
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNoble-metal-free catalysts for the oxygen evolution reaction in acids
Junwei
Han
a,
Qian
Liu
 a,
Yue
Yang
a and
Hao Bin
Wu
*ab
a,
Yue
Yang
a and
Hao Bin
Wu
*ab
aSchool of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China
bState Key Laboratory of Silicon and Advanced Semiconductor Materials, Zhejiang University, Hangzhou 310027, China. E-mail: hbwu@zju.edu.cn
First published on 5th February 2025
Abstract
Oxygen evolution catalysts are critical components of proton exchange membrane water electrolysers (PEMWEs), playing a decisive role in determining both the performance and cost of these devices. Non-noble metal-based oxygen evolution catalysts have recently drawn significant attention as potential alternatives to expensive noble metal catalysts. This review systematically summarizes the mechanism of non-noble metal catalysts for the oxygen evolution reaction in acids with respect to their activity and stability, incorporating theoretical calculations and the Pourbaix diagram. Advanced in situ techniques are highlighted as powerful tools for probing intermediate evolution and valence changes and further elucidating the catalytic mechanism. Furthermore, key strategies for enhancing catalytic activity and durability, such as elemental doping, the support effect, surface protection and novel phase design, are discussed. Finally, this review provides insights into the remaining challenges and emerging opportunities for advancing practical oxygen evolution catalysts in PEMWEs.
1. Introduction
In light of the growing global energy demand and the depletion of conventional energy sources, the world is now confronted with a significant challenge—the development of sustainable energy.1 Hydrogen energy possesses various advantages including high energy density and zero emissions, which enables large-scale and environmentally friendly utilization.2 Compared with other hydrogen production methods such as photolysis, gas reformation and biomass gasification, water electrolysis has gained significant attention for its cleanliness, compactness, and flexibility.3 Noteworthily, a proton exchange membrane water electrolyser (PEMWE) operating under acidic conditions shows advantages of high H2 purity and high current density compared to other electrolysers.4 Meanwhile, the anodic four-electron-transfer OER exhibits sluggish kinetics in a H+ concentrated environment. Considering the local acidic environment resulting from the use of a perfluorosulfonic acid (PFAS) membrane, a PEMWE places higher demands on the acid resistance of an anode catalyst.5 Hence, developing highly active and stable OER catalysts has become an urgent need for large-scale application of the PEMWE.6Among the OER catalysts for PEMWEs, IrO2 (ref. 7 and 8) and RuO2 (ref. 9–11) present ultra-high activity and decent stability for the OER under acidic conditions, and are yet limited by the scarcity and high cost of noble metal resources. Therefore, the exploration of noble metal-free catalysts for acidic OER has gained significant attention.12,13 Numerous noble metal-free alternatives have been proposed, including transition metal oxides,14 nitrides,15 phosphides,16 sulfides,17 and other complex compounds (such as polyoxometalates,18 phosphates,19 and metal–organic frameworks20). Compared with noble metal catalysts, it is even more challenging to achieve both high activity and durability for noble metal-free OER catalysts.21 Despite some promising candidates and modification strategies reported so far, systematic and comprehensive guidance for the development of high-performance acidic OER catalysts has not been well established.22 Thanks to the rapid advances in computational materials science, high-throughput material screening that points out future directions has become possible.23
In this review, we summarize the development of OER catalysts operating under acidic conditions, which could potentially be used for PEMWEs and other related applications. Starting from the mechanism, this review introduces the widely accepted adsorbate evolution mechanism (AEM) and lattice oxygen mechanism (LOM), as well as the nascent oxide path mechanism (OPM) and proton donor–acceptor mechanism (PDAM). Then, by incorporating high-throughput computational approaches, the calculated Pourbaix diagrams provide guidance for the screening of acid-stable catalysts within the anodic potential window. Advanced in situ characterization methods as powerful tools to analyze the catalytically active sites and the reaction mechanism have also been summarized. On the basis of mechanism analysis and theoretical calculations, experimental strategies to develop advanced noble metal-free catalysts for acidic OER are classified, such as elemental doping, the support effect, surface protection and new phase design, especially in cobalt- and manganese-based oxide systems. Finally, bottlenecks and future directions in the search for highly active and durable noble metal-free catalysts are discussed.
2. Proton exchange membrane water electrolyzers, thermodynamics, Pourbaix diagrams and in situ/operando techniques
First, the basic structure and challenges of proton exchange membrane water electrolyzers (PEMWEs) are introduced. The traditional thermodynamic mechanisms of the OER on the oxide surface include the AEM and LOM, while alternative mechanisms such as the OPM and PDAM have been proposed recently. In addition, the Pourbaix diagram is widely adopted to explain the catalyst stability during the OER in acids. Along with these theoretical approaches, in situ/operando characterization techniques are indispensable to reveal the evolution of catalytically active sites and shed light on the microscopic reaction mechanism.2.1 Proton exchange membrane water electrolysers
The basic structure of PEMWEs is the membrane electrode assembly (MEA) connected in between monopolar/bipolar plates.24 The MEA is the core component of PEMWEs where the electrochemical reactions (OER/HER) occur, while monopolar/bipolar plates carry current/heat and transport reactants/products from MEA.25 The MEA is an assembled stack of two electrodes and a proton exchange membrane, and the catalyst layers are generally coated on both sides of the membrane and sandwiched between two porous transport layers (PTLs). The choices of PTLs are typically platinized titanium felt for the anode and carbon paper for the cathode, allowing facile liquid/gas transport and electrical connection.26 Such a design helps to mitigate the energy penalties related to electronic/ionic conduction and mass transport, resulting in rapid electrochemical reactions at high current density.For the PEMWE system, the deviation of the cell voltage from the thermodynamic value originates from three sources: the activation overpotentials from electrochemical reactions, ohmic overpotentials associated with proton and electron conduction, and concentration overpotential due to mass transport limitation.27–29 The polarization of anodic reactions (OER) represents a major source of the voltage penalty due to the sluggish four-electron water oxidation process.30 In terms of the techno-economic consideration of PEMWEs, the Ir-based OER catalyst typically accounts for around 25% of the total cost of the MEA in commercial PEMWEs due to its high cost (∼174 $ per g, year of 2024) and high mass loading (∼4–12 g m−2).31,32 Therefore, searching for highly active and low-cost acid OER catalysts has been a major task in the field.
2.2 Thermodynamic mechanism of the OER in acids
The AEM was originally proposed by Rossmeisl et al.,33–36 who used density functional theory to calculate the binding energy of surface adsorbed intermediates as a criterion of catalyst activity. As shown in Fig. 1a, the AEM undergoes a proton–electron coupling process, involving three intermediates OH*, O*, and OOH* generated by the evolution of water molecules adsorbed at active centers. Specifically, in an acidic system, the water molecule is first adsorbed on the metal active site and undergoes a dehydrogenation step to produce the M–OH* species; the M–OH* species undergoes another dehydrogenation step to produce the M–O* species, which then binds to another water molecule to produce M–OOH* species; finally, another dehydrogenation reaction is carried out to finish the cycling with the breaking of the M–O bond and the release of O2. However, according to the AEM theory, the binding energies of M–OH* and M–OOH* intermediates have been constrained by a scaling relationship. Specifically, the gap between the adsorption energies of OH* and OOH* is theoretically fixed as 3.2 eV, independent of the binding energy of M–O*, which provides an immutable overpotential of the OER catalysts obeying the AEM theory.37 However, many catalysts have been experimentally observed to exhibit lower overpotentials that overcome the limit proposed by the AEM theory, suggesting alternative reaction pathways.38 Meanwhile, in oxide-based catalyst systems,39,40 the generation of surface lattice oxygen and oxygen vacancies provides a new perspective to explain the OER with higher activity, and thus the LOM was established. | ||
| Fig. 1 Schematics of OER mechanisms: the (a) adsorbate evolution mechanism (AEM), (b) lattice oxygen mechanism (LOM), (c) oxide path mechanism (OPM), and (d) proton donor–acceptor mechanism (PDAM). | ||
The LOM is not thermodynamically favorable as it involves the breaking of surface lattice metal–oxygen bonds.41 However, the involvement of lattice oxygen can be promoted by modulating the catalyst surface electronic structure and increasing the overlap between the 3d orbitals of metal and the 2p orbitals of oxygen.42 As shown in Fig. 1b, a typical LOM undergoes a similar process to the AEM until the formation of M–O*. In the LOM pathway, M–O* species directly combine with lattice oxygen in the crystal structure to release O2, generating an oxygen vacancy (VO); subsequently, another H2O molecule is adsorbed and deprotonated to fill the oxygen vacancy. The proposed LOM breaks the limitation caused by the scaling effect of OH* and OOH* species in the AEM and satisfactorily explains the experimentally observed enhanced catalytic activity. However, the involvement of lattice oxygen further induces the dissolution of transition metals and compromises the structural stability of catalysts.43
Overall, the LOM and AEM benefit the activity and stability of OER catalysts, respectively.44 However, the above two mechanisms are not well adapted to catalysts with low overpotential and high stability, and thus the oxide path mechanism (OPM) and proton donor–acceptor mechanism (PDAM) have been recently proposed.45 Unlike the AEM and LOM in which the electrooxidation of water molecules occurs at a single active site, the OPM and PDAM are based on the synergy of two adjoining catalytically active sites. The OPM (Fig. 1c) begins with water adsorption and dehydrogenation to produce an M–OH* intermediate, which is subjected to direct coupling with another M–OH* intermediate to produce O2.14 Catalysts with high density of metal active sites would theoretically benefit the OPM that involves the coupling step of two intermediates. Since the OPM does not involve the generation of M–OOH* intermediates, the typical scaling limitation in the AEM does not apply to the OPM, which potentially warrants the OER with a lower overpotential. In terms of stability, lattice oxygen does not participate in the OER through the OPM, which favors the stabilization of catalysts. Similar to the OPM, the PDAM (Fig. 1d) also relies on the formation of M–OO* intermediates. However, the two active centers in the PDAM are typically different atoms, with one serving as the primary reaction center and the other functioning as an indirect center to accept protons.46 The PDAM emphasizes the asynchronous transfer of protons and electrons between these distinct active centers, often facilitated by electron modulation induced by oxygen or metal vacancies. Compared to the AEM and LOM, the OPM and PDAM theoretically promise OER catalysts with both high stability and intrinsic activity. However, how to design OER catalysts that catalyze water oxidation through a desirable mechanism and how to experimentally verify the reaction mechanisms remain key challenges in the field.
2.3 Stability of catalysts and the Pourbaix diagram
Most noble-metal-free OER catalysts are unstable in acidic electrolytes. Searching for potential catalysts with high stability under acidic OER conditions (e.g., pH < 3 and potential > 1.23 V vs. RHE) has become the research priority.47 The Pourbaix diagram, also known as a potential–pH diagram, is an electrochemical equilibrium diagram illustrating stable species or the equilibrium state of a system in a specific potential range and pH environment. The Pourbaix diagram can conveniently reflect the possibility of corrosion/dissolution of metals and metal compounds from a thermodynamic perspective, and is thus used to screen candidates as stable acidic OER catalysts.48 Combining the Gibbs free energy gap (ΔGpbx) under specific pH–potential conditions, the calculated band gap (Eg) for considering the electronic properties, and convex hull (Ehull) for estimating the phase stability, 68 possible acid-stable candidates for oxygen evolution were identified.23Co-based and Mn-based oxides such as Co3O4 and MnO2 are promising OER catalysts, which however exhibit distinct Pourbaix diagrams.49 For Co3O4, the high potential and low pH range only allow the stable existence of Co2+ species,50 and the stability under specific conditions can be regulated by introducing other elements to change the phase region distribution in the Pourbaix diagram. For example, the Pourbaix diagrams of La-modified and pristine Co3O4 (ref. 51) showed the pH–potential effect on the form of elements present on the surface and the hydroxyl stabilization effect. It is generally believed that Mn-based catalysts present better stability in acidic OER, attributed to the fact that MnO2 can remain stable under pH < 2 conditions and in the 1.2 to 1.8 V potential window according to the Mn–H2O Pourbaix diagram.52 Nevertheless, as a thermodynamic steady-state methodology, the Pourbaix diagram cannot reflect the kinetic factors in acidic OER.
The stability of non-noble metal catalysts has been a key challenge in the field, which is generally associated with dissolution of metal ions at high voltages. Although the Pourbaix diagram provides a thermodynamic prediction of stability, the degradation of catalysts is rather complicated, and is related to the catalytic mechanism and the structure of catalysts. Several strategies have been proposed to improve the stability of acidic OER catalysts, including the enhancement of metal–oxygen bonding, interfacial protection, crystallinity design, etc.49 Moreover, catalysts may exhibit different degradation behaviors in MEA-based electrolyzers compared to liquid electrochemical cells. Therefore, effective performance assessment protocols are highly needed to bridge the gap between in-lab catalyst development and practical application scenarios.
