably emerge as split resonances due to rotational freezing in the solid state.
CS2) and the CS2 carbon atoms. While these resonances were found for the neutral carbene-CS2 adducts 1a–g at values in the range of 146.7–187.5 ppm (carbenoid, C1) and 219.7–229.2 ppm (CS2, C2),14b,h,14i,16,17 reductive coordination of the ligand led to shifts down to 130.0–141.5 ppm for the C1 carbon atom of the NHC and to 78.5–113.4 ppm for the C2 carbon atom of the CS2 group. Moreover, these values differ clearly from values obtained for analogous 1,1-ene-dithiolate nickel complexes of the type [NiL2(S2CCR2)] (R = ketyl) bearing electronegative ketyl groups at the C2 carbon atom, which typically give rise to resonances at ca. 120 ppm (R2CCS2, C1) and 210 ppm (CS2, C2).2113C NMR values for comparable carbene-ligated CE2 dianions (E = O, S) have so far not been reported.17,22 Furthermore, in the 15N CP/MAS NMR spectra of 2f–g, two almost identically shifted low field resonances were detected at −173.5 ppm and −175.2 ppm for 2f and at −173.3 ppm and −175.4 ppm for 2g, which must be assigned to the nitrogen atoms of the NHC ligands coordinated to nickel, as the nitrogen atoms of the dianionic ligands [IiPr-CS2]2− and [IiPrMe-CS2]2− in 2f and 2g give rise to resonances at significantly more negative chemical shifts of −267.0 ppm in 2f and −268.6 ppm in 2g. This is in the typical signal range observed for delocalized enamines, in line with the formal Lewis structure of the dianionic redox state C (Scheme 2).23 The nitrogen atoms of the NHC ligands attached to nickel most probably emerge as split resonances due to rotational freezing in the solid state.
In accordance with the spectroscopic data, the molecular structures of 2a–d clearly indicated that these complexes are best described as bis-NHC nickel(II) complexes coordinated by the reduced dianionic [carbene-CS2]2− ligands in a κ2-S,S′ mode (Fig. 1 and Table 1).
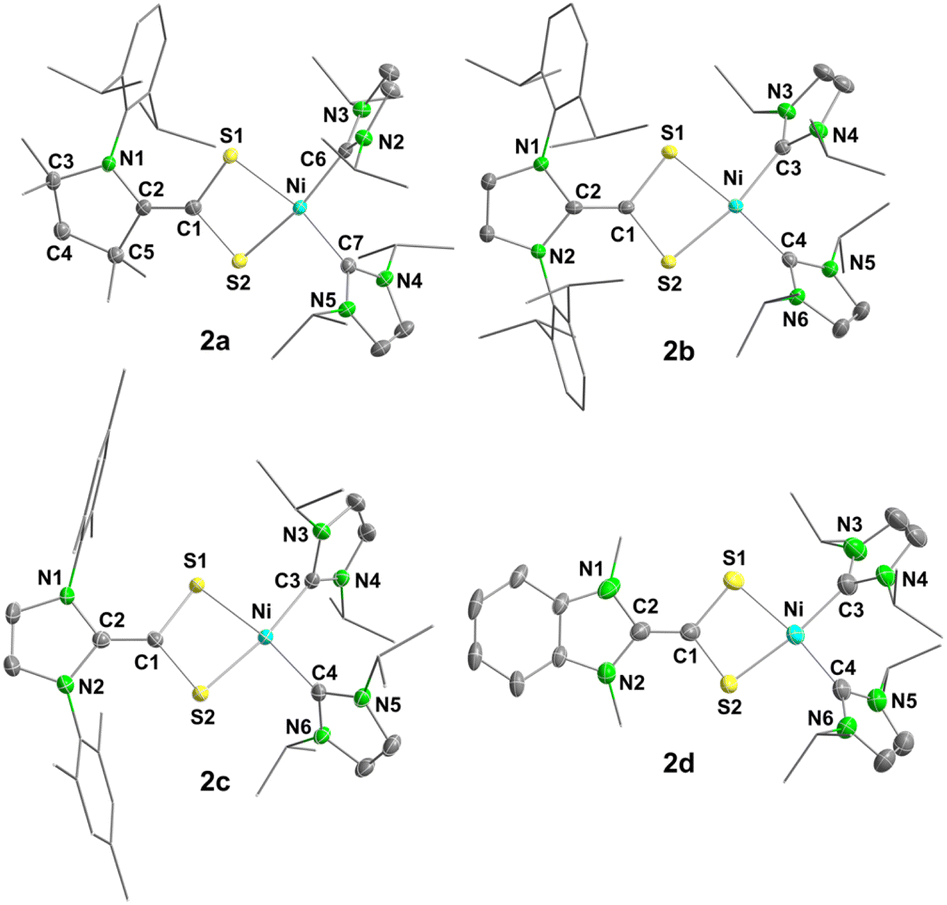 | ||
| Fig. 1 Molecular structures of [Ni(IiPr)2(cAACMe-CS2)] (2a, top left), [Ni(IiPr)2(IDipp-CS2)] (2b, top right), [Ni(IiPr)2(IMes-CS2)] (2c, bottom left), and [Ni(IiPr)2(BIMe-CS2)] (2d, bottom right) in the solid-state (ellipsoids set at the 50% probability level). The hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and angles [°] are provided within Table 1 or in the captions of Fig. S72–S75 of the ESI.‡ | ||
| d(C–C)a | d(C–N) | d(C–S) | d(S–M) | ∡SCS | ∡SMS | ∡LML | Twist-angle (∡NCCS) | |
|---|---|---|---|---|---|---|---|---|
| a Bond between the CS2 group and the NHC carbene atom. b Bond to the NHC carbene carbon atom. c Dihedral angle. d Average value. e Considering the phosphorus or carbon atoms of the L-type ligands. f As the carbene-CS2 moiety is not coordinated in a κ2-S,S′ mode, no value is provided here. g Considering the sulfur atoms of the same ligand. h M = K. | ||||||||
| [Ni(IiPr)2(cAACMe-CS2] (2a) | 1.343(2) | 1.415(2) | 1.776(1), 1.775(1) | 2.188(1), 2.186(1) | 102.7(1) | 78.7(1) | 101.3(1) | 3.5(2) |
| [Ni(IiPr)2(IDipp-CS2] (2b) | 1.343(2) | 1.417(2), 1.422(2) | 1.768(1), 1.777(1) | 2.179(1), 2.199(1) | 103.8(1) | 79.2(1) | 96.3(1) | 0.6(2), 5.4(2) |
| [Ni(IiPr)2(IMes-CS2] (2c) | 1.336(4) | 1.421(3), 1.418(3) | 1.776(3), 1.769(3) | 2.180(1), 2.192(1) | 103.6(1) | 79.1(1) | 97.4(1) | 1.6(4), 0.4(4) |
| [Ni(IiPr)2(BIMe-CS2] (2d) | 1.341(3) | 1.421(3), 1.417(3) | 1.780(2), 1.769(2) | 2.198(6), 2.185(1) | 103.3(1) | 78.8(1) | 93.9(1) | 8.2(4), 9.8(4) |
| [Ni(cAACMe-CS2)2] (3a) | 1.391(2) | 1.361(2) | 1.740(1), 1.732(1) | 2.190(1), 2.161(1) | 105.4(1) | 78.8(1) | — | 5.9(2) |
| [Ni(IDipp-CS2)2] (3b) | 1.395(2) | 1.380(2), 1.378(2) | 1.732(1), 1.730(1) | 2.177(1), 2.181(1) | 107.3(1) | 79.6(1) | — | 4.7(2), 3.6(2) |
| [Ni(IMes-CS2)2] (3c) | 1.399(2) | 1.377(2), 1.379(2) | 1.734(1), 1.734(1) | 2.174(1), 2.181(1) | 106.8(1) | 79.5(1) | — | 6.5(2), 8.7(2) |
| K[Ni(cAACMe-CS2)2] (4aK) | 1.360(3), 1.353(3) | 1.396(2), 1.399(2) | 1.749(2), 1.763(2), 1.759(2), 1.763(2) | 2.163(1), 2.187(1), 2.170(1), 2.180(1); 3.459(1), 3.163(6) | 104.7(1), 104.2(1) | 79.4(2), 79.5(1) | — | 5.1(3), 3.9(3) |
| K[Ni(IDipp-CS2)2] (4bK) | 1.362(5), 1.378(5) | 1.402(4), 1.401(4), 1.389(4), 1.389(4) | 1.751(3), 1.766(3), 1.746(3), 1.757(3) | 2.178(1), 2.184(1), 2.180(1), 2.199(1); 3.221(1), 3.242(1) | 104.8(2), 105.8(2) | 79.4(3), 79.3(3) | — | 10.3(5), 11.9(5), 10.1(5), 13.4(5) |
| IMes-CS2 (1c)14h | 1.501(7) | 1.315(4) | 1.662(2) | —f | 130.3(3) | — | — | 87.5(3) |
| [Pd(PPh3)2(IMes-CS2)]2+15h | 1.452(3) | 1.351(3), 1.352(3) | 1.692(2), 1.683(2) | 2.334(1), 2.372(1) | 113.5(1) | 73.7(1) | 101.1(1) | 10.8(3), 12.0(3) |
| [K(18-crown-6)(IMes-CS2)]16 | 1.407(5) | 1.388(4), 1.387(4) | 1.734(3), 1.733(3) | 3.216(1), 3.212(8) | 124.0(2) | 56.9(1) | — | 31.2(4), 31.8(4) |
| [IiPrMe-CS2]2−17 | 1.375(13) | 1.455(10), 1.454(15) | 1.803(8), 1.764(11) | 3.156d,h | 117.6(5) | —f, | — | 4.8(14), 8.4(14) |
The complexes 2a–d adopt a distorted square planar geometry with bond angles ∡CNiC between the carbene carbon atoms at the central nickel atom ranging between 93.9(1)° and 102.7(1)° and ∡SNiS between the sulfur atoms of the azolium-2-dithiocarboxylate ligands at nickel in a narrow range of 78.8(1)° to 79.2(1)°. The carbene-CS2 ligands undergo a series of structural changes upon coordination and internal reduction, i.e. electron transfer from the nickel atom to the ligand. The bond parameters indicate the presence of [carbene-CS2]2− dianions (Scheme 2, form C) coordinated to the Ni(II) centers. For instance, the central dihedral angle ∡NCCS (twist-angle) in 2a–d ranges from 0.4(4)−9.8(4)°, indicating that the CS2 group is almost ideally aligned co-planar to the NHC carbene plane, as reported previously for the dianion [IiPrMe-CS2]2− (∡NCCSavg = 6.6°).17 In neutral azolium-2-dithiocarboxylates, these moieties are perpendicular to each other, and the radical monoanions such as [K(18-crown-6)(IMes-CS2)] typically adopt twisted angles in-between (“twisted co-planar conformation”; for the potassium salt with ∡NCCSavg = 31.5°).16 The reduction of the ligand system is accompanied by strengthening of the central C–C and weakening of the C–S and C–N bonds of the azolium-2-dithiocarboxylate ligands. This effect has also been observed for the one electron reduction as realized in [K(18-crown-6)(IMes-CS2)],16 and is expected to be even more pronounced in the complexes 2a–d. The resulting bond lengths of 2a–d are d(C–C) = 1.336(4)–1.343(2) Å, d(C–S) = 1.780(2)–1.768(1) Å and d(C–N) = 1.415(2)–1.422(2) Å, which are in line with the presence of CC double bonds as well as C–N and C–S single bonds.24 DFT geometry optimization of the complexes [Ni(IiPr)2(cAACMe-CS2)] (2a), [Ni(IiPr)2(IDipp-CS2)] (2b), [Ni(IiPr)2(IMes-CS2)] (2c), and [Ni(IiPr)2(IiPr-CS2)] (2f) at the PBE0-D3(BJ)/def2-TZVP(Ni)/def2-SVP(C,H,N,S) level of theory resulted in minimum structures with CC distances of 1.352 Å (2a), 1.356 Å (2b), 1.355 Å (2c), and 1.365 Å (2f). In each case the HOMO is the CC π-bonding orbital (see Fig. 2 and also Fig. S2 of the ESI‡), which confirms the presence of the dianionic ligand redox state C.16 The differences in the geometrical data between coordinated and not coordinated ligands are also corroborated by calculated Wiberg bond indices (WBIs) obtained from the DFT-optimized (PBE0-D3BJ//def2-TZVP(Ni)/def2-SVP(C,H,N,S)) complexes [Ni(IiPr)2(cAACMe-CS2)] (2a), [Ni(IiPr)2(IDipp-CS2)] (2b), [Ni(IiPr)2(IMes-CS2)] (2c), and [Ni(IiPr)2(IiPr-CS2)] (2f) as well as the ligands cAACMe-CS2 (1a), IDipp-CS2 (1b), and IMes-CS2 (1c). The WBIs for the Ccarbene–CCS2 bonds in 2a (1.83), 2b (1.73), 2c (1.76), and 2f (1.73) are much larger than those calculated for the pure ligands 1a–c (0.95) and are consistent with double bond character (see Table S3 of the ESI‡). Accordingly, the WBIs for the C–N and C–S bonds decrease upon coordination from 1.25–1.44 (C–N) for 1a–c to 1.02–1.07 for 2a–f and from 1.48/1.49 (C–S) for 1a–c to 1.00–1.03 for 2a–f, respectively. All these factors are consistent with the transfer of two negative charges to the ligand.
 | ||
| Fig. 2 Highest occupied molecular orbitals (HOMOs) of [Ni(IiPr)2(cAACMe-CS2)] (2a, top left), [Ni(IiPr)2(IDipp-CS2)] (2b, top right), [Ni(IiPr)2(IMes-CS2)] (2c, bottom left) and [Ni(IiPr)2(IiPr-CS2)] (2f, bottom right) calculated at the PBE0-D3(BJ)/def2-TZVPP(Ni)/def2-TZVP(C,H,N,S) level of theory. | ||
Reductive ligation of azolium-2-dithiocarboxylates also leads, in general, to a decrease in the bite angle ∡SCS relative to the neutral free ligands (e.g. IMes-CS2: ∡SCS = 130.3(3)°), which results in ∡SCS angles in a range of 102.7(1)–103.8(1)° for complexes 2a–d. Interestingly, the bonding parameters observed for the azolium-2-dithiocarboxylate ligands in 2a–d differ significantly from those reported for the related square planar dicationic complex [Pd(PPh3)2(κ2-S,S′-IMes-CS2)]2+15h with (average) bond lengths of C–C: 1.452 Å, C–N: 1.352 Å and C–S: 1.688 Å, which closely resemble those of the neutral non-ligated IMes-CS2 molecule (d(C–C) = 1.452(3) Å, davg(C–N) = 1.315(4) Å; davg(C–S) = 1.662(2) Å).14h This deviation can be attributed to the difference in the total charge of both complexes (0 vs. +II), which does not affect the oxidation state of the central metal atoms (Ni(II) vs. Pd(II)) but influences the redox state of the IMes-CS2 ligand, which is dianionic in 2a–d and neutral in [Pd(PPh3)2(κ2-S,S′-IMes-CS2)]2+.
As the ligation of azolium-2-dithiocarboxylates with nickel(0) complexes bearing two additional NHC ligands (IiPr) led to the internal reduction of the carbene-CS2 ligands forming 1,1-dithiolate complexes of nickel(II), we were interested in investigating how two of such ligands would behave in homoleptic nickel complexes in the absence of additional co-ligands. Therefore we reacted [Ni(COD)2] (COD = 1,5-cyclooctadiene) with the CS2 adducts of the N-aryl substituted carbenes cAACMe-CS2 (1a), IDipp-CS2 (1b) and IMes-CS2 (1c). Solutions (1a) or suspensions (1b–c) of azolium-2-dithiocarboxylates in toluene react cleanly with [Ni(COD)2] at room temperature, leading to immediate dissolution and a color change in the reaction mixtures from yellow to green-purple (3a) or to blue and then dark-green (3b–c). The homoleptic bis-azolium-2-dithiocarboxylate complexes [Ni(cAACMe-CS2)2] (3a), [Ni(IDipp-CS2)2] (3b) and [Ni(IMes-CS2)2] (3c) were isolated as microcrystalline powders in yields of 78–80% after workup (Scheme 5).
 | ||
| Scheme 5 Synthesis of homoleptic bis-azolium-2-dithiocarboxylate nickel complexes 3a–c. | ||
Compounds 3a–c were characterized using elemental analysis, HRMS, NMR, IR and UV/VIS/NIR spectroscopy, as well as cyclic voltammetry (CV) and SC-XRD analysis using single crystals obtained from saturated benzene or THF solutions. The homoleptic bis-(carbene-CS2) complexes 3a–c in general were less sensitive towards air or moisture than their heteroleptic relatives 2a–g, with 3a being the most stable complex.
NMR spectroscopy of solutions of the complexes 3a–c revealed characteristic sets of signals of the carbene scaffolds in the 1H NMR spectra, which were shifted relative to the resonances of the free ligands 1a–c to values characteristic for diamagnetic compounds. Interestingly, the 13C{1H} NMR spectra recorded for 3a–c did not show resonances for the carbenoid carbon atom and the CS2 carbon atoms of the carbene-CS2 ligands, whereas all the peripheral carbon resonances were detected in the range expected (see Fig. S27, S29, S31 of the ESI‡). We attribute this behavior to considerable electronic mixing of an energetically low-lying triplet state into the singlet ground state of the molecule (see below).