2.4 In situ and operando characterization techniques
Monitoring catalyst evolution during electrochemical reactions is of great importance to understand the catalytic mechanisms, which relies on various characterization techniques.53,54Ex situ techniques analyze the difference between pre- and post-reaction samples, while in situ techniques monitor real-time reaction processes. Notably, real-time monitoring of structure changes55 and reaction intermediates56 can be accomplished by in situ/operando characterization techniques. Compared with in situ techniques that provide on-site monitoring under specific reaction conditions, operando techniques focus on real-time monitoring during the reaction. Several in situ and operando characterization techniques have been widely used in the studies of the OER, such as synchrotron X-ray absorption spectroscopy (XAS), Raman spectroscopy, Fourier-transform infrared spectroscopy (FTIR) and differential electrochemical mass spectrometry (DEMS), dedicated to determine the true active species and reaction intermediates.57Raman spectroscopy is based on the Raman scattering phenomenon. During the Raman measurement, a small fraction of incident laser photons experience inelastic scattering and transfer energy to the tested sample, leading to vibrational or rotational transitions of chemical bonds.58 The frequency gap between scattered light and incident light known as a Raman shift is related to many chemical characteristics such as molecular structures, chemical bonding and lattice vibration.59 By measuring the Raman shift and intensity of Raman scattered light, species transformation and interfacial evolution during electrochemical reactions can be monitored in real time. Raman spectroscopy can not only detect the behavior of water molecules,55 but also the formation of metal–oxygen bonds14 at low chemical shifts during the catalytic process. Taking the Raman spectrum of Co3O4 as an example, typical Raman peaks at 480 cm−1 (Eg), 520 cm−1 (F2g), 620 cm−1 (F2g), and 690 cm−1 (A1g) can be probed. During an in situ Raman measurement, the full width at half maximum (FWHM) of the A1g peak increases with operating time, attributed to the formation of an amorphous hydrated layer on the surface of Co3O4 that hinders the focusing of the laser light.60 The characteristic Raman peaks of Co3O4 can also be seen in the composite of Co3O4/CeO2.61 As the potential increased, another Raman signal at 600 cm−1 attributed to CoOOH species can be observed at 1.2 V (vs. RHE) and gradually disappears after reaching the OER onset potential (Fig. 2a). Meanwhile, the A1g Raman peak was found to be red-shifted after reaching the OER onset potential, which was ascribed to Co4+ species (Fig. 2b). Thus, the in situ Raman spectroscopy measurement suggests that high-valence Co species rather than CoOOH might be the catalytically active species for acidic OER.
 | ||
| Fig. 2 (a) In situ Raman spectra and (b) Raman A1g peaks of Co3O4 and Co3O4/CeO2 at various constant potentials (vs. RHE). Reproduced with permission.61 Copyright 2021, Springer Nature. (c) Scheme of an in situ XAS instrument. Reproduced with permission.62 Copyright 2023, Elsevier. (d) Fluorescence XANES spectra and (e) R-space EXAFS spectra collected at the Co K-edge. Reproduced with permission.51 Copyright 2023, The American Association for the Advancement of Science. Operando differential electrochemical mass spectrometry signals of O2 products for (f) Co3−xBaxO4 and (g) Co3O4. Reproduced with permission.14 Copyright 2023, American Chemical Society. | ||
Raman spectroscopy has been used to study various cobalt oxide-based OER catalysts. The Raman spectrum of Ba-doped Co3O4 (Co3−xBaxO4) showed a peak at 456 cm−1 at 1.45 V (vs. RHE), attributed to the formation of OH* on the surface.14 Liu et al.56 investigated a CeO2-loaded CoNiPOx (CeO2/Co2NiP0.03Ox) hetero-structured catalyst using Raman spectroscopy, which exhibited two weak peaks at 465 and 525 cm−1 at 1.2 V (vs. RHE), ascribed to O–Ce–O and CoII–O bonds, respectively. Compared with Co2NiP0.03Ox, CeO2/Co2NiP0.03Ox manifested the CoIII–O bonding signal at a lower potential of 1.2 V (vs. RHE), suggesting a transition from CoII to CoIII during acidic OER. In addition to Co3O4, CoO2 is also proved to be active during acidic OER and stable at high potential according to the Pourbaix diagram. Zhang et al.63 prepared Co loaded MnO2 by reacting MnO2 with molten cobalt salt and tested it in an electrolyte containing Co2+ to ensure the stable deposition of CoO2 during electrolysis. The 90-Co–MnO2 catalyst was characterized by in situ Raman spectroscopy over a potential range of 0 to 1.8 V (vs. RHE) in 0.1 M HClO4 containing 2.4 mg per mL Co2+. Two peaks corresponding to MnO2 were observed at 624 cm−1 and 579 cm−1, and a peak associated with CoOOH was observed at 497.8 cm−1 within the potential range of 1.2–1.7 V (vs. RHE). CoOOH was then converted to CoO2 above 1.7 V, as indicated by the peak at 460.9 cm−1. Although in situ Raman spectroscopy enables the real-time monitoring of the evolutionary process during operation, the weak signal intensity of intermediates highlights the need for advanced spectroscopic techniques with higher detection sensitivity, such as surface-enhanced Raman spectroscopy (SERS), tip-enhanced Raman spectroscopy (TERS), etc.64
X-ray absorption spectroscopy (XAS) is a characterization technique based on the interaction of materials and high-energy X-rays, such as those generated by synchrotron light sources65 (Fig. 2c). By analyzing the scattering, absorption and emission phenomena, detailed information about the local atomic arrangement can be obtained, including bond lengths, coordination numbers and so on. When the X-ray energy matches the binding energy of electrons in the irradiated sample, resonance absorption occurs, causing a sudden increase in X-ray absorption, referred to as the adsorption edge. This adsorption edge gives rise to the X-ray absorption near edge structure (XANES), which provides insights into the electronic configuration and local atomic structure around the absorbing centers. XANES allows determination of valence states, d-band properties, orbital hybridization, symmetry, and other structural information. At higher X-ray energies, absorbing-center electrons are excited to continuum states, creating wave interference patterns known as extended X-ray absorption fine structures (EXAFS), providing information on the local atomic arrangement and coordination numbers surrounding the absorbing centers.
Chong et al.51 investigated a self-supported LaMn-doped ZIF-67 fiber (LMCF) catalyst using XAS. In situ XANES of the Co K-edge showed a slight red-shift and reduced intensity as the potential increased, indicating a lower oxidation state in LMCF with a smaller O coordination number (Fig. 2d). At the same time, the enhanced intensity of the 1s–3d orbital transition peak suggests a less symmetric coordination environment for cobalt of LMCF, verifying oxide lattice distortion caused by oxygen vacancies in the lattice structure (Fig. 2e). The higher white line intensity of La in LMCF than that of La2O3 suggests a higher coordination number of La in LMCF. Similarly, Wang et al.62 analyzed the structural stability of FeCoSbOx using the in situ XAS technique. They observed that the Co pre-edge intensity of FeCoOx increased with higher potentials, whereas that of FeCoSbOx remained almost unchanged up to 1.8 V (vs. RHE). This suggests that Sb doping alleviated structural distortion during operation.
In situ differential electrochemical mass spectrometry (DEMS) utilizes mass spectrometry to identify species with different m/z ratios and their relative abundances, providing insights into reaction intermediates and pathways.66 For example, when evaluating Ba-doped Co3O4 in H218O and H216O electrolytes, Co3−xBaxO4 consistently produced 32O2, 34O2, and 36O2 during each LSV cycle (Fig. 2f), while Co3O4 produced only 34O2 and 36O2 (Fig. 2g), supporting the proposed OPM mechanism.14
Advancements in advanced spectroscopy and analytical techniques have significantly enhanced the ability to explore reaction intermediates, active sites and real-time reaction pathways in catalytic reactions. In situ Raman spectroscopy is mainly applied to detect chemical structure changes on electrode surfaces during reactions, and is particularly sensitive to oxygen reaction intermediates. In situ XAS collects bulk information of the catalysts and reflects changes in the overall valence state and coordination environment. In situ DEMS provides real-time monitoring of ion dissolution, offering experimental information for catalyst failure analysis. The combination of various in situ characterization techniques is essential for providing a comprehensive understanding of the catalytic process. Nevertheless, experimental confirmation of the catalytic mechanism with both high spatial and temporal resolution remains highly challenging. Therefore, developing in situ/operando techniques is essential for monitoring critical intermediates, understanding the reaction processes, and further elucidating the acidic OER catalytic mechanism.
3. Transition metal oxides as OER catalysts in acids
First-row d-block metal oxides, especially Co and Mn oxides, have been widely investigated as promising noble-metal-free OER catalysts. The intrinsic activity and catalyst evolution of Co-based and Mn-based oxides as acid OER catalysts with various modification methods, such as atomic doping, the support effect, protection coating and phase design, have been studied.3.1 Co-based oxides
 | ||
| Fig. 3 (a) Crystal structure of Co3O4. Reproduced with permission.60 Copyright 2021, American Chemical Society. (b) In situ Raman spectra of Co3O4, Co3O4-VO and Co3O4-VCo in the voltage window from 0.9 to 1.55/1.6 V (vs. RHE). Reproduced with permission.38 Copyright 2023, American Chemical Society. (c) Time-resolved in situ confocal Raman spectra of Co3O4 at 1.72 V (vs. RHE) and (d) the LOER mechanism and the degradation route of Co3O4 during LOER. Reproduced with permission.60 Copyright 2021, American Chemical Society. | ||
Determining the surface structure of the catalyst under operating conditions is the key to analyze the catalytic activity. A recent study by Zhang et al.38 investigated the vacancy-induced effect on the evolution of Co3O4 during the oxygen evolution process by constructing Co and O vacancies. Co3O4 undergoes a two-step reconstruction during the OER process, from oxide to a hydroxide analogue and then to an oxyhydroxide analogue, which is accompanied by adsorption of hydroxyl and deprotonation. The potentials at which the Eg peaks appear or disappear in Raman spectroscopy demonstrate the difference in hydroxyl adsorption and deprotonation capacities of various structures. It has been found that O vacancies lead to easier hydroxyl adsorption and Co vacancies lead to easier deprotonation (Fig. 3b). Moreover, Co vacancies contribute to a shorter Co–Co distance, which show the highest OER activity.
Similar to studies on the alkaline OER, Natarajan et al.60 synthesized Co3O4 with different types of defects and investigated the reconstruction of surface hydrous oxide layers under acidic conditions. Their findings showed that Co3O4 with higher crystallinity tends to form a thinner hydrous oxide layer during the OER. By in situ Raman analysis (Fig. 3c), the hydrous oxide layer thickens with operation time, and a direct correlation was also found between the ratio of Co(III)/Co(VI) and layer thickness. However, a thick surface hydrous oxide layer does not promote the catalytic activity but leads to the deactivation of the catalyst due to the loss of contact and degradation of active surface sites. The dissolution of Co species is closely linked to the LOM. This suggests that with the formation of oxygen vacancies around the metal sites, H2O would possibly occupy the oxygen vacancies and form soluble high-valent Co hydrates (Fig. 3d).
Another important issue is the effects of an acidic environment on catalyst stability and activity. The study by Huang et al.61 shed light on this issue by testing the Co3O4 and Co3O4/CeO2 catalysts in H2SO4 solutions with different pH values. LSV curves show that the catalyst activity is less impacted by the concentration of protons. Therefore, when developing OER catalysts for acidic systems, stability might be the first consideration rather than activity. Over the past decade, many studies have provided insights into stability issues67 in view of the degradation mechanism, and various strategies have been conducted including atomic doping, constructing hetero-structures, surface engineering and phase design. The following sections will focus on the modulation of Co3O4 catalysts to improve catalyst activity and stability.