The complexes 3a–c crystallize in the space groups P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , P21/n and P21/c. The results of the X-ray analyses reveal that these complexes are homoleptic coordinated bis-azolium-2-dithiocarboxylate complexes of Ni(II) in a distorted square planar environment (Fig. 3 and Table 1). Both azolium-2-dithiocarboxylates are attached via two κ2-S,S′ interactions to nickel and the molecular units crystallize without considerable intermolecular interactions between the molecular entities of 3a–c (compare Fig. S77, S78, S80, S81, S83 and S84‡ for unit cell packing), which is most probably hindered by the steric bulk of the N-aryl groups of 3a–c. Thus, the intermolecular distances from the nickel atom of one molecular entity to the NiS4 plane of the next parallelly aligned molecular unit are 6.258(1) Å (3a), 10.473(1) Å (3b) and 6.680(1) Å (3c).
, P21/n and P21/c. The results of the X-ray analyses reveal that these complexes are homoleptic coordinated bis-azolium-2-dithiocarboxylate complexes of Ni(II) in a distorted square planar environment (Fig. 3 and Table 1). Both azolium-2-dithiocarboxylates are attached via two κ2-S,S′ interactions to nickel and the molecular units crystallize without considerable intermolecular interactions between the molecular entities of 3a–c (compare Fig. S77, S78, S80, S81, S83 and S84‡ for unit cell packing), which is most probably hindered by the steric bulk of the N-aryl groups of 3a–c. Thus, the intermolecular distances from the nickel atom of one molecular entity to the NiS4 plane of the next parallelly aligned molecular unit are 6.258(1) Å (3a), 10.473(1) Å (3b) and 6.680(1) Å (3c).
 | ||
| Fig. 3 Molecular structures of [Ni(cAACMe-CS2)2] (3a, top left), [Ni(IDipp-CS2)2] (3b, bottom left) and [Ni(IMes-CS2)2] (3c, middle right) in the solid-state (ellipsoids set at the 50% probability level). The hydrogen atoms and co-crystallized solvent molecules are omitted for clarity. Selected bond lengths [Å] and angles [°] are provided within Table 1 or in the captions of Fig. S76, S79 and S82 of the ESI.‡ | ||
Similar to 2a–d, the redox state of the carbene-CS2 ligands in 3a–c is altered upon coordination to nickel, leading to internal reduction and structural changes within the ligand system. In 3a–c, the present oxidation state of the central nickel ions may therefore be deduced by considering the changes in the bond parameters of the attached carbene-CS2 ligands, which indicate the presence of d8-nickel(II) ions, which is also in line with the adoption of a square planar geometry. These nickel(II) ions are coordinated by two radical monoanionic [carbene-CS2]˙− ligands (redox state B, Scheme 2) as a result of internal reduction (for a theoretical assessment, see below). This is corroborated by C–C bond strengthening, as well as C–N and C–S bond weakening compared to the non-coordinated azolium-2-dithiocarboxylates. This happens to a degree similar to that observed for the radical monoanions of the type [K(18-crown-6)(carbene-CS2)] reported recently,16 but is less pronounced than that observed for the complexes 2a–d presented above. The bond lengths observed for 3 range in between 1.391(2) Å and 1.399(2) Å (C–C), 1.361(2) Å and 1.380(2) Å (C–N) as well as 1.730(1) Å and 1.740(1) Å (C–S). The [carbene-CS2]˙− ligands adopt almost ideal co-planar conformations with dihedral angles ∡NCCS in the range of 3.6(2)–8.7(2)°. Thus, co-planarity extends across both radical anionic 1,1-dithiolate ligands and the central nickel atom. Furthermore, the bite angles ∡SCS of 105.4(1)–107.3(1)° observed are slightly smaller than those observed in complexes 2a–d, where the carbene-CS2 ligands carry a dianionic charge. These metric data are also corroborated by the calculated WBIs obtained from DFT-optimized (PBE0-D3BJ//def2-TZVP(Ni)/def2-SVP(C,H,N,S)) complexes [Ni(cAACMe-CS2)2] (3a), [Ni(IDipp-CS2)2] (3b) and [Ni(IMes-CS2)2] (3c). The WBIs for the Ccarbene–CCS2 bonds of 1.45 (3a), 1.40 (3b), and 1.44 (3c) lie between those calculated for the pure ligands 1a–c (0.95) and the NHC-ligated complexes [Ni(IiPr)2(carbene-CS2)] 2a–c, 2f (1.73–1.83); similarly the WBIs for the C–N and C–S bonds align with this trend (see Table S3 of the ESI‡), which is consistent with the transfer of one negative charges to each ligand.
DFT calculations have been performed at different levels of theory to elucidate the electronic structure of the bis-(carbene-CS2)-ligated nickel complexes 3. A molecular orbital diagram of the frontier orbitals of D2h-symmetric [Ni(IMes-CS2)2] (3c), calculated at the B3-LYP//def2-TZVP(Ni)/def2-SVP(C,H,N,S) level of theory, is provided in Fig. 4 and S3 of the ESI.‡
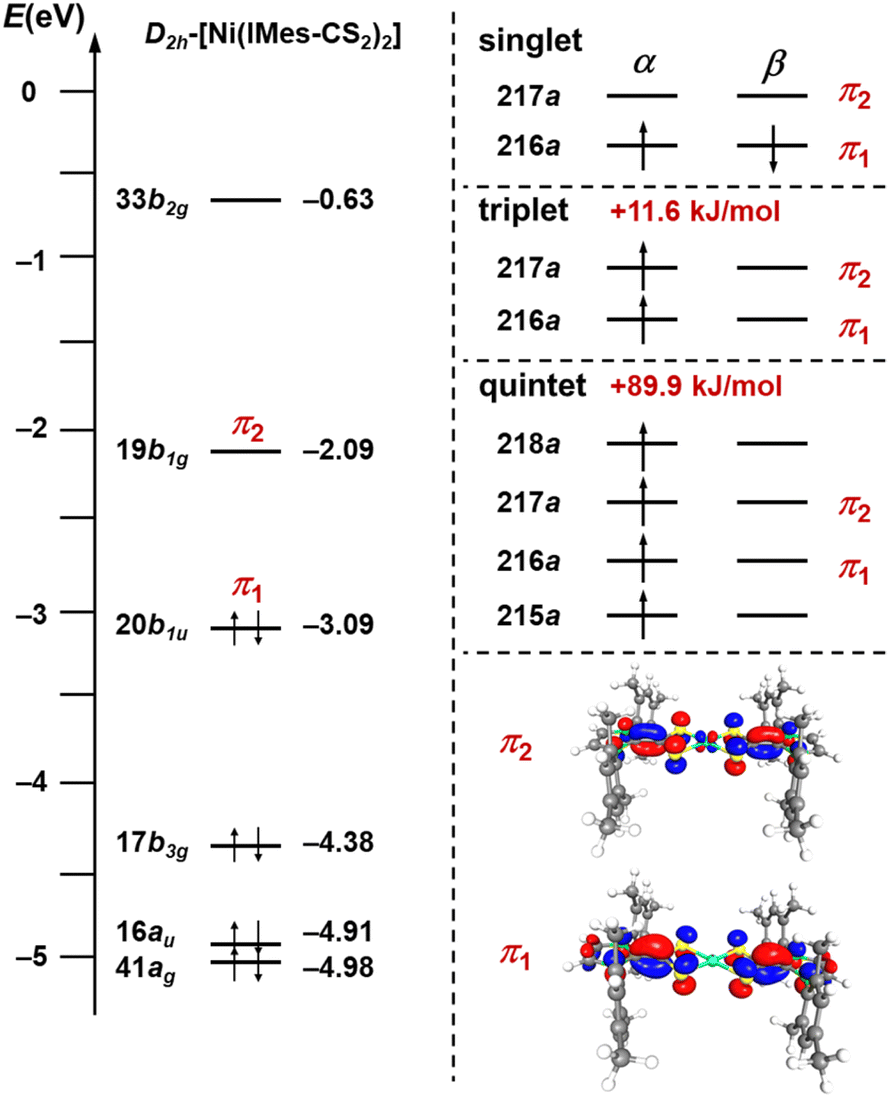 | ||
| Fig. 4 Molecular orbital diagram obtained from DFT calculations on geometry-optimized, D2h-symmetric [Ni(IMes-CS2)2] (3c) at the B3-LYP//def2-TZVP(Ni)/def2-SVP(C,H,N,S) level of theory and the relative energies of the singlet, triplet, and quintet state of this complex. The MOs and an overview of the changes in selected geometric parameters of 3c in different electronic states are provided in Fig. S3 and S4 of the ESI.‡ | ||
Within the D2h-symmetry restraint, the orbitals in the region between −4.3 eV and −6.5 eV are either located on the sulfur atoms and can be described as lone pairs on sulfur or are the typical metal d-centered orbitals of a square planar complex. The unoccupied orbital 33b2g at −0.63 eV is the σ*-antibonding Ni-centered x2 − y2 orbital of the square plane. The electronic structure of D2h-[Ni(IMes-CS2)2] is dominated by the 20b2u orbital at −3.09 eV, which is the HOMO of the complex and the 19b1g orbital at −2.09 eV, which corresponds to the LUMO. Both orbitals are in principle linear combinations of the Ccarbene=CCS2 π-orbitals mentioned earlier. The occupied orbital 20b1u (denoted as π1 in Fig. 4) is the symmetric linear combination with both π-bonds in phase, whereas the unoccupied orbital 19b1g (denoted as π2 in Fig. 4) is the asymmetric linear combination of these π-bonds with some antibonding admixture of a metal 3d orbital. The gap between both orbitals is merely 1.00 eV. Due to this small HOMO/LUMO gap a triplet state is relevant for 3c, which lies only 11.6 kJ mol−1 above the singlet ground state, where the orbitals π1 and π2 are singly occupied by electrons of the same spin. In addition to the triplet state, a quintet state, where one electron of the 17b3g orbital is formally promoted to MO 33b2g, lies 89.9 kJ mol−1 above the singlet diradical state.