Nwanebu et al.70 prepared NixCo1−x–oxide catalysts on titanium plates by direct nitrate pyrolysis and investigated the effects of Ni/Co ratio on the catalytic OER performance. When the Ni molar percentage was lower than 50%, the crystal structure was dominated by the Co3O4 phase, and vice versa by the NiO phase. Meanwhile, the catalytic performance showed a volcano distribution since Ni doping induces the formation of high-valence active Co species whereas excessive Ni results in NiO with low OER activity. Wang et al.14 synthesized a Ba-doped Co3O4 catalyst (Co3−xBaxO4) through facile electrodeposition and heat treatment, which exhibited an overpotential of 278 mV at 10 mA cm−2 in 0.5 M H2SO4 (Fig. 4a) and stable operation for over 110 h. Through Ba doping, the Co–Co bond length was shortened, creating conditions for direct dehydrogenation O–O coupling (Fig. 4b). The in situ synchrotron FT infrared (FTIR) spectra directly detected the generation of O–O bonds, which is consistent with the oxygen bridge intermediate in the OPM (Fig. 4c). DFT calculations showed that the O–O coupling step was considered as the rate determining step which promoted the OPM pathway under acidic conditions. Chong et al.51 synthesized a Mn/La co-doped Co3O4 fiber catalyst (LMCF) using zeolitic imidazolate frameworks (ZIF-67) as a precursor through electrospinning and annealing treatment. The doping of Mn cations inside the ZIF lattice triggered a higher valence of Co active sites and shorter Co–Co band length, which contribute to promoted acidic oxygen evolution activity. Meanwhile, the La cations with strong affinity to *OH groups concentrated at the surface of cobalt oxide, provide excellent acid-proof ability (Fig. 4d). The Pourbaix diagrams of different crystal planes such as (111), (110) and (100) in La-modified and pristine Co3O4 were calculated and compared as shown in Fig. 4e. Among the five regions, regions I and II contain La ions, so the structure is unstable and soluble. Regions III, IV, and V exhibit relative stability due to the presence of Co2+, Co3+, and La3+ surfaces connected with OH*, OOH*, and O*. The LMCF catalyst was evaluated in a PEMWE under potentiostatic conditions, demonstrating stable operation over 100 h at a cell voltage of 1.65 V (Fig. 4f). Doping Co3O4 with Fe69 and Ag75 has also been proven to improve the catalytic activity and durability of acidic OER.
 | ||
| Fig. 4 (a) LSV and (b) in situ EXAFS spectra of the Co K-edge of Co3−xBaxO4 and (c) the OPM vs. the AEM for catalysts in acidic electrolytes. Reproduced with permission.14 Copyright 2023, American Chemical Society. (d) STEM image and the corresponding La distributions (scale bar, 2 nm) of LMCF and the surface (e) Pourbaix diagram for the La-doped Co3O4 (111) facet obtained from the DFT+U calculations. (f) Potentiostatic measurement of a PEMWE using the LMCF catalyst at 1.65 V. Reproduced with permission.51 Copyright 2023, The American Association for the Advancement of Science. | ||
Doping Co oxides with non-metal elements such as carbon, nitrogen, sulfur and phosphorus would have profound effects on the catalytic activity and stability. Yang and co-workers73 demonstrated nanostructured Co3O4/CN deposited on carbon paper as a self-standing electrode using ZIF-67 as a precursor. FT-EXAFS shows that Co–O/N was established in the Co3O4/CN structure and the doping of N induced O vacancies by partially replacing lattice oxygen. In addition, the nitrogen-doped carbon structure improves the acid resistance of Co3O4, which guarantees stability for 80 h at 10 mA cm−2 in 0.5 M H2SO4. Shang et al.74 used the solvothermal method to introduce P into the Co3O4 lattice, where P5+ replaces Co3+ located in the 16d sites with an octahedral coordination. The unique PO6 units increased the percentage of Co2+ in the Co3O4 lattice, which suppresses the leaching of Co ions and increases the stability during acidic OER.
Constructing a protective surface layer has been another widely used strategy to mitigate the performance degradation caused by catalyst detachment as well as direct proton attack on metal–oxygen bonding in strong acidic environments.81 Carbon coatings, especially highly graphitized carbon, have been considered as preferred choices due to their high conductivity, decent stability and convenient preparation, despite the possible corrosion at high potential. Lai et al.82 prepared carbon protected Co3O4 by spraying cobalt nitrate on carbon paper with a gas diffusion layer fixed on a hot plate. The carbon coated Co3O4 catalyst achieved an overpotential of 460 mV for more than 80 hours in 0.5 M H2SO4. Yang et al.83 also reported a Co3O4 catalyst with a 3.5 nm thick carbon layer, synthesized using glucose as the carbon source. LSV curves demonstrated the negative impact of the carbon layer on catalytic activity, with the overpotential increased by 150 mV. However, the durability of the catalyst improved, with the duration time extending by 20% to 86.8 h at 10 mA cm−2. The incorporation of carbon into electrocatalysts or electrodes might also modulate hydrophobicity, which could help to prevent excessive solvation of metal oxides, thus minimizing their dissolution. For example, Yu et al.84 synthesized a nano-Co3O4@C composite, and introduced graphite and paraffin oil during electrode preparation. This process created partially hydrophobic zones that effectively suppressed the dissolution of active Co3O4.
Apart from carbon coating, acid stable metal oxides such as TiO2 (ref. 76) and F doped tin oxide (FTO)85 have also been investigated as coating materials. Tran-Phu et al.76 explored the effect of a TiO2 coating layer on the catalytic properties of Co3O4. Coating layers with a suitable thickness would contain pitting channels, which maintain a balance between protection and exposure of the catalytic surface. Yeh et al.85 synthesized FTO coated and loaded Co3O4 samples with a low Co3+/Co2+ ratio and oxygen vacancies, exhibiting enhanced stability.
Selection of materials with good electrical conductivity, acid resistance and mechanical strength is critical for effective coating strategies. However, achieving both high acid resistance and electrical conductivity is challenging due to the limitation of intrinsic properties of available materials. As a result, catalysts designed through coating strategies often require compromises in catalytic activity. Thus, maintaining both catalytic activity and stability presents a significant challenge in these approaches. Besides, the structure and OER mechanism of the active sites at the coating interface remains unclear and requires further investigation.
According to the Pourbaix diagram, Sb2O5 is an acid-resistant and potential-endurance oxide that can help stabilize active centers in an acidic medium during the OER.91 As a result, introducing Sb into Co-oxide systems as a stabilizer can extend the stability of catalysts. However, the poor electrical conductivity of metal–oxide catalysts may diminish the catalytic performance. Evans et al.92 synthesized rutile CoSb2O6via electrodeposition, demonstrating stability for over 24 h at a current density of 10 mA cm−2 in 0.5 M H2SO4. Wang et al.91 synthesized pyrochloryl Co2Sb2O7, which exhibited an ultra-low overpotential of 288 mV and stability for over 40 h at 1 mA cm−2. After the constant potential test, the XRD pattern showed peaks corresponding to tetragonal CoSb2O6, indicating the transformation of Co2Sb2O7 into CoSb2O6 during the OER process. The evolved CoSb2O6 acted as a stable protective layer to improve the stability.
However, the introduction of Ni into the CoSb2Ox system did not improve the performance, though the reasons have not been thoroughly discussed.88 Wang et al.62 prepared FeCoSbOx catalysts on nickel foam via direct pyrolysis, reducing the onset potential by 271 mV at a current density of 10 mA cm−2 compared with that of FeOx prepared by the same method (Fig. 5a). The in situ ICP-MS test revealed that Co dissolution of FeCoSbOx was significantly suppressed (Fig. 5b). X-ray absorption near-edge structure (XANES) spectral analysis indicated that Fe maintained a stable electronic structure, while the Co K-edge XANES peaks for CoOx, FeCoOx and FeCoSbOx electrodes shifted toward higher photon energies. The pre-edge peak, which represents structural symmetry, shifted notably for FeCoOx, whereas FeCoSbOx showed only a slight change. Overall, Sb contributed positively to structural symmetry and stability.
 | ||
| Fig. 5 (a) Scheme of the crystal structure of FeCoSbOx. (b) Scheme of in situ/operando ICP-MS analysis and Co dissolution profiles during cyclic voltammetry conducted by using an on-line ICP-MS. Reproduced with permission.62 Copyright 2023, Elsevier. (c) Long-term stability of Co2MnO4 during the OER in acid. (d) Two possible pathways of dissolution and correlation between experimental and theoretical stability. Reproduced with permission.71 Copyright 2022, Springer Nature. | ||
For inert components with poor conductivity, elements with catalytic activity and strong metal–oxygen bonds, such as Mn, are better choices for balancing stability and catalytic activity in acidic OER catalysts. Li et al.71 designed a spinel Co/Mn oxide film (Co2MnO4) by pyrolyzing nitrate at 300 °C on an FTO support, achieving 200 mA cm−2 for over 2000 h in H3PO4 electrolyte with pH = 1 (Fig. 5c). Bader charge analysis revealed enhanced electron transfer from Mn to O on the Co2MnO4 surface, confirming the formation of stronger Mn–O bonds (Fig. 5d). Additionally, it is worth noting that the type of electrolyte can have a huge impact on stability, likely due to the effect of anion adsorption on the electrode surface. Notably, in phosphate electrolytes, the stability of Co-based catalysts was significantly enhanced according to previous studies. Therefore, further emphasis should be placed on the study of catalyst–electrolyte interfaces.
3.2 Mn-based oxides
Manganese-based oxides are characterized by rich valences and variable structures, such as MnO2, Mn2O3, Mn3O4, etc.93 According to the Pourbaix diagram of Mn–H2O, MnO2 is the most stable phase in the OER potential range under acidic conditions, which makes it a promising candidate for acidic OER.49Differing superposition and connection of the basic repeating unit [MnO6] results in structural variants of MnO2,94 such as cryptomelanes (α-MnO2) with a 2 × 2 tunnel structure (Fig. 6a), pyrolusites (β-MnO2) with a 1 × 1 tunnel structure (Fig. 6b), birnessites (δ-MnO2) with a layered structure (Fig. 6c), and nsutite-type γ-MnO2 (Fig. 6d) composed of 1 × 1 and 1 × 2 tunnels. Hayashi et al.95 evaluated the OER activity of various MnO2 (δ-MnO2, α-MnO2, γ-MnO2, and β-MnO2) and ranked their OER activity in PEMWEs as α-MnO2 > β-MnO2 > δ-MnO2 > γ-MnO2. This difference in activity may stem from the specific adsorption of intermediates within the different-sized tunnels formed by the distinct arrangement of [MnO6] units. Kong et al.98 synthesized MnO2 by electrodepositing a manganese sulfate solution onto a Ti felt substrate and modulated the ratio of Mn–O structural units of the crystal structure through different heat treatment temperatures. As the heat treatment temperature increases, the proportion of planar oxygen increased, leading to enhanced catalyst stability. Pair distribution function analysis revealed that planar oxygen exhibits stronger Mn–O binding compared to pyramidal oxygen. This finding was also supported by DFT calculations, which showed that the dissolution from planar oxygen sites is energetically less favorable than that from pyramidal oxygen sites.
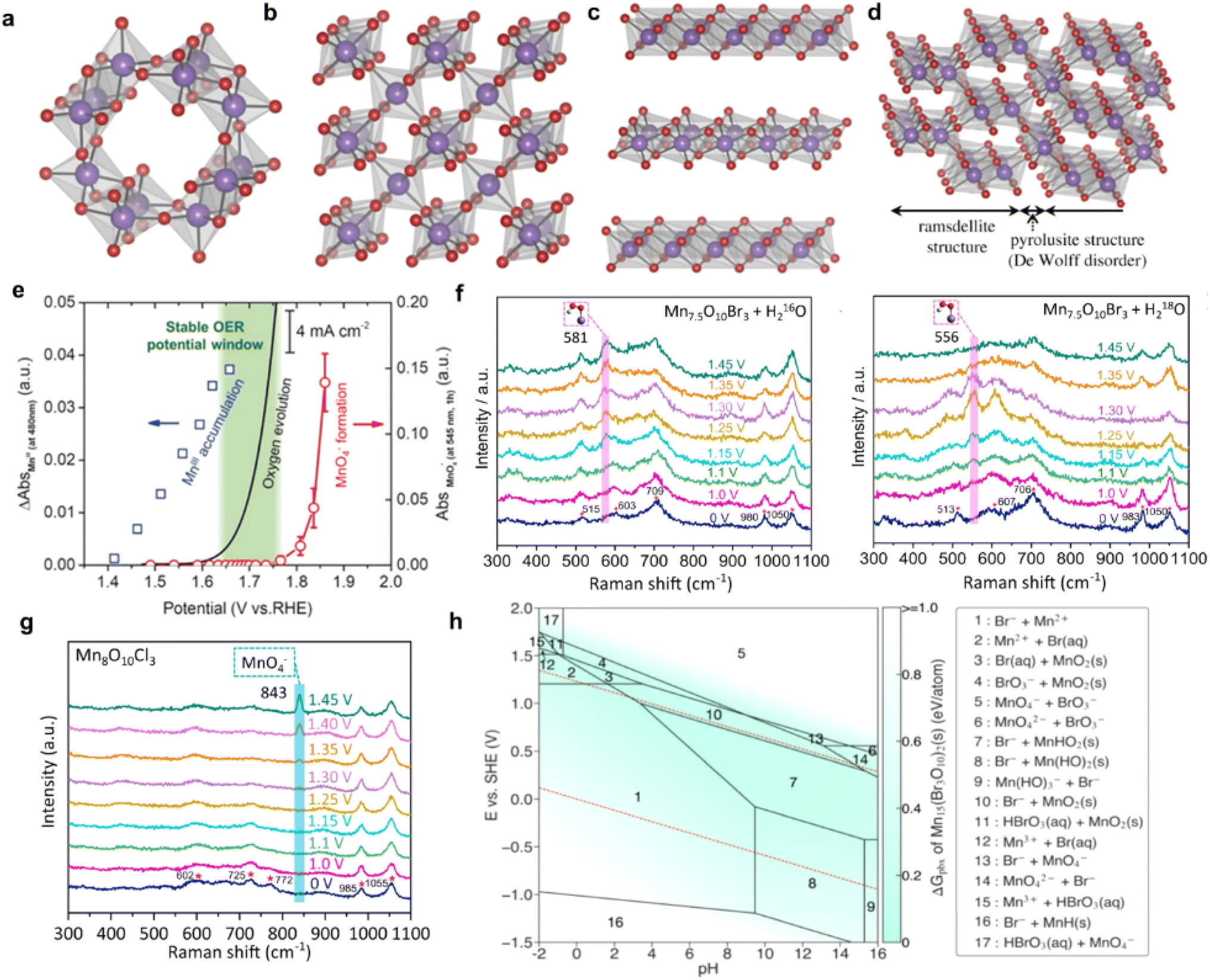 | ||
| Fig. 6 Structure of (a) α-MnO2, (b) β-MnO2, (c) δ-MnO2 and (d) γ-MnO2. Reproduced with permission.95 Copyright 2019, Royal Society. (e) Potential dependence of Mn3+ accumulation, oxygen evolution current, and MnO4− generation of γ-MnO2. Reproduced with permission.96 Copyright 2019, Wiley-VCH. In situ Raman spectra of (f) Mn7.5O10Br3 and (g) Mn8O10Cl3. Reproduced with permission.97 Copyright 2022, Springer Nature. (h) Calculated Mn–O–Br Pourbaix diagrams generated with an aqueous ion concentration of 10−4 M at 25 °C. Reproduced with permission.97 Copyright 2022, Springer Nature. | ||
Further studies have identified the high-spin electron configuration of Mn3+ as a critical active species that facilitates the formation and transformation of unstable Mn–O intermediate bonds. Yan et al.99 reported that the use of ionic liquid additives can induce the phase transition from β-MnO2 to α-MnO2, leading to alterations in both phase composition and morphology. TEM, EDX, and FTIR analyses collectively showed that the surface of α-MnO2 is coated with ionic liquid cations, which further adsorb anions, such as OH−. In contrast to Mn 2p, the analysis of Mn 3s peak splitting (ΔE3s) provides a more accurate characterization of the Mnn+ valence state. The peak area revealed a higher Mn3+/Mn4+ ratio in the ionic liquid-modified α-MnO2. Other than the impact of ionic liquid additives, variations in electrodeposition procedures might also affect the transformation of MnO2 during electrocatalysis. δ-MnO2 deposited via constant potential was transformed into the α-Mn3O4 phase after cathodization, and then into disordered δ-MnO2 through a multi-step anodic oxidation or oxygen generation process, which exhibited enhanced activity.100 XPS analysis shows a mixed Mn3+/Mn4+ valence state in the disordered δ-MnO2 phase, a result of defect generation during the spinel-to-layered structure conversion that stabilizes Mn3+. Li et al.101 employed the stochastic surface walking (SSW) pathway sampling method to elucidate the atomic-level mechanism underlying the transformation from α-Mn3O4 into δ-MnO2, and validated the role of defects in the phase transformation process through theoretical calculations. Li et al.96 investigated the stabilization voltage window of γ-MnO2 under acidic conditions, demonstrating that the catalyst dissolves into MnO4− when the voltage exceeds the stabilization voltage window. UV/vis absorption spectra showed that Mn3+ formed at 1.4 V during positive voltage sweeps, which catalyzed the onset of the OER at 1.6 V vs. RHE. As the potential reached 1.8 V, MnO4− was detected, suggesting that the dissolution of γ-MnO2 mainly produces MnO4− (Fig. 6e). Consequently, a stabilization window between 1.6 V and 1.75 V was established for γ-MnO2 during acidic OER. γ-MnO2 exhibited stability for over 1800 h at 10 mA cm−2 and 1.73 V, whereas the stability decreased to only 120 h at 100 mA cm−2 and 1.8 V.