If the symmetry constraints are removed, the HOMO 20b1u becomes MO 216a and the LUMO 19b1g becomes MO 217a, and the ground state of this molecule can be described as a singlet diradical, which is in accordance with the electronic structure of closely related palladium complexes reported recently.15s However, significant mixing of the singlet and the triplet state occurs, which stabilizes the molecule. Geometry optimizations of [Ni(IMes-CS2)2] (3c) for the closed shell and open shell complexes at different levels of theory (functional B3-LYP and PBE0-D3BJ; basis sets def2-TZVP(Ni)/def2-SVP(C,H,N,S) and def2-TZVPP(Ni)/def2-TZVP(C,H,N,S)) reveal that the unrestricted open shell singlet diradical ground state is energetically favored compared to the restricted closed shell state, but it is heavily spin-contaminated (S2 statistics of 0.752–0.867 for 3c, depending of the level of theory, for details see Tables S1 and S2 of the ESI‡). For example, the open shell singlet diradical is favored by 21.2 kJ mol−1 compared to the closed shell singlet at the B3-LYP//def2-TZVP(Ni)/def2-SVP(C,H,N,S) level. If spin-contamination of the singlet diradical state originates exclusively from the mixing of the low-lying triplet state into the singlet diradical state an S2 value of 0.80 would account for roughly 40% admixture of the triplet into the singlet state. Geometry optimization of the closed shell singlet, open shell singlet, triplet, and quintet state leads to slight changes in the geometries of 3c (see Fig. S4 of the ESI‡). Especially the Ni–S distances widen upon going from the closed shell singlet (2.2201 Å) to the triplet state (2.2607 Å), and the calculated bond lengths for the open shell singlet diradical lie in-between (2.2477 Å).
The same features as described in detail for [Ni(IMes-CS2)2] (3c) also apply to the complexes [Ni(cAACMe-CS2)2] (3a) and [Ni(IDipp-CS2)2] (3b), which show singlet diradical ground states with significant admixtures of energetically low-lying triplet states (see Tables S1 and S2 of the ESI‡). The spin orbitals of the π1 orbital and the spin densities of the singlet diradical ground states of [Ni(cAACMe-CS2)2] (3a), [Ni(IDipp-CS2)2] (3b), and [Ni(IMes-CS2)2] (3c), calculated at the B3-LYP//def2-TZVP(Ni)/def2-SVP(C,H,N,S) level, are presented in Fig. 5 (3c) and S5 (3a and 3b, see ESI).‡
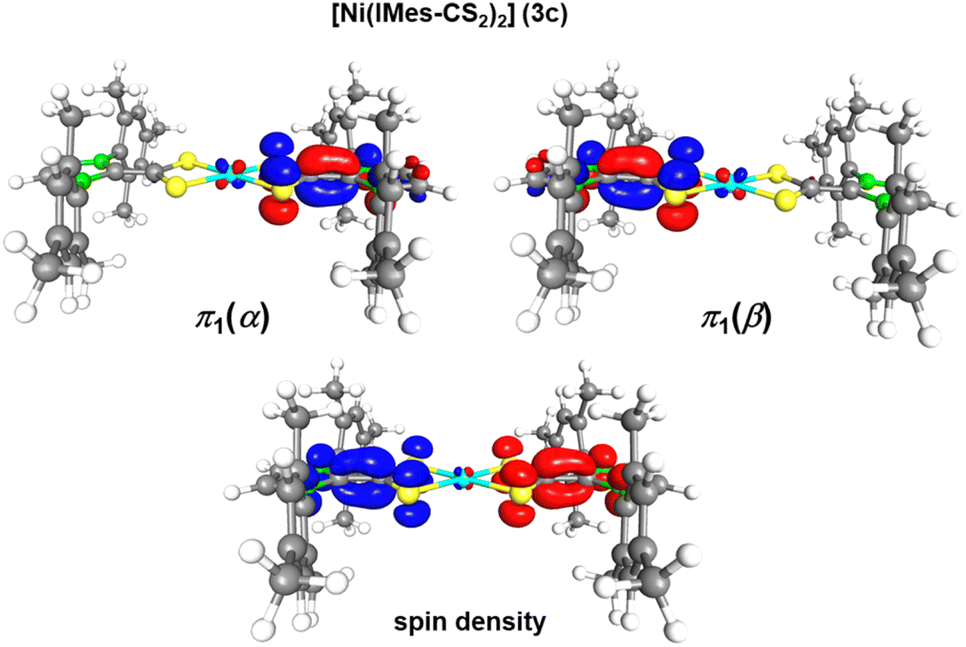 | ||
| Fig. 5 π1 spin orbitals π1(α) (216a,α) and π1(β) (216a,β) and the spin densitiy (blue: spin up and red: spin down) of the singlet diradical ground states of [Ni(IMes-CS2)2] (3c) calculated at the B3-LYP//def2-TZVP(Ni)/def2-SVP(C,H,N,S) level of theory. The spin-densities calculated for 3a and 3b are provided in Fig. S5 of the ESI.‡ | ||
In each of the congeners 3a–c the orbital π1 is the HOMO of the complex and each spin orbital π1(α) and π1(β) is centered on a different dithiolato ligand. Thus, the spin density plot of each complex clearly reveals that the spin densities for up and down spins are located on different dithiolato ligands. The open-shell singlet ground state therefore demonstrates that SOMO1 and SOMO2 (SOMO = singly occupied molecular orbital) are localized on different ligands, which agrees with a description of the azolium-2-dithiocarboxylates in 3a–c as radical monoanionic ligands and a nickel atom in the oxidation state +II.
The nickel complexes ligated with the anionic carbene-CS2 ligands reported herein should, in principle, exhibit distinctly different C–C vibrations in comparison to the neutral ligands due to C–C bond strengthening upon reductive complexation. However, DFT calculations at the B3-LYP//def2TZVP(Ni)/def2SVP(C,H,N,S) level of theory predicted for 3b several vibrations at 1353 cm−1 (asymmetric stretching vibration + CN coupling), 1323 cm−1 (symmetric stretching vibration + CN coupling), 1259 cm−1 and 1243 cm−1 (C–N stretches C–C coupling), which showed significant coupling of the C–C stretches with the vibrations of the imidazolium ligand frame. As the relevant range in the experimental IR spectra were additionally overlayed with other vibrations of similar intensities, the C–C stretches of these complexes were not unambiguously allocated and are thus not discussed herein.
Time-dependent (TD) DFT calculations at the PBE0-D3BJ//def2-TZVPP(Ni)/def2-TZVP(C,H,N,S)/COSMO(benzene) level using the singlet ground state of the complexes predicted characteristic intense absorptions of the complexes 3a–c in the near-IR region at 1008 nm (3a), 1034 nm (3b) and 1056 nm (3c) (Fig. S6 of the ESI‡), which were assigned as ligand-to-ligand charge transfer (LLCT) transitions from the π1 to the π2 orbital. This was also confirmed by experimental UV/VIS/NIR spectroscopic measurements of complexes 3a–c in benzene (see Fig. 6).
 | ||
| Fig. 6 Experimental UV/VIS/NIR spectra of [Ni(cAACMe-CS2)2] (3a, green), [Ni(IDipp-CS2)2] (3b, blue) and [Ni(IMes-CS2)2] (3c, red) in benzene and TD-DFT (PBE0-D3BJ//def2-TZVPP(Ni)/def2-TZVP(C,H,N,S)/COSMO(benzene)) calculated absorption maxima λcalc for the NIR absorption (ω1). | ||
The UV/VIS/NIR spectra of all three compounds 3a–c reveal the same absorption pattern with similar values of λabs featuring two intense absorptions in the ultra-violet region (ca.: 292 nm and 367 nm), two less-intense, broad absorptions in the visible region (ca.: 462 nm and 711 nm) and an intense absorption in the NIR region (ω1) at almost identical values of 1076 nm (3a), 1077 nm (3b) and 1064 nm (3c), which is qualitatively in line with the predicted values obtained from TD-DFT calculations (see discussion above, λcalc = 1008 nm (3a), 1034 nm (3b), and 1056 nm (3c)). These NIR absorptions ω1 of the nickel complexes 3a–c are absent in the complexes 2 with formally dianionic ligands [carbene-CS2]2− (see Fig. S54–S56 of the ESI‡) and lie at almost the same energies as those observed for some palladium congeners reported previously, such as [Pd(IDipp-CS2)2],15s which absorbs at 1014 nm. This NIR absorption is an intrinsic feature of these neutral complexes [M(IDipp-CS2)2] and seems to be independent of substitutional variations in the system.