The introduction of halogen atoms is an effective strategy to enhance the metallic bond strength and regulate the electronic structure of the active centers. By substituting oxygen atoms in MnO2 with halogen ions, such as F,102,103 Cl,97 or Br,97 halogen–Mn bonds are formed, which not only shift the d-band center but also create a stronger metallic bond with improved stability. Patel et al.103 prepared crystalline F-doped Cu1.5Mn1.5O4 nanoparticles by adding CuCl2 and NH4F to amorphous MnO2. A linear downshift of the d-band center was achieved with increasing F-doping, which improved the overall electrochemical catalytic activity. Ghadge et al.102 observed that fluorine doping in Mn0.8Nb0.2O2 induced a positive shift in the Nb 3d and Mn 2p peaks, corresponding to the formation of high-valence active sites. Lemoine et al.104 prepared a hydrated MnFe fluoride and found that the dehydrated MnFeF4.6O0.2 with an amorphous structure showed remarkable stability under highly acidic conditions. Pan et al.97 prepared a manganese oxy-bromide catalyst via pyrolysis of manganese bromide, achieving an OER overpotential of 295 mV at 10 mA cm−2 and stability for over 500 h. Comparative studies of halogen doping effects revealed notable differences. Isotopic analysis in H216O and H218O electrolytes showed a negative shift of 26 cm−1 at 1.25 V, indicating that the Mn–18O18OH intermediate is produced by the exchange of two 16O atoms (Fig. 6f). Raman spectroscopy of Mn8O10Cl3 displayed a distinct MnO4− peak at 843 cm−1 after reaching 1.35 V (vs. RHE), indicating catalyst degradation through the formation of MnO4− (Fig. 6g). Mn7.5O10Br3 and Mn8O10Cl3 can be stabilized by forming a MnOx (e.g., MnO2) passivation layer on the surface at potentials of 1.40–1.60 V (vs. RHE) under acidic conditions (pH = 0) (Fig. 6h).
Another common modification strategy involves incorporating inert, acid-resistant components into MnO2, such as PbO2,105,106 TiO2,107 and SiO2.108 However, this strategy typically reduces catalytic performance due to the poor electrical conductivity and non-catalytic nature of these acid-resistant substances. Fryfendal et al.107 prepared a multilayer MnO2 catalyst with surface-modified TiO2 by sputtering deposition and predicted the catalyst lifetime by a mass loss method. At 1.8 V, the expected lifetime of MnO2 was 140 h, whereas TiO2-modified MnO2 extended this to 265 hours. Directly combining acid-resistant elements with catalytically active elements to form new phases, rather than heterogeneous structures, has proven to be a superior modification method. For example, Moreno-Hernandez et al.109 synthesized a NiMnSb ternary rutile oxide that exhibited a stability of 168 h at 10 mA cm−2 due to the synergistic effect of the Ni–Mn elements and the stabilizing effect of Sb during acidic OER. Zhou et al.110 explored the origin of the catalytic activity in the rutile phase Mn–Sb–O and demonstrated that the Mn–Sb–O system stabilizes Mn3+. XPS results showed that the Mn valence remained between +2 and +3. Interestingly, increasing the Mn content in the Mn–Sb–O system elevated the valence state of Mn while that of Sb remained unchanged, suggesting that abundant Mn may facilitate the formation of oxygen vacancies or covalent Mn–O bonding. Ifkovits et al.111 coupled the OER with the Ce4+/Ce3+ redox reaction in the MnySb1−yOx system and found a similar correlation between Mn3+ concentration and catalytic activity in the Mn-rich system, with Tafel slopes up to 100 mV dec−1. Furthermore, an inverse relationship between catalytic activity and stability with increasing Sb content was observed.
4. Transition metal nitrides, sulfides and phosphides
In addition to oxides, transition metal nitrides, sulfides, and phosphides have also been considered as acidic OER catalysts with optimized electronic structures and acidic stability. Note that transition metal carbides112 have been generally considered unstable during acidic OER due to carbon corrosion. Lei et al.113 developed a self-supported iron nitride catalyst loaded on polyaniline-electrodeposited exfoliated graphene (FeN4/NF/EG) and explored the effect of different annealing temperatures on catalytic performance. Polyaniline provided attachment sites for the loaded iron nitride and also exhibited oxygen evolution activity. The catalyst stayed stable at a current density of 20 mA cm−2 for more than 24 h, which was attributed to the enhanced conductivity from graphitic N and highly active catalytic sites induced by pyridine N. Nitridation often modifies the valence states of active metal sites. Liu et al.114 synthesized a NiCo–nitride/oxide catalyst forming a heterogeneous structure between a NiCo–nitride shell and a NiCo–oxide core by annealing NiCo2O4 in an NH3 atmosphere. XPS analysis showed negative shifts in the binding energy peaks of Ni 2p and Co 2p, indicating decreased valence states of Ni and Co after nitridation. Additionally, nitridation enhances metallic bonding and produces stronger Co–Co/Ni–Ni bonds, thus enhancing corrosion-resistant ability. Besides boosting catalytic activity, nitridation also serves as a protective coating for supports such as Ti mesh and carbon paper. Guo et al.15 developed an OER catalyst operated across a wide pH range by constructing CoxN nanoparticles on TiN coated titanium mesh by atomic layer deposition (ALD) (Fig. 7a). CoxN and TiN evolved into an active and protective CoTi layered double hydroxide, contributing to the long-term stability. The catalyst required only 398 mV overpotential to achieve a current density of 50 mA cm−2 in 1 M HClO4. | ||
| Fig. 7 (a) Proposed self-transformation of Ti/TiN @ Co5.47N. Reproduced with permission.15 Copyright 2020, Wiley-VCH. Operando Co K-edge extended X-ray absorption fine structure (EXAFS) spectra of (b) Co3(PO4)2 and (c) Co3(PO4)2·8H2O at different potentials. Reproduced with permission.19 Copyright 2020, Wiley-VCH. (d) Schematic illustration of the dissolution path of Co ions in Co3O4 and CWO-del-48. Reproduced with permission.115 Copyright 2024, The American Association for the Advancement of Science. (e) Linear-sweep voltammetry of CWO-del-based catalysts at 5 mV s−1 in 0.5 M H2SO4 electrolyte. Reproduced with permission.115 Copyright 2024, The American Association for the Advancement of Science. | ||
Transition metal sulfides also show great potential in the OER due to their unique electronic structure and diverse modulation methods. However, only a few sulfide-based OER catalysts under acidic conditions have been reported, such as CoS2,17 MoS2 (ref. 116) and Ni4Fe5S8.117 Wu et al.116 investigated the catalytic activity of MoS2 with different crystalline phases and found that the octahedral 1T phase exhibited higher catalytic activity than the trigonal prismatic 2H phase. Meanwhile, activation of 1T MoS2 was observed during potential cycling between 1.2 V and 2.0 V, attributed to the rearrangement of MoS2 nanosheets by oxygen molecules generated during the OER process, leading to better exposure of active sites. Heterogeneous structures formed between MoS2 and other metal sulfides can modulate the valence states and promote the generation of high-valence active sites. For example, Yang et al.17 grew Co9S8 and MoS2 nanosheets on Ni3S2 nanorods as a bifunctional overall water splitting catalyst and found that the heterogeneous interface facilitated electron transfer through up shifting Co 2p binding energy and down shifting Mo 3d binding energy.
The choice of electrolyte such as phosphoric acid, sulfuric acid, or perchloric acid, greatly influences the stability of acidic OER catalysts, potentially due to different anion adsorption states at the reaction interface and catalyst evolution.71 Therefore, the introduction of phosphorus could improve stability by forming phosphorus oxide at the interface. Liu et al.118 prepared cobalt–manganese phosphide arrays grown on nickel phosphide nanoplates (CoMnP/Ni2P nanosheet-based microplate arrays). The doping of Mn induced charge transfer from CoP to MnP, creating high-valent cobalt active centers. At the same time, the catalyst maintained superhydrophilic and supergas-phobic characters, which mitigated the negative effect of gas bubbles on mass transfer at high current density. In addition to CoMnP/Ni2P, NiFe based phosphide catalysts also showed good stability in acidic OER. Hu et al.16 designed an amorphous NiFeP catalyst that maintained an overpotential of 540 mV for over 30 h in 0.05 M H2SO4 at 10 mA cm−2.
5. Other transition metal compounds
Similar to noble metal molecular complexes, complex transition-metal compounds have been investigated for water oxidation in acidic media, including polyoxometalates18,119,120 (POMs), MOF-supported catalysts,20 cobalt phosphates19,121,122 and cobalt tungstate.115 However, practical application remains distant due to challenges such as limited conductivity, restricted current density and high overpotentials. Meanwhile, alloys containing acid stable elements, such as Ni2Ta,123 Fe5Si3,124 Mn5Si3 (ref. 125) and TiMn2,126 might be candidates for acidic OER. Yet the electrochemical performance and structural evolution of these alloys have been poorly understood at this stage and need further studies.Kanan et al.127 developed an in situ-synthesized KCoPO amorphous film electrocatalyst with self-healing properties, prepared through electro-cycling in aqueous solution containing phosphoric acid and cobalt ions. This study demonstrated that an amorphous composite of cobalt and phosphoric ions would be a potential catalyst for the OER in strong acid. While insoluble cobalt phosphate salts with octahedral and tetrahedral sites have been studied, the catalytic mechanism of these two structures needs further exploration. Qi et al.19 synthesized cobalt phosphate catalysts featuring either exclusively tetrahedral or octahedral sites, allowing for direct comparison of their catalytic performance. Co3(PO4)2 with tetrahedral sites exhibits higher activity than its octahedral counterpart, attributed to the more facile formation of high valence (hydro)oxides on the dehydrated tetrahedral Co3(PO4)2 surface. EXAFS analysis showed minimal change in the spectra of the octahedral cobalt phosphate at positive potentials, indicating the difficulty in forming high-valent active intermediates (Fig. 7b and c). This might be related to the six-coordination of cobalt atoms in octahedral cobalt phosphate, requiring ligand removal to form Co![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species. Ram et al.115 prepared layered cobalt tungstate (CWO-del-48) by alkali etching of high-valent tungsten, a process that facilitates the incorporation of water and hydroxide at the surface, thereby mitigating the leaching of active cobalt during the reaction (Fig. 7d). The CWO-del-48 catalyst demonstrated an overpotential of 288 mV at 10 mA cm−2 and maintained stable operation at 1 A cm−2 for over 600 h in a PEMWE (Fig. 7e).
O species. Ram et al.115 prepared layered cobalt tungstate (CWO-del-48) by alkali etching of high-valent tungsten, a process that facilitates the incorporation of water and hydroxide at the surface, thereby mitigating the leaching of active cobalt during the reaction (Fig. 7d). The CWO-del-48 catalyst demonstrated an overpotential of 288 mV at 10 mA cm−2 and maintained stable operation at 1 A cm−2 for over 600 h in a PEMWE (Fig. 7e).
Polyoxometalates (POMs), composed of oxygen-rich ligands coordinated with metal cations, offer several advantages, including acid resistance, structural tunability and ease of synthesis. The introduction of different cations into POM systems has tremendous effects on their properties, and elements such as Fe,120 Ba and Cs18 have been explored. Blasco-Ahicart et al.18 found that introducing barium ions into [Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]16− (Co-POM) remarkably enhances the OER performance by exposing more active sites. 40% Ba[Co-POM] exhibits an overpotential of 361 mV at 10 mA cm−2 in 1 M H2SO4, outperforming the noble-metal catalyst IrO2. However, the low conductivity of POMs restricts the maximum current density to less than 20 mA cm−2. Thus, further research is required to develop POMs that can operate at higher current densities. Additionally, the mechanistic understanding of POM-based catalysis is still in its early stages, lacking theoretical frameworks to guide the enhancement of these catalysts.
Compared to heterostructures, constructing chemical bonds between active components and acid-resistant substrates is a more effective strategy to mitigate corrosion. Gao et al.20 developed a semi-rigid single-metal-site catalyst anchored on a Th-MOF, which featured open metal sites and rotatable anionic sites, providing greater flexibility for reaction intermediates. The CoCl2@Th-MOF catalyst showed an overpotential of 388 mV to reach 10 mA cm−2 in 0.1 M HClO4 electrolyte.
6. Conclusion and perspectives
In recent years, significant efforts have been directed toward the development of low-cost OER catalysts in acids with well-balanced activity and stability. This review summarizes recent key advances in this field, including the mechanistic insights, in situ/operando techniques, and synthetic/modification strategies for non-noble metal OER catalysts under acidic conditions. By discussing the conventional mechanisms such as the AEM and LOM, alongside newly proposed ones such as the OPM and PDAM, the mechanistic framework for acidic OER has been enriched.Unlike alkaline OER, acidic OER catalysts must withstand high potentials and low pH environments, which poses unique stability challenges. High-throughput screening methods combined with Pourbaix diagrams offer a promising alternative to conventional experimental approaches for identifying stable catalyst compositions. Also, advanced in situ/operando techniques that monitor surface evolution and intermediate species during reactions are crucial for elucidating detailed mechanisms. Despite these efforts, most acidic OER catalysts remain at the laboratory testing stage due to insufficient stability, which is far from meeting the requirements for practical application (Fig. 7). In addition, the lack of standardized evaluation methods hinders the precise evaluation of the intrinsic activity of catalysts. Finally, testing OER catalysts in a membrane electrode assembly (MEA) setup would be critical to evaluate the catalytic performance under practical working conditions. The following sections provide perspectives on overcoming these challenges for non-noble metal acidic OER catalysts.
6.1 Breaking the activity–stability tradeoff
Stability is a critical challenge for non-noble metal catalysts under acidic conditions, and prioritizing activity at the expense of stability is not a sustainable strategy (Fig. 8). The stability issues mainly come from the inherent instability of the lattice structure in acidic solutions and at high potentials. Modification strategies such as element doping, surface coating and phase engineering have shown promise in addressing these limitations. Mechanistic studies indicate that OER processes often involve multiple pathways simultaneously. Enhancing the contributions of the AEM, OPM, and PDAM pathways while minimizing the lattice oxygen involvement might significantly improve acid stability. | ||
| Fig. 8 Benchmarking of noble-metal-free OER electrocatalysts tested in acidic media. Overpotentials and stability of the catalysts at a current density of 10 mA cm−2. See detailed information in Table 1 for noble-metal-free catalysts. | ||
Besides intrinsic stability, external factors also play a critical role in catalyst durability. These include detachment of catalyst active sites caused by bubble generation at high current density, peeling of catalysts from the current collector due to week binder adhesion, and corrosion of the carbon current collector at high potentials. Addressing these issues requires innovative structural design, such as hydrophilic/hydrophobic interfaces to facilitate bubble removal, binder-free self-supported catalysts, and high-strength binders to improve catalyst adhesion.
6.2 Establishing unified catalyst evaluation criteria
The lack of standardized evaluation methods for acidic OER catalysts complicates the comparison of performance across studies. Differences in preparation methods, characterization techniques, and testing parameters can significantly affect key performance metrics, such as the onset potential and operational stability (Table 1). For powder catalysts, a mixed slurry of catalyst and binder is generally sprayed on carbon/titanium substrates or drop cast on glassy carbon electrodes to evaluate their performance. The measurements of onset potential and catalytic activity often ignore the impact of mass loading or true catalyst surface area. Mass activity or electrochemical active surface area (ECSA) normalization should be incorporated to better reflect intrinsic activity of catalysts.| Catalyst | Morphology | Electrolyte | Support | Loading (mg cm−2) | Overpotential @ 10 mA cm−2 (mV) | Stability | Max current density (mA) |
|---|---|---|---|---|---|---|---|
| 40-Co3O4@C/GPO75 | Nanoparticles | 1 M H2SO4 | Pocket working electrode | 20 | 356 | 43 h @ 10 mA cm−2 | 100 |
Co3O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 117 117 |
Film | 0.5 M H2SO4 | FTO | 0.182 | 570 | 12 h @ 10 mA cm−2 | 50 |
| Ag–Co3O466 |
Nanosheets | 0.5 M H2SO4 | Glassy carbon electrode | 0.2 | 470 | — | 60 |
| Co3−xBaxO414 |
Nanosheets | 0.5 M H2SO4 | Carbon paper | 1.6 | 278 | 110 h @ 10 mA cm−2 | 500 |
| Fe–Co3O4@C60 | Nanoparticles | 0.5 M H2SO4 | FTO | 1.1 | 396 | 50 h @ 10 mA cm−2 | 50 |
| LaMnCo-fiber42 | Fiber | 0.1 M HClO4 | RDE | 0.26 | 353 | 360 h @ 10 mA cm−2 | 70 |
| 3D Co3O4/NC-25062 |
Nanoarrays/cubes | 0.5 M H2SO4 | Carbon paper | 0.5 | 225 | 80 h @ 10 mA cm−2 | 100 |
| P–Co3O465 |
Nanowires | 0.1 M HClO4 | RDE | 0.07 | 400 | 30 h @ 10 mA cm−2 | 50 |
| CeO2/Co–Ni–P–Ox47 | Nanorods | 0.5 M H2SO4 | Carbon paper | 0.6 | 262 | 50 h @ 5 mA cm−2 | 10 |
| Co3O4/CeO252 |
Nanosheets | 0.5 M H2SO4 | Carbon paper | — | 347 | 50 h @ 10 mA cm−2 | 120 |
| TRGO@ZnO/Co3O470 |
Nanoparticles | 0.5 M H2SO4 | Carbon paper | 0.01 | 160 | 50 h @ 10 mA cm−2 | 100 |
| PdO@Co3O469 |
Nanorods/nanospheres | 0.1 M HNO3 | RDE | 1.36 | 523 | 2 h @ 10 mA cm−2 | 50 |
| Co/29BC73 | Nanoparticles | 0.5 M H2SO4 | Carbon paper | — | 450 | 12 h @ 10 mA cm−2 | 40 |
| TiO2/Co3O467 |
Nanorods | 0.5 M H2SO4 | Pt/Ti | — | 510 | 80 h @ 10 mA cm−2 | 60 |
| Co3O4@C74 | Nanosheets | 0.5 M H2SO4 | Carbon paper | 5 | 370 | 86.8 h @ 100 mA cm−2 | 200 |
| FTO@Co3O476 |
Nanoparticles | 0.5 M H2SO4 | Carbon paper | 3 | 511 | 21.5 h @ 10 mA cm−2 | 100 |
| Co2MnO463 |
Film | pH = 1H2SO4 | FTO | 10 | 395 | 400 h @ 200 mA cm−2 | 2000 |
| Co2Sb2O782 |
Bulk | 0.5 M H2SO4 | Glassy carbon electrode | 0.28 | 288 | 40 h @ 1 mA cm−2 | 20 |
| Co2TiO478 |
Nanoparticles | 0.5 M H2SO4 | Carbon paper | — | 513 | — | 60 |
| γ-MnO288 |
Film | 1 M H2SO4 | Carbon paper | 3.5 | 428 | 8000 h at 10 mA cm−2 | 100 |
| M94%87 |
Nanoarrays | 1 M H2SO4 | Pt/Ti mesh | 7.2 (Mn loading) | 440 | 3200 h at 200 mA cm−2 | 200 |
| R20-Mn118 | Nanoplates | 0.5 M H2SO4 | Carbon paper | 1 | 210 | 20 h at 10 mA cm−2 | 50 |
| MnFeF4.6O0.295 |
Amorphous | 0.5 M H2SO4 | Carbon paper | 1 | 500 | 20 h @ 10 mA cm−2 | 40 |
| Mn7.5O10Br389 |
Film | 0.5 M H2SO4 | Carbon paper | 7.2 | 295 | 500 h @ 10 mA cm−2 | 100 |
| Si–MnO299 |
Film | 0.1 M H2SO4 | FTO | — | 640 | ∼3 h @ 10 mA cm−2 | 15 |
| Pb/Pb–MnO298 |
Film | 1.63 M H2SO4 | Lead substrate | — | 655 (50 mA cm−2) | — | 150 |
| MnxSb1−xOz102 | Film | 1 M H2SO4 | Pt/Ti/SiO2/Si substrates | (Sb + Mn loading) 20.47 nmol mm−2 | 580 | 25 h @1 mA cm−2 | 140 |
| Cu1.5Mn1.5O4:10F94 | Nanoparticles | 0.5 M H2SO4 | Porous Ti | 1 | 320 (9.15 mA cm−2) | 24 h @ 16 mA cm−2 | 20 |
| Ni0.5Mn0.5Sb1.7Oy100 | Film | 1 M H2SO4 | ATO | (Ni + Sb + Mn loading) 13 μmol cm−2 | 672 | 168 h @ 10 mA cm−2 | 10 |
| (Mn0.8Nb0.2)O2:10F93 | Nanorods | 0.5 M H2SO4 | Titanium foils | 0.3 | 680 | 22 h @ 1.9 V (vs. RHE) | 25 |
| 1T MoS2105 |
Nanosheets | 0.5 M H2SO4 | Carbon paper | 1 | 420 | 120 h @ 10 mA cm−2 | 60 |
| P-NSC/Ni4Fe5S8-1000106 |
Nanoparticles | 0.5 M H2SO4 | Glassy carbon electrode | 0.4 | 300 (1 mA cm−2) | — | 15 |
| MoS2/Co9S8/Ni3S2/Ni17 | Nanorods/nanosheets | 0.5 M H2SO4 | Ni foam | 1.86 | 255 | 1.3 h @ 1.53 V (vs. RHE) | 100 |
| NiFeP16 | Amorphous | 0.05 M H2SO4 | Self-standing | — | 540 | 30 h @ 10 mA cm−2 | 350 |
| CoMnP/Ni2P107 | Nanosheets/microplate arrays | 0.5 M H2SO4 | Ni foam | 1.68 | 165 | 12 h @ 10 mA cm−2 | 200 |
| TiN@Co5.47N15 | Film | 0.1 M HClO4 | Ti mesh | — | 398 (50 mA cm−2) | 20 h @ 100 mA cm−2 | 450 |
| FeN4/NF/EG103 | Nanofibers | 0.5 M H2SO4 | Exfoliated graphene | 5.7 | 294 | 24 h @ 20 mA cm−2 | 30 |
| Mn-doped FeP/Co3(PO4)2111 |
Nanosheets | 0.5 M H2SO4 | Carbon paper | 1.2 | 390 | — | 50 |
| CWO-del-48112 |
Delaminated nanoparticles | 0.5 M H2SO4 | Carbon paper | 1.4 | 300 | 175 h @ 10 mA cm−2 | 80 |
| CoCl2@Th-BPYDC20 | Nanoparticles | 0.1 M HClO4 | Carbon paper | 0.075 | 388 | 25 h @ 1.68 V (vs. RHE) | 100 |
| Ba[Co-POM]18 | Nanoparticles | 1 M H2SO4 | Carbon paper | (Ba + Co loading) 16.13 μmol cm−2 | 361 | 24 h @ 1.48 V (vs. RHE) | 15 |
Performance assessments, typically conducted in a H-cell or single chamber three-electrode system, do not fully reflect the real-world scenario under PEMWE conditions. Firstly, liquid electrochemical cells and MEA-based solid electrolyzers exhibit a distinct catalyst–electrolyte interface structure. Interfacial chemical environments have been known to profoundly affect the electrocatalytic reactions. Secondly, mass transport processes are different in liquid cells and solid electrolyzers. A properly designed MEA would favor the supply of water to the catalyst layer and ensure rapid release of oxygen gas at the anode, which supports water electrolysis at A cm−2-level high current density. Thirdly, degradation mechanisms might be different. For example, in liquid electrochemical cells, violent gas release at high current density might lead to detachment of catalysts from the current collector. While in solid electrolyzers, dissolution of metal ions from catalysts might cause more severe activity degradation. These differences result in notable performance discrepancies when evaluating the same catalysts in a three-electrode liquid cell and PEMWE (Fig. 9). Additionally, incorporating a reference electrode ensures the precise measurement of the overpotential of an individual electrode, which is a conventional practice in liquid electrochemical cells but technically challenging in MEA-type electrolyzers. Finally, evaluations in PEMWE systems are essential but remain underreported due to the challenges associated with the assembly of these systems. Addressing this gap is crucial to bridge the disparity between laboratory-scale testing and practical applications.