However, the choice of the central metal atom affects the redox-chemical properties in the systems [M(carbene-CS2)2], which are governed by (quasi)reversible one-electron processes corresponding to oxidation or reduction reactions addressing the π1 or π2 ligand centered orbitals. Cyclic voltammetric measurements performed on the complexes 3a–c (Fig. 7; all potentials referenced vs. the ferrocenium/ferrocene (Fc+/Fc) redox couple) in THF thus revealed two redox events (EII/III, EIII/IV) corresponding to step-wise one-electron oxidations of the nickel(II)-ligated radical monoanions [carbene-CS2]˙− to the respective neutral forms, which leads to mono- (EII/III) and dicationic (EIII/IV) complexes [Ni(carbene-CS2)]+/2+ (3a: EII/III = −1.19 V and EIII/IV = −0.29 V; 3b: EII/III = −2.14 V and EIII/IV = −1.09 V; 3c: EII/III = −1.96 V and EIII/IV = −1.05 V). While the oxidation step EII/III is reversible for all complexes 3a–c, the second oxidation step to yield the dication EIII/IV shows decreasing reversibility in the order 3b < 3a < 3c. In comparison, the one-electron oxidation processes EII/III and EIII/IV in 3a–c occur at significantly more negative potentials than those observed for the palladium congeners. For example, for [Pd(IDipp-CS2)2], two reversible processes were reported at −0.97 V (EII/III; vs.2b: −2.14 V) and −0.325 V (EIII/IV; vs.2b: −1.09 V) in total.15s Moreover, for the nickel complexes 3a–c we observed an additional third reversible redox event in the cyclic voltammograms at even more negative potentials, which may be assigned to a one-electron reduction, yielding the corresponding nickelates [Ni(carbene-CS2)2]˙− (3a: EI/II = −1.71 V; 3b: EI/II = −2.52 V; 3c: EI/II = −2.31 V), where the low-lying π2 LUMO is involved. All three redox events EI/II, EII/III and EIII/IV in general occur at more positive potentials for the cAAC-based compound 3a, i.e., the formation of the nickelate anion, as well as the mono- and dications is easier to achieve for the cAAC derivative 3a, which also reflects the better π-accepting properties of cAACs compared to NHCs.19i,26
 | ||
| Fig. 7 Cyclic voltammograms of [Ni(cAACMe-CS2)2] (3a, green, bottom), [Ni(IDipp-CS2)2] (3b, blue, middle) and [Ni(IMes-CS2)2] (3c, red, top) in THF (concentrations ca. 1 mM) with added [TBA][PF6] (TBA = tetra(n-butyl)ammonium; 0.1 M). Potentials are internally referenced relative to the ferrocenium/ferrocene (Fc+/Fc) couple with negative sweep directions. Scan velocities v = 25 mV s−1 (3a, 3c), 50 mV s−1 (3b). | ||
In light of these results, the synthesis and isolation of monoanionic, radical nickelates of the type [Ni(carbene-CS2)2]˙− posed a challenge, which is achievable by reduction of the neutral nickel complexes 3a–c with metallic reductants.
If solutions of 3a–c in THF were treated with an excess of KC8 or [CoCp2] (for 3a) at room temperature the reaction mixtures immediately changed their color from dark green to yellow-green (3a) or red (3b–c). Workup of the reaction mixtures led to isolation of salts of the radical monoanionic nickelates [Cat]+[Ni(cAACMe-CS2)2]− (Cat = K: 4aK, CoCp2: 4aCo) and K+[Ni(IDipp-CS2)2]− (4bK) in 70% (4aK), 71% (4aCo) and 41% (4bK) yield (Scheme 6).
 | ||
| Scheme 6 Synthesis of the nickelates [Cat][Ni(carbene-CS2)2] (4a–bK/Co; Cat = K, CoCp2). | ||
The isolated paramagnetic nickelate salts 4a–b were characterized by EPR, IR and UV/VIS/NIR spectroscopy as well as SC-XRD, elemental analysis and HRMS. High resolution mass spectrometry of THF solutions of 4a–bK proved that the central [Ni(carbene-CS2)2] moiety remains intact upon reduction, as the [Ni(carbene-CS2)2]˙− molecular ion peaks (m/z = 780.3161 (4aK) and 986.4016 (4bK)) were detected. Instantaneous re-formation of the neutral complexes 3a–b was observed upon exposure of the complexes to air, as indicated by the characteristic dark green (3b) or green-purple (3a) color of the neutral compounds and the corresponding NMR and UV/VIS/NIR spectra, which revealed the characteristic resonances or absorptions of 3a–b.
Single crystals of 4a–bK were obtained from saturated THF solutions and used for SC-XRD analyses, which confirmed the molecular structures as the potassium salts of nickelate anions [Ni(carbene-CS2)2]˙− (Fig. 8 and Table 1).
 | ||
| Fig. 8 Molecular structures of [K(THF)3][Ni(cAACMe-CS2)2] (4aK, left) and [K(THF)2][Ni(IDipp-CS2)2] (4bK, right) in the solid-state (ellipsoids set at the 50% probability level). The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] are given in Table 1 or in the captions of Fig. S85 and S88.‡ The unit cell packing of these complexes are provided in Fig. S86–S90 of the ESI.‡ | ||
The salts 4aK and 4bK crystallize in the space groups P21/n and P212121 and contain intact, non-stacked monoanionic square planar nickelates [Ni(carbene-CS2)2]˙−. The potassium cations are attached to two of the sulfur atoms (d(S–K) = 3.163(6)–3.459(1) Å), with a separation from the central nickel atoms of d(Ni–K) = 3.615(1) Å and 3.358(1) Å). The potassium cations are coordinatively saturated by complexation of three THF ligands in 4aK or two THF ligands in 4bK. As observed in the molecular structures of the carbene-CS2 compounds 2a–d and 3a–c, the difference in the ligands' redox states for the nickelates 4a–bK can be deduced from the change in bond parameters which can be attributed to ligand-centered reductions and occupation of the π2 LUMOs of the neutral complexes (Fig. 4). In both nickelates 4a–bK, the two carbene-CS2 ligands are found with almost identical bond lengths (d(C–C) = 1.353(3)–1.378(5) Å, d(C–N) = 1.389(4)–1.402(4) Å, and d(C–S) = 1.746(3)–1.766(3) Å), which are between those of the dianionic (C; e.g.2a–d) and the radical monoanionic (B; e.g.3a–c) ligand forms (see Table 1). This geometrical change is, again, also reflected in the Wiberg bond indices calculated for the DFT-optimized (PBE0-D3BJ//def2-TZVP(Ni)/def2-SVP(C,H,N,S)) nickelates [Ni(cAACMe-CS2)2]˙− (4a) and [Ni(IDipp-CS2)2]˙− (4b). The WBIs for the Ccarbene–CCS2 bonds of 1.70 (4a) and 1.59 (4b) are higher than those calculated for the neutral complexes 3a (1.45) and 3b (1.40) and almost reach the values calculated for the related NHC-ligated complexes [Ni(IiPr)2(carbene-CS2)] 2a (1.83) and 2b (1.73). Similarly, the WBIs calculated for the C–N and C–S bonds of 4a and 4b reach values close to those calculated for 2a and 2b (see Table S3 of the ESI‡), which is consistent with the additional negative charge delocalized over both ligands.
Reduction with coordination of the potassium cations comes along with a slight distortion of the overall square planar structure of the complexes. This distortion is visualized in Fig. S7 of the ESI,‡ which provides side-on perspectives of the experimentally obtained molecular structures of the neutral compounds 3a–bvs. the anions 4a–bK. Thus, in 4aK and 4bK, the angles ∡SNiS between sulfur atoms in trans positions deviate from the ideal 180° angle as observed in the square planar neutral compounds 3a and 3b, with coordination to nickel at angles of 173.5(1)° or 178.2(1)° (4aK) and 173.6(1)° or 177.0(1)° (4bK). Furthermore, minor curving of the overall planar structures of 3a–b is observed in 4a–bK with the ligand frames bending towards the attached potassium ions. The latter is reflected by an increase in the angles between the C–C vectors of the bonds between the CS2 groups and the carbenoid carbon atoms with respect to the central NiS4 square planes. Thus, in the nickelate 4bK the angles ∡(vector(CC)−plane(NiS4)) are 9.2(1)° and 13.8(1)° for both ligands, while for the neutral form 3b, values of 3.7(1)° are found. In the cAAC derived nickelate 4aK, analogous bending of the ligand frame in one direction is found, although to a smaller degree as in 4bK, which is reflected by respective angles ∡(vector(CC)−plane(NiS4)) of 1.7(1)° and 9.1(1)°. Contrastingly, in the neutral compound 3a, both ligands are bent in opposite directions away from the central NiS4 square plane by 7.3(1)°. Furthermore, the (average) twist angles ∡NCCS of the CS2 group relative to the attached carbene increase to 11.4° for the nickelate 4bK relative to the neutral form 3b, while in 4aK this twisting slightly decreases to 4.5° relative to neutral 3a which has an angle of 5.9(2)°.
Quantum chemical geometry optimizations (see Fig. S8 of the ESI,‡ top) resulted in a similar distorted geometry for the free nickelate 4b (calc.: ∡(vector(CC)–plane(NiS4)) = 20.9°, 27.7°; ∡SNiS = 171.1°, 171.7°), while for 4a an almost undistorted structure was obtained (calc.: ∡(vector(CC)–plane(NiS4)) = 0.9°; ∡SNiS = 180°). We attribute the observed structural distortion of the nickelates 4a–bK to the additional electron, which is delocalized over both ligands and results in [carbene-CS2]1.5− ligands, while the oxidation state of the nickel atom remains unchanged. DFT calculations (see Fig. S8 of the ESI,‡ bottom) confirm that the spin density of the nickelates 4a and 4b is delocalized over the whole molecule. Thus, due to the almost indistinguishable nature of both carbene-CS2 ligands in 4a–bK, the metalla-anions should be considered as ligand based mixed-valent (LBMV) systems which feature strong electronic coupling between both ligands, similar to Robin-Day class III systems.27
The additional electron in the nickelates 4a–bK leads to paramagnetic character of the metallates, which was confirmed via X-band EPR spectroscopy (Fig. 9) performed on THF solutions of the complexes at 70 K. These EPR spectra revealed broad resonances without resolved hyperfine coupling (Fig. 9).
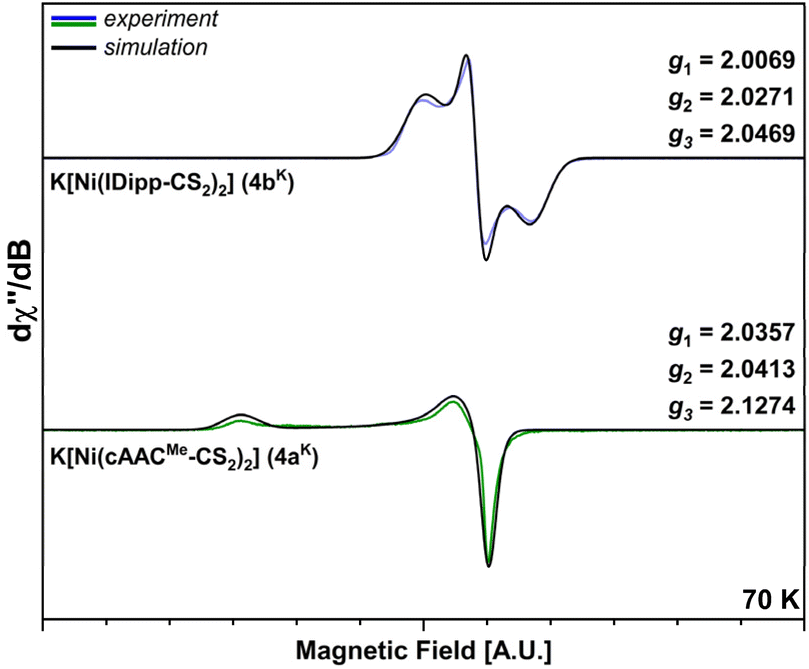 | ||
| Fig. 9 βX-band EPR spectra of K[Ni(cAACMe-CS2)2] (4aK, bottom, green) and K[Ni(IDipp-CS2)2] (4bK, top, blue) at 70 K with normalized x- and y-axes (exact values are provided in Fig. S32–S34 of the ESI;‡ resonances are detected at ca. 310–340 mT; black lines: simulated spectra; regular, colored lines: experimental spectra). | ||
For both compounds 4aK and 4bK, rhombic g-tensors were found (4aK: g1 = 2.0357, g2 = 2.0413, and g3 = 2.1274; 4bK: g1 = 2.0069, g2 = 2.0271, and g3 = 2.0469), both matching simulations. The obtained EPR data are essentially in line with spin systems of square planar symmetry28 and other EPR-characterized nickel bis-dithiolene monoanions reported earlier.25a The significant differences in g anisotropy between 4aK and 4bK can be attributed to the different degrees of structural distortion observed in the anions, as well as to different spin distributions of the anions (see Fig. S8 of the ESI,‡ bottom), with 4aK exhibiting a more localized unpaired spin, while 4bK features a highly delocalized spin across both ligand moieties.
UV/VIS/NIR spectroscopy on THF solutions of the nickelate compounds 4aK and 4bK were performed to evaluate the absorption properties of the bis-(carbene-CS2) complexes 3 upon reduction (Fig. 10). Intriguingly, while the neutral complexes 3a–c absorb at ca. 1076 nm, in the respective anionic compounds K[Ni(cAACMe-CS2)2] (4aK) and K[Ni(IDipp-CS2)2] (4bK) these absorptions vanish. Instead, 4aK and 4bK display significantly red shifted broad NIR-absorptions at wavelengths of 1499 nm for 4aK and at even lower energies for 4bK, which absorbs at 1909 nm. These absorption maxima are in line with TD-DFT calculations (PBE0-D3BJ//def2-TZVPP(Ni)/def2-TZVP(C,H,N,S)/COSMO(THF)) performed on [Ni(cAACMe-CS2)2]˙− (4a) and [Ni(IDipp-CS2)2]˙− (4b), which predict characteristic intense absorptions (λcalc) of these metalates at 1573 nm (4a) and 1947 nm (4b) (see Fig. S9 of the ESI‡). These absorptions are shifted to higher energies compared to the neutral nickel complexes 3a (λcalc = 1008 nm) and 3b (λcalc = 1034 nm) and are due to electronic transitions between the doubly occupied π1 orbital and the singly occupied π2 orbital, i.e. π1(β) → π2(β), and are thus essentially IVCT bands.
 | ||
| Fig. 10 Experimental UV/VIS/NIR absorption spectra of K[Ni(cAACMe-CS2)2] (4aK, green) and K[Ni(IDipp-CS2)2] (4bK, blue) in THF in the NIR region and TD-DFT (PBE0-D3BJ//def2-TZVPP(Ni)/def2-TZVP(C,H,N,S)/COSMO(THF)) calculated absorption maxima λcalc. | ||
The observed drastic differences in the NIR-absorptions of 4aK and 4bK demonstrate that small changes in the carbene-CS2 scaffold strongly influence the absorption spectra of these reduced nickelates, which allows tuning of the redox state dependent NIR absorptivity in these systems by substitutional variation. These basic observations may lay the foundation for further development towards application of such nickel bis-(carbene-CS2) complexes as tunable and switchable electrochromic NIR dyes, enabled by the distinct redox activity of carbene-CS2 ligands.
Conclusions
In this study we demonstrated that azolium-2-dithiocarboxylates are redox active ligands in nickel complexes and that the redox activity of these carbene-CS2 adducts enables access to a variety of formal ligand based redox states. Using nickel(0) precursors and depending on other ancillary co-ligands, bonding of carbene-CS2 adducts through κ2-S,S′ coordination led to the formation of mono- or bis-(carbene-CS2) complexes of the types [NiL2(carbene-CS2)] (2a–g) or [Ni(carbene-CS2)2] (3a–c), in which the ligands were reduced upon coordination to the central nickel atom. Reduction by external redox reagents afforded anionic nickelates [Ni(carbene-CS2)2]˙− (4a–bK). The ligand redox states in these compounds range from radical monoanionic (Scheme 2; B: 3a–c) and dianionic (Scheme 2; C: 2a–g) to a redox state with a non-integer negative charge of −1.5 (4a–bK). SC-XRD analytic evaluation of the structural parameters such as CCarbene–CCS2, C–N, and C–S distances or ∡NCCS torsion angles of the ligated carbene-CS2 adducts proved that these parameters are valuable tools for the determination of the ligands' redox state and thus also for the evaluation of the formal oxidation state of the central nickel atom, which in this study was exclusively Ni(II). Moreover, the strong electronic coupling of the two radical anionic carbene-CS2 ligands in the homoleptic complexes 3a–c led to the description of the electronic structure as a singlet diradical with admixtures from a triplet state that is only slightly higher in energy, which was indicated by quantum chemical calculations and also experimentally corroborated by the diamagnetic character of the ligands in these complexes, except for the central CCarbene and CCS2 carbon atoms, as well as by the presence of characteristic intense ligand-to-ligand charge transfer (LLCT) transitions in the NIR region of their electronic absorption spectra at 1076 nm (3a), 1077 nm (3b) and 1064 nm (3c). In the nickelates 4a–bK the added electron is equally delocalized across the π-system of both carbene-CS2 ligands, leading to a ligand-based mixed-valent (LBMV) redox-state corresponding to the Robin-Day class III. Consequently, these nickelates exhibit characteristic IVCT transitions, which are strongly red-shifted relative to those of 3a–c, with experimental values of 1499 nm (4aK) and 1909 nm (4bK). This work lays the foundation to understand the interesting chemical reactivity and physical properties of redox-active carbene-CS2 ligands in general and of their transition metal complexes and thus paves the way for the application of azolium-2-dithiocarboxylate ligands in different applications, for example, as redox catalysts, as electronically switchable materials or as NIR dyes.Data availability
The data supporting this article have been included as ESI.‡ Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB21EZ, UK.Author contributions
M. S. L. and U. R. conceived of the project; M. S. L., T. M. F. and C. S. C. conducted most of the experiments, K. O. performed CV analyses, I. K. performed EPR spectroscopy, U. R. carried out quantum-chemical calculations, and R. B. conducted solid-state NMR spectroscopy; M. S. L., U. R., M. F., and H. B. wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by funds from the Julius-Maximilians-Universität Würzburg.References
- (a) P. J. Chirik, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed; (b) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS; (c) V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem. Int. Ed., 2012, 51, 10228–10234 ( Angew. Chem. , 2012 , 124 , 10374–10380 ) CrossRef CAS PubMed; (d) J. R. Khusnutdinova and D. Milstein, Angew. Chem. Int. Ed., 2015, 54, 12236–12273 ( Angew. Chem. , 2015 , 127 , 12406–12445 ) Search PubMed; (e) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 Search PubMed; (f) A. Nakada, T. Matsumoto and H.-C. Chang, Coord. Chem. Rev., 2022, 473, 214804 CrossRef CAS; (g) S. B. H. Karnbrock and M. Alcarazo, Chem.–Eur. J., 2024, 30, e202302879 CrossRef CAS PubMed; (h) Redox-Active Ligands. Concepts and Catalysis, ed. M. Desage-El Murr, Wiley-VCh, Weinheim, 2024 Search PubMed.
- U. Rastetter, A. Jacobi von Wangelin and C. Herrmann, J. Comput. Chem., 2023, 44, 468–479 CrossRef CAS PubMed.
- G. G. Briand, Dalton Trans., 2023, 52, 17666–17678 RSC.
- R. Mondal, A. K. Guin, G. Chakraborty and N. D. Paul, Org. Biomol. Chem., 2022, 20, 296–328 RSC.
- (a) B. Singh and A. Indra, Inorg. Chim. Acta, 2020, 506, 119440 Search PubMed; (b) T. Benkó, D. Lukács, M. Li and J. S. Pap, Environ. Chem. Lett., 2022, 20, 3657–3695 CrossRef.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 Search PubMed.
- (a) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC; (b) Z. Niu, L. Yang, Y. Xiao, M. Xue, J. Zhou, L. Zhang, J. Zhang, D. P. Wilkinson and C. Ni, Electrocatalysis, 2022, 13, 230–241 CrossRef CAS.
- A. Hinz, J. Bresnien, F. Breher and A. Schulz, Chem. Rev., 2023, 123, 10468–10526 Search PubMed.
- (a) J. A. McCleverty, Prog. Inorg. Chem., 1968, 10, 49–221 CrossRef CAS; (b) R. Eisenberg, Coord. Chem. Rev., 2011, 255, 825–836 Search PubMed; (c) R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS PubMed.
- (a) C. K. Jørgensen, Coord. Chem. Rev., 1966, 1, 164–178 CrossRef; (b) M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC.
- (a) F. Bigoli, P. Deplano, F. A. Devillanova, J. R. Ferraro, V. Lippolis, P. J. Lukes, M. L. Mercuri, M. A. Pellinghelli, E. F. Trogu and J. M. Williams, Inorg. Chem., 1997, 36, 1218–1226 CrossRef PubMed; (b) Dithiolene Chemistry: Synthesis, Properties, and Applications, ed. E. I. Stiefel, John Wiley & Sons, Hoboken, New Jersey, 2004, vol. 52 Search PubMed; (c) P. Deplano, L. Pilia, D. Espa, M. L. Mercuri and A. Serpe, Coord. Chem. Rev., 2010, 254, 1434–1447 CrossRef CAS; (d) B. Garreau-de Bonneval, K. I. Moineau-Chane Ching, F. Alary, T.-T. Bui and L. Valade, Coord. Chem. Rev., 2010, 254, 1457–1467 CrossRef CAS.
- (a) U. T. Mueller-Westerhoff, B. Vance and D. I. Yoon, Tetrahedron, 1991, 47, 909–932 CrossRef CAS; (b) P. Deplano, M. L. Mercuri, G. Pintus and E. F. Trogu, Comments Inorg. Chem., 2001, 22, 353–374 CrossRef CAS; (c) D. Natali, M. Sampietro, M. Arca, C. Denotti and F. A. Devillanova, Synth. Met., 2003, 137, 1489–1490 CrossRef CAS; (d) M. Caironi, D. Natali, M. Sampietro, M. Ward, A. Meacham, F. A. Devillanova, M. Arca, C. Denotti and L. Pala, Synth. Met., 2005, 153, 273–276 CrossRef CAS; (e) M. C. Aragoni, M. Arca, F. A. Devillanova, F. Isaia, V. Lippolis, A. Mancini, L. Pala, G. Verani, T. Agostinelli, M. Caironi, D. Natali and M. Sampietro, Inorg. Chem. Commun., 2007, 10, 191–194 CrossRef CAS; (f) Y. Liu, Z. Zhang, X. Chen, S. Xu and S. Cao, Dyes Pigm., 2016, 128, 179–189 Search PubMed; (g) F. Camerel and M. Fourmigué, Eur. J. Inorg. Chem., 2020, 508–522 Search PubMed; (h) K. Chen, W. Fang, Q. Zhang, X. Jiang, Y. Chen, W. Xu, Q. Shen, P. Sun and W. Huang, ACS Appl. Bio Mater., 2021, 4, 4406–4412 CrossRef CAS PubMed.
- (a) J. P. Fackler and D. Coucouvanis, Chem. Commun., 1965, 556–557 RSC; (b) J. P. Fackler, Jr. and D. Coucouvanis, J. Am. Chem. Soc., 1966, 88, 3913–3920 Search PubMed; (c) D. Coucouvanis, Prog. Inorg. Chem., 1970, 233–371 CAS; (d) F. J. Hollander, M. L. Caffery and D. Coucouvanis, J. Am. Chem. Soc., 1974, 96, 4682–4684 CrossRef CAS; (e) D. Coucouvanis, F. J. Hollander and M. L. Caffery, Inorg. Chem., 1976, 15, 1853–1860 CrossRef CAS; (f) S. Sproules, C. I. Macleod, O. D. Keramidas and H. N. Miras, Chem.–Eur. J., 2024, 30, e202401710 CrossRef PubMed.
- (a) H. E. Winberg and D. D. Coffman, J. Am. Chem. Soc., 1965, 87, 2776–2777 CrossRef CAS; (b) W. S. Sheldrick, A. Schönberg, E. Singer and P. Eckert, Chem. Ber., 1980, 113, 3605–3609 CrossRef CAS; (c) W. Krasuski, D. Nikolaus and M. Regitz, Liebigs Ann. Chem., 1982, 1451–1465 CrossRef CAS; (d) N. Kuhn, H. Bohnen and G. Henkel, Z. Naturforsch., B, 1994, 49, 1473–1480 CrossRef CAS; (e) J. Nakayama, T. Kitahara, Y. Sugihara, A. Sakamoto and A. Ishii, J. Am. Chem. Soc., 2000, 122, 9120–9126 CrossRef CAS; (f) G. Kuchenbeiser, M. Soleilhavoup, B. Donnadieu and G. Bertrand, Chem.–Asian J., 2009, 4, 1745–1750 CrossRef CAS PubMed; (g) L. Delaude, Eur. J. Inorg. Chem., 2009, 1681–1699 CrossRef CAS; (h) L. Delaude, A. Demonceau and J. Wouters, Eur. J. Inorg. Chem., 2009, 1882–1891 CrossRef CAS; (i) A. R. Katritzky, D. Jishkariani, R. Sakhuja, C. D. Hall and P. J. Steel, J. Org. Chem., 2011, 76, 4082–4087 CrossRef CAS PubMed; (j) U. Siemeling, H. Memczak, C. Bruhn, F. Vogel, F. Träger, J. E. Baio and T. Weidner, Dalton Trans., 2012, 41, 2986–2994 RSC; (k) T. F. Beltrán, G. Zaragoza and L. Delaude, Dalton Trans., 2017, 46, 9036–9048 RSC; (l) F. Mazars, M. Hrubaru, N. Tumanov, J. Wouters and L. Delaude, Eur. J. Org Chem., 2021, 2025–2033 CrossRef CAS; (m) N. Touj, F. Mazars, G. Zaragoza and L. Delaude, Beilstein J. Org. Chem., 2023, 19, 1947–1956 CrossRef CAS PubMed.