 | ||
| Fig. 9 PEMWE and three-electrode electrochemical cell performance comparison of reported non-noble metal OER catalysts. Representative Ir-based catalysts and DOE target 2026 are given for comparison. | ||
6.3 Overcoming application challenges in PEM water electrolysis
PEMWE systems face unique challenges compared with conventional open electrolysis systems due to their distinct device structures, assembly procedures, proton conduction mechanisms, and gas transmission processes. Key challenges in PEMWE systems are typically associated with electrolyte conditions, catalyst-collector bonding stability, membrane–electrolyte–catalyst interface interactions, and metal cation toxicity to proton membranes.Firstly, pure water is supplied to PEMWEs rather than acidic electrolyte, which requires a higher overpotential. Secondly, titanium-based substrates, such as titanium felt or mesh, are commonly used but suffer from passivation due to the formation of titanium oxide layers, hampering interfacial electron transfer. Free-standing catalysts typically require pre-treatments of titanium substrates with hazardous chemicals (e.g., hydrofluoric acid, nitric acid, and oxalic acid) and/or at high temperatures, which remains a significant hurdle. Alternative surface modification approaches, such as platinum or gold plating and nitridation with plasma, have shown promise in improving interfacial stability. Thirdly, the interaction between catalysts and PEMs remains poorly understood. Real-time monitoring of these inorganic–organic interfaces under operating conditions is critical for understanding their behavior and optimizing device performance.128,129
Overall, PEMWE systems with multiple interfaces and a complex operational environment face greater challenges compared to conventional three-electrode systems. Deeper exploration of these interfaces and their interactions is essential to clarify the connection between fundamental mechanisms and practical applications. By addressing these challenges, future research can enable the rational design of non-noble metal acidic OER catalysts with improved performance and scalability, paving the way for their widespread adoption in renewable energy technologies.
Data availability
The data that support the findings of this study are available from the corresponding author, H. B. Wu, upon reasonable request.Author contributions
Junwei Han: conceptualization, methodology, writing – original draft, visualization; Qian Liu: writing – review & editing, visualization; Yue Yang: writing – review & editing; Hao Bin Wu: investigation, funding acquisition, writing – review & editing, validation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. B. W. acknowledges the funding support from the National Key R&D Program of China (2023YFB4203900) and the Fundamental Research Funds for the Central Universities (226-2024-00075).References
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, The role of hydrogen and fuel cells in the global energy system, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- S. Aslam, S. Rani, K. Lal, M. Fatima, T. Hardwick, B. Shirinfar and N. Ahmed, Electrochemical hydrogen production: sustainable hydrogen economy, Green Chem., 2023, 25, 9543–9573 RSC.
- T. Ioroi, Z. Siroma, S. I. Yamazaki and K. Yasuda, Electrocatalysts for PEM Fuel Cells, Adv. Energy Mater., 2018, 9, 1801284 CrossRef.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, A comprehensive review on PEM water electrolysis, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- H. Y. Lin, Z. X. Lou, Y. Ding, X. Li, F. Mao, H. Y. Yuan, P. F. Liu and H. G. Yang, Oxygen Evolution Electrocatalysts for the Proton Exchange Membrane Electrolyzer: Challenges on Stability, Small Methods, 2022, 6, e2201130 CrossRef PubMed.
- A. Minguzzi, O. Lugaresi, E. Achilli, C. Locatelli, A. Vertova, P. Ghigna and S. Rondinini, Observing the oxidation state turnover in heterogeneous iridium-based water oxidation catalysts, Chem. Sci., 2014, 5, 3591–3597 RSC.
- H. Zhu, Y. Wang, Z. Jiang, B. Deng, Y. Xin and Z. J. Jiang, Defect Engineering Promoted Ultrafine Ir Nanoparticle Growth and Sr Single-Atom Adsorption on TiO2 Nanowires to Achieve High-Performance Overall Water Splitting in Acidic Media, Adv. Energy Mater., 2024, 14, 2303987 CrossRef CAS.
- J. He, X. Zhou, P. Xu and J. Sun, Regulating Electron Redistribution of Intermetallic Iridium Oxide by Incorporating Ru for Efficient Acidic Water Oxidation, Adv. Energy Mater., 2021, 11, 2102883 CrossRef CAS.
- H. Jin, S. Choi, G. J. Bang, T. Kwon, H. S. Kim, S. J. Lee, Y. Hong, D. W. Lee, H. S. Park, H. Baik, Y. Jung, S. J. Yoo and K. Lee, Safeguarding the RuO2 phase against lattice oxygen oxidation during acidic water electrooxidation, Energy Environ. Sci., 2022, 15, 1119–1130 RSC.
- N. Yao, H. Jia, J. Zhu, Z. Shi, H. Cong, J. Ge and W. Luo, Atomically dispersed Ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation, Chem, 2023, 9, 1882–1896 CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, Electrocatalysts for acidic oxygen evolution reaction: Achievements and perspectives, Nano Energy, 2020, 78, 105392 CrossRef CAS.
- C. Wang, R. Deng, M. Guo and Q. Zhang, Recent progress of advanced Co3O4-based materials for electrocatalytic oxygen evolution reaction in acid: from rational screening to efficient design, Int. J. Hydrogen Energy, 2023, 48, 31920–31942 CrossRef CAS.
- N. Wang, P. Ou, R. K. Miao, Y. Chang, Z. Wang, S.-F. Hung, J. Abed, A. Ozden, H.-Y. Chen, H.-L. Wu, J. E. Huang, D. Zhou, W. Ni, L. Fan, Y. Yan, T. Peng, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, Doping Shortens the Metal/Metal Distance and Promotes OH Coverage in Non-Noble Acidic Oxygen Evolution Reaction Catalysts, J. Am. Chem. Soc., 2023, 145, 7829–7836 CrossRef CAS.
- D. Guo, Z. Wan, Y. Li, B. Xi and C. Wang, TiN @ Co5.47N Composite Material Constructed by Atomic Layer Deposition as Reliable Electrocatalyst for Oxygen Evolution Reaction, Adv. Funct. Mater., 2020, 31, 2008511 CrossRef.
- F. Hu, S. Zhu, S. Chen, Y. Li, L. Ma, T. Wu, Y. Zhang, C. Wang, C. Liu, X. Yang, L. Song, X. Yang and Y. Xiong, Amorphous Metallic NiFeP: A Conductive Bulk Material Achieving High Activity for Oxygen Evolution Reaction in Both Alkaline and Acidic Media, Adv. Mater., 2017, 29, 1606570 CrossRef PubMed.
- Y. Yang, H. Yao, Z. Yu, S. M. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao, G. Sun, H. Li, S. Ma, P. Zapol and M. G. Kanatzidis, Hierarchical Nanoassembly of MoS2/Co9S8/Ni3S2/Ni as a Highly Efficient Electrocatalyst for Overall Water Splitting in a Wide pH Range, J. Am. Chem. Soc., 2019, 141, 10417–10430 CrossRef CAS.
- M. Blasco-Ahicart, J. Soriano-López, J. J. Carbó, J. M. Poblet and J. R. Galan-Mascaros, Polyoxometalate electrocatalysts based on earth-abundant metals for efficient water oxidation in acidic media, Nat. Chem., 2017, 10, 24–30 CrossRef PubMed.
- J. Qi, Y. P. Lin, D. Chen, T. Zhou, W. Zhang and R. Cao, Autologous Cobalt Phosphates with Modulated Coordination Sites for Electrocatalytic Water Oxidation, Angew. Chem., Int. Ed., 2020, 59, 8917–8921 CrossRef CAS PubMed.
- Z. Gao, Y. Lai, L. Gong, L. Zhang, S. Xi, J. Sun, L. Zhang and F. Luo, Robust Th-MOF-Supported Semirigid Single-Metal-Site Catalyst for an Efficient Acidic Oxygen Evolution Reaction, ACS Catal., 2022, 12, 9101–9113 CrossRef CAS.
- R. Y. Fan, Y. S. Zhang, J. Y. Lv, G. Q. Han, Y. M. Chai and B. Dong, The Promising Seesaw Relationship Between Activity and Stability of Ru-Based Electrocatalysts for Acid Oxygen Evolution and Proton Exchange Membrane Water Electrolysis, Small, 2023, 20, 2304636 CrossRef PubMed.
- T. Nishimoto, T. Shinagawa, T. Naito and K. Takanabe, Microkinetic assessment of electrocatalytic oxygen evolution reaction over iridium oxide in unbuffered conditions, J. Catal., 2020, 391, 435–445 CrossRef CAS.
- Z. Wang, Y.-R. Zheng, I. Chorkendorff and J. K. Nørskov, Acid-Stable Oxides for Oxygen Electrocatalysis, ACS Energy Lett., 2020, 5, 2905–2908 CrossRef CAS.
- G. Mirshekari, R. Ouimet, Z. Zeng, H. Yu, S. Bliznakov, L. Bonville, A. Niedzwiecki, C. Capuano, K. Ayers and R. Maric, High-performance and cost-effective membrane electrode assemblies for advanced proton exchange membrane water electrolyzers: Long-term durability assessment, Int. J. Hydrogen Energy, 2021, 46, 1526–1539 CrossRef CAS.
- X. Li, Y. Yao, Y. Tian, J. Jia, W. Ma, X. Yan and J. Liang, Recent advances in key components of proton exchange membrane water electrolysers, Mater. Chem. Front., 2024, 8, 2493–2510 RSC.
- T. Zhang, L. Meng, C. Chen, L. Du, N. Wang, L. Xing, C. Tang, J. Hu and S. Ye, Similarities and Differences between Gas Diffusion Layers Used in Proton Exchange Membrane Fuel Cell and Water Electrolysis for Material and Mass Transport, Adv. Sci., 2024, 11, e2309440 CrossRef PubMed.
- R. T. Liu, Z. L. Xu, F. M. Li, F. Y. Chen, J. Y. Yu, Y. Yan, Y. Chen and B. Y. Xia, Recent advances in proton exchange membrane water electrolysis, Chem. Soc. Rev., 2023, 52, 5652–5683 RSC.
- C. Biaku, N. Dale, M. Mann, H. Salehfar, A. Peters and T. Han, A semiempirical study of the temperature dependence of the anode charge transfer coefficient of a 6kW PEM electrolyzer, Int. J. Hydrogen Energy, 2008, 33, 4247–4254 CrossRef CAS.
- B. Mohamed, B. Alli and B. Ahmed, Using the hydrogen for sustainable energy storage: Designs, modeling, identification and simulation membrane behavior in PEM system electrolyser, J. Energy Storage, 2016, 7, 270–285 CrossRef.
- K. Ayers, High efficiency PEM water electrolysis: enabled by advanced catalysts, membranes, and processes, Curr. Opin. Chem. Eng., 2021, 33, 100719 CrossRef.
- A. Makhsoos, M. Kandidayeni, B. G. Pollet and L. Boulon, A perspective on increasing the efficiency of proton exchange membrane water electrolyzers– a review, Int. J. Hydrogen Energy, 2023, 48, 15341–15370 CrossRef CAS.
- K. Ayers, The potential of proton exchange membrane–based electrolysis technology, Curr. Opin. Electrochem., 2019, 18, 9–15 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Electrolysis of water on (oxidized) metal surfaces, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, H. A. Hansen, I. C. Man, J. Rossmeisl and T. Bligaard, Volcano Relation for the Deacon Process over Transition-Metal Oxides, ChemCatChem, 2010, 2, 98–102 CrossRef CAS.
- K. L. Svane and J. Rossmeisl, Theoretical Optimization of Compositions of High-Entropy Oxides for the Oxygen Evolution Reaction, Angew. Chem., Int. Ed., 2022, 134, e202201146 CrossRef.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- R. Zhang, L. Pan, B. Guo, Z.-F. Huang, Z. Chen, L. Wang, X. Zhang, Z. Guo, W. Xu, K. P. Loh and J.-J. Zou, Tracking the Role of Defect Types in Co3O4 Structural Evolution and Active Motifs during Oxygen Evolution Reaction, J. Am. Chem. Soc., 2023, 145, 2271–2281 CrossRef CAS PubMed.
- H. M. A. Amin and H. Baltruschat, How many surface atoms in Co3O4 take part in oxygen evolution? Isotope labeling together with differential electrochemical mass spectrometry, Phys. Chem. Chem. Phys., 2017, 19, 25527–25536 RSC.
- H. M. A. Amin, P. Königshoven, M. Hegemann and H. Baltruschat, Role of Lattice Oxygen in the Oxygen Evolution Reaction on Co3O4: Isotope Exchange Determined Using a Small-Volume Differential Electrochemical Mass Spectrometry Cell Design, Anal. Chem., 2019, 91, 12653–12660 CrossRef CAS PubMed.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Anionic redox processes for electrochemical devices, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- L. Lin, Q. Fu, R. Wang, T. Yao, X. Wang and B. Song, Spinel-Type Oxides for Acidic Oxygen Evolution Reaction: Mechanism, Modulation, and Perspective, Adv. Energy Sustainability Res., 2023, 4, 2300075 CrossRef CAS.
- X. Rong, J. Parolin and A. M. Kolpak, A Fundamental Relationship between Reaction Mechanism and Stability in Metal Oxide Catalysts for Oxygen Evolution, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS.
- Q. Liang, G. Brocks and A. Bieberle-Hütter, Oxygen evolution reaction (OER) mechanism under alkaline and acidic conditions, J. Phys.: Energy, 2021, 3, 026001 CAS.
- I. Barlocco, L. A. Cipriano, G. Di Liberto and G. Pacchioni, Does the Oxygen Evolution Reaction follow the classical OH*, O*, OOH* path on single atom catalysts?, J. Catal., 2023, 417, 351–359 CrossRef CAS.
- Y. Hao, S.-F. Hung, W.-J. Zeng, Y. Wang, C. Zhang, C.-H. Kuo, L. Wang, S. Zhao, Y. Zhang, H.-Y. Chen and S. Peng, Switching the Oxygen Evolution Mechanism on Atomically Dispersed Ru for Enhanced Acidic Reaction Kinetics, J. Am. Chem. Soc., 2023, 145, 23659–23669 CrossRef CAS PubMed.
- A. M. Patel, J. K. Nørskov, K. A. Persson and J. H. Montoya, Efficient Pourbaix diagrams of many-element compounds, Phys. Chem. Chem. Phys., 2019, 21, 25323–25327 RSC.
- A. Minguzzi, F.-R. F. Fan, A. Vertova, S. Rondinini and A. J. Bard, Dynamic potential-pH diagrams application to electrocatalysts for wateroxidation, Chem. Sci., 2012, 3, 217–229 RSC.
- S. Cherevko, Stabilization of non-noble metal electrocatalysts for acidic oxygen evolution reaction, Curr. Opin. Electrochem., 2023, 38, 101213 CrossRef CAS.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, Theoretical Investigation of the Activity of Cobalt Oxides for the Electrochemical Oxidation of Water, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X.-M. Lin, H. Xu, L.-W. Wang and D.-J. Liu, La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis, Science, 2023, 380, 609–616 CrossRef CAS.
- B.-R. Chen, W. Sun, D. A. Kitchaev, K. H. Stone, R. C. Davis, G. Ceder, L. T. Schelhas and M. F. Toney, Kinetic origins of the metastable zone width in the manganese oxide Pourbaix diagram, J. Mater. Chem. A, 2021, 9, 7857–7867 RSC.
- Y. Shi, H. Wu, J. Chang, Z. Tang and S. Lu, Progress on the mechanisms of Ru-based electrocatalysts for the oxygen evolution reaction in acidic media, J. Energy Chem., 2023, 85, 220–238 CrossRef CAS.
- Z. Shi, X. Wang, J. Ge, C. Liu and W. Xing, Fundamental understanding of the acidic oxygen evolution reaction: mechanism study and state-of-the-art catalysts, Nanoscale, 2020, 12, 13249–13275 RSC.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, In situ Raman spectroscopy reveals the structure and dissociation of interfacial water, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- J. Liu, T. Wang, X. Liu, H. Shi, S. Li, L. Xie, Z. Cai, J. Han, Y. Huang, G. Wang and Q. Li, Reducible Co3+-O Sites of Co-Ni-P-Ox on CeO2 Nanorods Boost Acidic Water Oxidation via Interfacial Charge Transfer-Promoted Surface Reconstruction, ACS Catal., 2023, 13, 5194–5204 CrossRef CAS.
- Z. Chen, Q. Fan, J. Zhou, X. Wang, M. Huang, H. Jiang and H. Cölfen, Toward Understanding the Formation Mechanism and OER Catalytic Mechanism of Hydroxides by In Situ and Operando Techniques, Angew. Chem., Int. Ed., 2023, 62, 101213 Search PubMed.
- P. M. Radjenovic, R.-Y. Zhou, J.-C. Dong and J.-F. Li, Watching Reactions at Solid-Liquid Interfaces with in Situ Raman Spectroscopy, J. Phys. Chem. C, 2021, 125, 26285–26295 CrossRef CAS.
- M. Chen, D. Liu, L. Qiao, P. Zhou, J. Feng, K. W. Ng, Q. Liu, S. Wang and H. Pan, In-situ/operando Raman techniques for in-depth understanding on electrocatalysis, Chem. Eng. J., 2023, 461, 141939 CrossRef CAS.
- K. Natarajan, E. Munirathinam and T. C. K. Yang, Operando Investigation of Structural and Chemical Origin of Co3O4 Stability in Acid under Oxygen Evolution Reaction, ACS Appl. Mater. Interfaces, 2021, 13, 27140–27148 CrossRef CAS PubMed.
- J. Huang, H. Sheng, R. D. Ross, J. Han, X. Wang, B. Song and S. Jin, Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid, Nat. Commun., 2021, 12, 3036 CrossRef CAS PubMed.
- J. Wang, H. Kim, H. Lee, Y.-J. Ko, M. H. Han, W. Kim, J. M. Baik, J.-Y. Choi, H.-S. Oh and W. H. Lee, Sb incorporated into oxides enhances stability in acid during the oxygen evolution reaction by inhibiting structural distortion, Nano Energy, 2023, 110, 108355 CrossRef CAS.
- X. Zhang, C. Feng, B. Dong, C. Liu and Y. Chai, High-Voltage-Enabled Stable Cobalt Species Deposition on MnO2 for Water Oxidation in Acid, Adv. Mater., 2023, 35, 2207066 CrossRef CAS.
- S. Schlucker, Surface-enhanced Raman spectroscopy: concepts and chemical applications, Angew Chem. Int. Ed. Engl., 2014, 53, 4756–4795 CrossRef.
- L. Fang, S. Seifert, R. E. Winans and T. Li, Understanding Synthesis and Structural Variation of Nanomaterials Through In Situ/Operando XAS and SAXS, Small, 2022, 18, e2106017 CrossRef PubMed.
- A. M. Ullman, C. N. Brodsky, N. Li, S. L. Zheng and D. G. Nocera, Probing Edge Site Reactivity of Oxidic Cobalt Water Oxidation Catalysts, J. Am. Chem. Soc., 2016, 138, 4229–4236 CrossRef CAS PubMed.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, Stability and deactivation of OER electrocatalysts: A review, J. Energy Chem., 2022, 69, 301–329 CrossRef CAS.
- S. R. Ede and Z. Luo, Tuning the intrinsic catalytic activities of oxygen-evolution catalysts by doping: a comprehensive review, J. Mater. Chem. A, 2021, 9, 20131–20163 RSC.
- D. Senthil Raja, P.-Y. Cheng, C.-C. Cheng, S.-Q. Chang, C.-L. Huang and S.-Y. Lu, In-situ grown metal-organic framework-derived carbon-coated Fe-doped cobalt oxide nanocomposite on fluorine-doped tin oxide glass for acidic oxygen evolution reaction, Appl. Catal., B, 2022, 303, 120899 CrossRef CAS.
- E. O. Nwanebu and S. Omanovic, The influence of NixCo1-x-oxide composition on its electrocatalytic activity in the oxygen evolution reaction, Mater. Chem. Phys., 2019, 228, 80–88 CrossRef CAS.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid, Nat. Catal., 2022, 5, 109–118 CrossRef CAS.
- I. Abidat, N. Bouchenafa-Saib, A. Habrioux, C. Comminges, C. Canaff, J. Rousseau, T. W. Napporn, D. Dambournet, O. Borkiewicz and K. B. Kokoh, Electrochemically induced surface modifications of mesoporous spinels (Co3O4−δ, MnCo2O4−δ, NiCo2O4−δ) as the origin of the OER activity and stability in alkaline medium, J. Mater. Chem. A, 2015, 3, 17433–17444 RSC.
- X. Yang, J. Cheng, H. Li, Y. Xu, W. Tu and J. Zhou, Self-supported N-doped hierarchical Co3O4 electrocatalyst with abundant oxygen vacancies for acidic water oxidation, Chem. Eng. J., 2023, 465, 142745 CrossRef CAS.
- F. Shang, H. He, P. Li, H. Cai, B. An, X. Li, S. Yang, Z. Sun and B. Wang, PO6 geometric configuration unit enhanced electrocatalytic performance of Co3O4 in acidic oxygen evolution, J. Colloid Interface Sci., 2023, 641, 329–337 CrossRef CAS PubMed.
- K.-L. Yan, J.-F. Qin, J.-H. Lin, B. Dong, J.-Q. Chi, Z.-Z. Liu, F.-N. Dai, Y.-M. Chai and C.-G. Liu, Probing the active sites of Co3O4 for the acidic oxygen evolution reaction by modulating the Co2+/Co3+ ratio, J. Mater. Chem. A, 2018, 6, 5678–5686 RSC.
- T. Tran-Phu, H. Chen, R. Daiyan, M. Chatti, B. Liu, R. Amal, Y. Liu, D. R. Macfarlane, A. N. Simonov and A. Tricoli, Nanoscale TiO2 Coatings Improve the Stability of an Earth-Abundant Cobalt Oxide Catalyst during Acidic Water Oxidation, ACS Appl. Mater. Interfaces, 2022, 14, 33130–33140 CrossRef CAS.
- M. Musiani, Anodic deposition of PbO2/Co3O4 composites and their use as electrodes for oxygen evolution reaction, Chem. Commun., 1996, 21, 2403–2404 RSC.
- G. S. Rocha, A. L. Silva, L. P. C. Silva, F. B. Passos and N. M. F. Carvalho, Improved Activity of PdO Supported over Co3O4 in the Electrocatalytic Oxygen Evolution Reaction in a Wide pH Range, Energy Fuels, 2022, 36, 12719–12728 CrossRef CAS.
- T. Niyitanga and H. Kim, Reduced graphene oxide supported zinc/cobalt oxide nanoparticles as highly efficient electrocatalyst for oxygen evolution reaction, Inorg. Chim. Acta, 2022, 539, 121008 CrossRef CAS.
- B. Rodríguez-García, Á. Reyes-Carmona, I. Jiménez-Morales, M. Blasco-Ahicart, S. Cavaliere, M. Dupont, D. Jones, J. Rozière, J. R. Galán-Mascarós and F. Jaouen, Cobalt hexacyanoferrate supported on Sb-doped SnO2 as a non-noble catalyst for oxygen evolution in acidic medium, Sustainable Energy Fuels, 2018, 2, 589–597 RSC.
- X. M. C. Ta, T. Trân-Phú, J. A. Yuwono, T. K. A. Nguyen, A. D. Bui, T. N. Truong, L. C. Chang, E. Magnano, R. Daiyan, A. N. Simonov and A. Tricoli, Optimal Coatings of Co3O4 Anodes for Acidic Water Electrooxidation, Small, 2023, 20, 2304650 CrossRef.
- Q. W. Lai, V. Vediyappan, K. F. Aguey-Zinsou and H. Matsumoto, One-Step Synthesis of Carbon-Protected CoO Nanoparticles toward Long-Term Water Oxidation in Acidic Media, Adv. Energy Sustainability Res., 2021, 2, 2100086 CrossRef CAS.
- X. Yang, H. Li, A.-Y. Lu, S. Min, Z. Idriss, M. N. Hedhili, K.-W. Huang, H. Idriss and L.-J. Li, Highly acid-durable carbon coated Co3O4 nanoarrays as efficient oxygen evolution electrocatalysts, Nano Energy, 2016, 25, 42–50 CrossRef CAS.
- J. Yu, F. A. Garces-Pineda, J. Gonzalez-Cobos, M. Pena-Diaz, C. Rogero, S. Gimenez, M. C. Spadaro, J. Arbiol, S. Barja and J. R. Galan-Mascaros, Sustainable oxygen evolution electrocatalysis in aqueous 1 M H2SO4 with earth abundant nanostructured Co3O4, Nat. Commun., 2022, 13, 4341 CrossRef CAS PubMed.
- Y.-X. Yeh, C.-C. Cheng, P.-S. Jhu, S.-H. Lin, P.-W. Chen and S.-Y. Lu, Core-shell FTO@Co3O4 nanoparticles as active and stable anode catalysts for acidic oxygen evolution reaction and proton exchange membrane water electrolysis, J. Mater. Chem. A, 2023, 11, 3399–3407 RSC.
- A. Yu, M. H. Kim, C. Lee and Y. Lee, Structural transformation between rutile and spinel crystal lattices in Ru-Co binary oxide nanotubes: enhanced electron transfer kinetics for the oxygen evolution reaction, Nanoscale, 2021, 13, 13776–13785 RSC.
- S. Anantharaj, K. Karthick and S. Kundu, Spinel Cobalt Titanium Binary Oxide as an All-Non-Precious Water Oxidation Electrocatalyst in Acid, Inorg. Chem., 2019, 58, 8570–8576 CrossRef CAS PubMed.