- (a) L. L. Borer, J. Kong and E. Sinn, Inorg. Chim. Acta, 1986, 122, 145–148 CrossRef CAS; (b) T. Fujihara, T. Sugaya, A. Nagasawa and J. Nakayama, Acta Cryst. Sect. E, 2004, 60, m282–m284 CrossRef CAS; (c) L. Delaude, X. Sauvage, A. Demonceau and J. Wouters, Organometallics, 2009, 28, 4056–4064 CrossRef CAS; (d) T. Sugaya, T. Fujihara, A. Nagasawa and K. Unoura, Inorg. Chim. Acta, 2009, 362, 4813–4822 CrossRef CAS; (e) S. Naeem, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Inorg. Chem., 2010, 49, 1784–1793 CrossRef CAS PubMed; (f) S. Naeem, A. L. Thompson, L. Delaude and J. D. E. T. Wilton-Ely, Chem.–Eur. J., 2010, 16, 10971–10974 CrossRef CAS PubMed; (g) S. Naeem, A. L. Thompson, A. J. P. White, L. Delaude and J. D. E. T. Wilton-Ely, Dalton Trans., 2011, 40, 3737–3747 RSC; (h) M. J. D. Champion, R. Solanki, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Dalton Trans., 2012, 41, 12386–12394 RSC; (i) A. Neuba, J. Ortmeyer, D. D. Konieczna, G. Weigel, U. Flörke, G. Henkel and R. Wilhelm, RSC Adv., 2015, 5, 9217–9220 RSC; (j) T. F. Beltrán, G. Zaragoza and L. Delaude, Dalton Trans., 2016, 45, 18346–18355 RSC; (k) T. F. Beltrán, G. Zaragoza and L. Delaude, Dalton Trans., 2017, 46, 1779–1788 RSC; (l) T. F. Beltrán, G. Zaragoza and L. Delaude, Dalton Trans., 2017, 46, 13002–13009 RSC; (m) J. Ortmeyer, U. Flörke, G. Henkel, R. Wilhelm and A. Neuba, Eur. J. Inorg. Chem., 2017, 3191–3197 CrossRef CAS; (n) T. F. Beltrán, G. Zaragoza and L. Delaude, Polyhedron, 2021, 197, 115055 CrossRef; (o) P. Rungthanaphatsophon, A. J. Gremillion, Y. Wang, S. P. Kelley, G. H. Robinson and J. R. Walensky, Inorg. Chim. Acta, 2021, 514, 120033 CrossRef CAS; (p) P. Pérez-Ramos, M. A. Mateo, D. Elorriaga, D. García-Vivó, R. G. Soengas and H. Rodríguez-Solla, Dalton Trans., 2024, 53, 9433–9440 RSC; (q) V. P. Carvalho, G. Zaragoza and L. Delaude, Dalton Trans., 2024, 53, 11436–11444 RSC; (r) F. Mazars, G. Zaragoza and L. Delaude, Organometallics, 2024, 43, 2284–2304 CrossRef CAS; (s) S. Park, J.-Y. Hwang, J. Shin and Y. Kim, J. Am. Chem. Soc., 2024, 146, 28508–28515 CAS; (t) M. Lee, H. Noh and Y. Kim, Chem. Commun., 2024, 60, 13867–13870 RSC.
- M. S. Luff, I. Krummenacher, K. Oppel, H. Braunschweig and U. Radius, Chem. Commun., 2024, 60, 14447–14450 RSC.
- N. Kuhn, G. Weyers and G. Henkel, Chem. Commun., 1997, 627–628 RSC.
- The X-ray structure of this compound, however, is reported, see P. G. Jones and I. Dix, CSD Commun., 2004, CCDC: 238439.
- (a) T. Schaub and U. Radius, Z. Anorg. Allg. Chem., 2006, 632, 807–813 CrossRef CAS; (b) T. Schaub and U. Radius, Z. Anorg. Allg. Chem., 2006, 632, 981–984 CrossRef CAS; (c) U. Radius and F. M. Bickelhaupt, Organometallics, 2008, 27, 3410–3414 CrossRef CAS; (d) T. Schaub, P. Fischer, T. Meins and U. Radius, Eur. J. Inorg. Chem., 2011, 3122–3126 CrossRef CAS; (e) T. Zell, P. Fischer, D. Schmidt and U. Radius, Organometallics, 2012, 31, 5065–5073 CrossRef CAS; (f) D. Schmidt, T. Zell, T. Schaub and U. Radius, Dalton Trans., 2014, 43, 10816–10827 RSC; (g) M. W. Kuntze-Fechner, H. Verplancke, L. Tendera, M. Diefenbach, I. Krummenacher, H. Braunschweig, T. B. Marder, M. C. Holthausen and U. Radius, Chem. Sci., 2020, 11, 11009–11023 RSC; (h) L. Tendera, M. Helm, M. J. Krahfuss, M. W. Kuntze-Fechner and U. Radius, Chem.–Eur. J., 2021, 27, 17849–17861 CrossRef CAS PubMed; (i) L. Tendera, M. S. Luff, I. Krummenacher and U. Radius, Eur. J. Inorg. Chem., 2022, e202200416 CrossRef CAS; (j) L. Tendera, F. Fantuzzi, T. B. Marder and U. Radius, Chem. Sci., 2023, 14, 2215–2228 RSC; (k) L. Tendera, L. Kuehn, T. B. Marder and U. Radius, Chem.–Eur. J., 2023, 29, e202302310 CrossRef CAS PubMed; (l) M. S. Luff, C. Kerpen, J. A. P. Sprenger, M. Finze and U. Radius, Inorg. Chem., 2024, 63, 2204–2216 CrossRef CAS PubMed; (m) M. S. Luff, L. Walther, M. Finze and U. Radius, Dalton Trans., 2024, 53, 5391–5400 RSC.
- (a) T. Schaub and U. Radius, Chem.–Eur. J., 2005, 11, 5024–5030 CrossRef CAS PubMed; (b) P. Fischer, T. Linder and U. Radius, Z. Anorg. Allg. Chem., 2012, 638, 1491–1496 CrossRef CAS; (c) J. H. J. Berthel, L. Tendera, M. W. Kuntze-Fechner, L. Kuehn and U. Radius, Eur. J. Inorg. Chem., 2019, 3061–3072 CrossRef CAS; (d) L. Kuehn, D. G. Jammal, K. Lubitz, T. B. Marder and U. Radius, Chem.–Eur. J., 2019, 25, 9514–9521 CrossRef CAS PubMed; (e) J. H. J. Berthel, M. J. Krahfuß and U. Radius, Z. Anorg. Allg. Chem., 2020, 646, 692–704 CrossRef CAS.
- Anamika, D. K. Yadav, K. K. Manar, C. L. Yadav, K. Kumar, V. Ganesan, M. G. B. Drew and N. Singh, Dalton Trans., 2020, 49, 3592–3605 RSC.
- (a) L. A. Freeman, A. D. Obi, H. R. Machost, A. Molino, A. W. Nichols, D. A. Dickie, D. J. D. Wilson, C. W. Machan and R. J. Gilliard, Chem. Sci., 2021, 12, 3544–3550 RSC; (b) N. Kuhn, G. Weyers, S. Dümmling and B. Speiser, Phosphorus Sulfur Silicon Relat. Elem., 1997, 128, 45–62 CrossRef CAS.
- G. J. Martin, J. P. Gouesnard, J. Dorie, C. Rabiller and M. L. Martin, J. Am. Chem. Soc., 1977, 99, 1381–1384 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc. Perk. Trans., 1987, 2, S1–S19 Search PubMed.
- (a) B. S. Lim, D. V. Fomitchev and R. H. Holm, Inorg. Chem., 2001, 40, 4257–4262 CrossRef CAS PubMed; (b) M. C. Aragoni, C. Caltagirone, V. Lippolis, E. Podda, A. M. Z. Slawin, J. D. Woollins, A. Pintus and M. Arca, Inorg. Chem., 2020, 59, 17385–17401 CrossRef CAS PubMed; (c) Z. M. Wong, X. Yong, T. Deng, W. Shi, G. Wu, N. Li, H.-K. Luo and S.-W. Yang, J. Phys. Chem. A, 2022, 126, 5552–5558 CrossRef CAS PubMed; (d) K. Youssef, C. Poidevin, A. Vacher, A. Fihey, Y. Le Gal, T. Roisnel and D. Lorcy, Dalton Trans., 2024, 53, 9763–9776 RSC.
- (a) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362–3375 CrossRef CAS; (b) U. S. D. Paul and U. Radius, Organometallics, 2017, 36, 1398–1407 CrossRef CAS; (c) U. S. D. Paul, M. J. Krahfuß and U. Radius, Chem. unserer Zeit, 2019, 53, 212–223 CrossRef CAS.
- (a) M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422 CrossRef; (b) A. Vogler and H. Kunkely, Comments Inorg. Chem., 1990, 9, 201–220 CrossRef CAS; (c) A. Vogler and H. Kunkely, Coord. Chem. Rev., 2007, 251, 577–583 CrossRef CAS.
- (a) M. M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC; (b) M. Ju, J. Kim and J. Shin, Bull. Korean Chem. Soc., 2024, 45, 835–862 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr Franc Meyer and Prof. Dr Christian Limberg on the occasion of their 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2409878–2409888. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08449g |
| This journal is © The Royal Society of Chemistry 2025 |