- A. Babaei and M. Rezaei, Development of a highly stable and active non-precious anode electrocatalyst for oxygen evolution reaction in acidic medium based on nickel and cobalt-containing antimony oxide, J. Electroanal. Chem., 2023, 935, 117319 CrossRef CAS.
- M. Chatti, J. L. Gardiner, M. Fournier, B. Johannessen, T. Williams, T. R. Gengenbach, N. Pai, C. Nguyen, D. R. MacFarlane, R. K. Hocking and A. N. Simonov, Intrinsically stable in situ generated electrocatalyst for long-term oxidation of acidic water at up to 80 °C, Nat. Catal., 2019, 2, 457–465 CrossRef CAS.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Design of template-stabilized active and earth-abundant oxygen evolution catalysts in acid, Chem. Sci., 2017, 8, 4779–4794 RSC.
- Y. Wang, H. Zhao, Y. Guo, J. Wu, X. Lu and X. Tang, Pyrochlore-type cobalt and manganese antimonate electrocatalysts with excellent activity and stability for OER in acidic solution, Nanoscale, 2023, 15, 9413–9422 RSC.
- T. A. Evans and K. S. Choi, Electrochemical Synthesis and Investigation of Stoichiometric, Phase-Pure CoSbO and MnSbO Electrodes for the Oxygen Evolution Reaction in Acidic Media, ACS Appl. Energy Mater., 2020, 3, 5563–5571 CrossRef CAS.
- B. Liu, Y. Sun, L. Liu, S. Xu and X. Yan, Advances in Manganese-Based Oxides Cathodic Electrocatalysts for Li-Air Batteries, Adv. Funct. Mater., 2018, 28, 1704973 CrossRef.
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Electrochemical water splitting by layered and 3D cross-linked manganese oxides: correlating structural motifs and catalytic activity, Energy Environ. Sci., 2013, 6, 2745 RSC.
- T. Hayashi, N. Bonnet-Mercier, A. Yamaguchi, K. Suetsugu and R. Nakamura, Electrochemical characterization of manganese oxides as a water oxidation catalyst in proton exchange membrane electrolysers, R. Soc. Open Sci., 2019, 6, 190122 CrossRef CAS PubMed.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Stable Potential Windows for Long-Term Electrocatalysis by Manganese Oxides Under Acidic Conditions, Angew. Chem., Int. Ed., 2019, 58, 5054–5058 CrossRef CAS.
- S. Pan, H. Li, D. Liu, R. Huang, X. Pan, D. Ren, J. Li, M. Shakouri, Q. Zhang, M. Wang, C. Wei, L. Mai, B. Zhang, Y. Zhao, Z. Wang, M. Graetzel and X. Zhang, Efficient and stable noble-meta-free catalyst for acidic water oxidation, Nat. Commun., 2022, 13, 2294 CrossRef CAS PubMed.
- S. Kong, A. Li, J. Long, K. Adachi, D. Hashizume, Q. Jiang, K. Fushimi, H. Ooka, J. Xiao and R. Nakamura, Acid-stable manganese oxides for proton exchange membrane water electrolysis, Nat. Catal., 2024, 7, 252–261 CrossRef CAS.
- G. Yan, Y. Lian, Y. Gu, C. Yang, H. Sun, Q. Mu, Q. Li, W. Zhu, X. Zheng, M. Chen, J. Zhu, Z. Deng and Y. Peng, Phase and Morphology Transformation of MnO2 Induced by Ionic Liquids toward Efficient Water Oxidation, ACS Catal., 2018, 8, 10137–10147 CrossRef CAS.
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, Nature of Activated Manganese Oxide for Oxygen Evolution, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS.
- Y.-F. Li and Z.-P. Liu, Active Site Revealed for Water Oxidation on Electrochemically Induced δ-MnO2: Role of Spinel-to-Layer Phase Transition, J. Am. Chem. Soc., 2018, 140, 1783–1792 CrossRef CAS.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, P. M. Shanthi, S. Tan and P. N. Kumta, Computational and Experimental Study of Fluorine Doped (Mn1–xNbx)O2 Nanorod Electrocatalysts for Acid-Mediated Oxygen Evolution Reaction, ACS Appl. Energy Mater., 2019, 3, 541–557 CrossRef.
- P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, K. Damodaran, P. Jampani, B. Gattu, P. M. Shanthi, S. S. Damle and P. N. Kumta, Noble metal-free bifunctional oxygen evolution and oxygen reduction acidic media electro-catalysts, Sci. Rep., 2016, 6, 28367 CrossRef CAS PubMed.
- K. Lemoine, Z. Gohari-Bajestani, R. Moury, A. Terry, A. Guiet, J.-M. Grenèche, A. Hémon-Ribaud, N. Heidary, V. Maisonneuve, N. Kornienko and J. Lhoste, Amorphous Iron-Manganese Oxyfluorides, Promising Catalysts for Oxygen Evolution Reaction under Acidic Media, ACS Appl. Energy Mater., 2021, 4, 1173–1181 CrossRef CAS.
- Y. Li, L. Jiang, F. Liu, J. Li and Y. Liu, Novel phosphorus-doped PbO2-MnO2 bicontinuous electrodes for oxygen evolution reaction, RSC Adv., 2014, 4, 24020–24028 RSC.
- Y. Lai, Y. Li, L. Jiang, W. Xu, X. Lv, J. Li and Y. Liu, Electrochemical behaviors of co-deposited Pb/Pb-MnO2 composite anode in sulfuric acid solution-Tafel and EIS investigations, J. Electroanal. Chem., 2012, 671, 16–23 CrossRef CAS.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Toward an Active and Stable Catalyst for Oxygen Evolution in Acidic Media: Ti-Stabilized MnO2, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- Z. Zand, M. R. Mohammadi, A. S. Sologubenko, S. Handschin, R. Bagheri, P. Chernev, Z. Song, H. Dau and M. M. Najafpour, Oxygen Evolution Reaction by Silicate-Stabilized Manganese Oxide, ACS Appl. Energy Mater., 2023, 6, 1702–1713 CrossRef CAS.
- I. A. Moreno-Hernandez, C. A. MacFarland, C. G. Read, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Crystalline nickel manganese antimonate as a stable water-oxidation catalyst in aqueous 1.0 M H2SO4, Energy Environ. Sci., 2017, 10, 2103–2108 RSC.
- L. Zhou, A. Shinde, J. H. Montoya, A. Singh, S. Gul, J. Yano, Y. Ye, E. J. Crumlin, M. H. Richter, J. K. Cooper, H. S. Stein, J. A. Haber, K. A. Persson and J. M. Gregoire, Rutile Alloys in the Mn-Sb-O System Stabilize Mn3+ To Enable Oxygen Evolution in Strong Acid, ACS Catal., 2018, 8, 10938–10948 CrossRef CAS.
- Z. P. Ifkovits, J. M. Evans, P. A. Kempler, M. B. Morla, K. H. Pham, J. A. Dowling, A. I. Carim and N. S. Lewis, Powdered MnySb1–yOx Catalysts for Cerium-Mediated Oxygen Evolution in Acidic Environments, ACS Energy Lett., 2022, 7, 4258–4264 CrossRef CAS.
- N. Han, K. R. Yang, Z. Lu, Y. Li, W. Xu, T. Gao, Z. Cai, Y. Zhang, V. S. Batista, W. Liu and X. Sun, Nitrogen-doped tungsten carbide nanoarray as an efficient bifunctional electrocatalyst for water splitting in acid, Nat. Commun., 2018, 9, 924 CrossRef.
- C. Lei, H. Chen, J. Cao, J. Yang, M. Qiu, Y. Xia, C. Yuan, B. Yang, Z. Li, X. Zhang, L. Lei, J. Abbott, Y. Zhong, X. Xia, G. Wu, Q. He and Y. Hou, FeN4 Sites Embedded into Carbon Nanofiber Integrated with Electrochemically Exfoliated Graphene for Oxygen Evolution in Acidic Medium, Adv. Energy Mater., 2018, 8, 1801912 CrossRef.
- Z. Liu, H. Tan, D. Liu, X. Liu, J. Xin, J. Xie, M. Zhao, L. Song, L. Dai and H. Liu, Promotion of Overall Water Splitting Activity Over a Wide pH Range by Interfacial Electrical Effects of Metallic NiCo-nitrides Nanoparticle/NiCo2O4 Nanoflake/graphite Fibers, Advanced Science, 2019, 6, 1801829 CrossRef.
- R. Ram, L. Xia, H. Benzidi, A. Guha, V. Golovanova, A. Garzon Manjon, D. Llorens Rauret, P. Sanz Berman, M. Dimitropoulos, B. Mundet, E. Pastor, V. Celorrio, C. A. Mesa, A. M. Das, A. Pinilla-Sanchez, S. Gimenez, J. Arbiol, N. Lopez and F. P. Garcia de Arquer, Water-hydroxide trapping in cobalt tungstate for proton exchange membrane water electrolysis, Science, 2024, 384, 1373–1380 CrossRef CAS PubMed.
- J. Wu, M. Liu, K. Chatterjee, K. P. Hackenberg, J. Shen, X. Zou, Y. Yan, J. Gu, Y. Yang, J. Lou and P. M. Ajayan, Exfoliated 2D Transition Metal Disulfides for Enhanced Electrocatalysis of Oxygen Evolution Reaction in Acidic Medium, Adv. Mater. Interfaces, 2016, 3, 1500669 CrossRef.
- Q. Hu, G. Li, X. Liu, B. Zhu, G. Li, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, Coupling pentlandite nanoparticles and dual-doped carbon networks to yield efficient and stable electrocatalysts for acid water oxidation, J. Mater. Chem. A, 2019, 7, 461–468 RSC.
- M. Liu, Z. Sun, S. Li, X. Nie, Y. Liu, E. Wang and Z. Zhao, Hierarchical superhydrophilic/superaerophobic CoMnP/Ni2P nanosheet-based microplate arrays for enhanced overall water splitting, J. Mater. Chem. A, 2021, 9, 22129–22139 RSC.
- J. T. Arens, M. Blasco-Ahicart, K. Azmani, J. Soriano-López, A. García-Eguizábal, J. M. Poblet and J. R. Galan-Mascaros, Water oxidation electrocatalysis in acidic media with Co-containing polyoxometalates, J. Catal., 2020, 389, 345–351 CrossRef CAS.
- X.-B. Han, D.-X. Wang, E. Gracia-Espino, Y.-H. Luo, Y.-Z. Tan, D.-F. Lu, Y.-G. Li, T. Wågberg, E.-B. Wang and L.-S. Zheng, Fe-substituted cobalt-phosphate polyoxometalates as enhanced oxygen evolution catalysts in acidic media, Chin. J. Catal., 2020, 41, 853–857 CrossRef CAS.
- L. G. Bloor, P. I. Molina, M. D. Symes and L. Cronin, Low pH Electrolytic Water Splitting Using Earth-Abundant Metastable Catalysts That Self-Assemble in Situ, J. Am. Chem. Soc., 2014, 136, 3304–3311 CrossRef CAS PubMed.
- H. Liu, X. Peng, X. Liu, G. Qi and J. Luo, Porous Mn-Doped FeP/Co3(PO4)2 Nanosheets as Efficient Electrocatalysts for Overall Water Splitting in a Wide pH Range, ChemSusChem, 2019, 12, 1334–1341 CrossRef CAS PubMed.
- J. S. Mondschein, K. Kumar, C. F. Holder, K. Seth, H. Kim and R. E. Schaak, Intermetallic Ni(2)Ta Electrocatalyst for the Oxygen Evolution Reaction in Highly Acidic Electrolytes, Inorg. Chem., 2018, 57, 6010–6015 CrossRef CAS PubMed.
- B. Shen, Y. He, Z. He, Z. Wang, Y. Jiang and H. Gao, Porous Fe(5)Si(3) intermetallic anode for the oxygen evolution reaction in acidic electrolytes, J. Colloid Interface Sci., 2022, 605, 637–647 CrossRef CAS PubMed.
- W. Li, B. Shen, J. Kang, Z. Wang and Y. He, Oxygen evolution and corrosion behaviours of the porous Mn5Si3 electrode in sulfuric acid, Mater. Res. Express, 2019, 6, 085542 CrossRef CAS.
- Z. Wang, Y. Jiang, L. Feng, Z. He, X. Kang, L. Yu, Y. He, Z. Qin, Q. Zhao, Y. Qiu and H. Gao, Synthesis and study of TiMn2 intermetallic compound anode materials with different structures for zinc electrowinning, Intermetallics, 2023, 161, 107989 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Wang, Y. Zou, L. Tao, Y. Wang, G. Huang, S. Du and S. Wang, Rational design of three-phase interfaces for electrocatalysis, Nano Res., 2019, 12, 2055–2066 CrossRef CAS.
- C. Yang, N. Han, Y. Wang, X.-Z. Yuan, J. Xu, H. Huang, J. Fan, H. Li and H. Wang, A Novel Approach to Fabricate Membrane Electrode Assembly by Directly Coating the Nafion Ionomer on Catalyst Layers for Proton-Exchange Membrane Fuel Cells, ACS Sustain. Chem. Eng., 2020, 8, 9803–9812 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |