
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneral access to furan-substituted gem-difluoroalkenes enabled by PFTB-promoted cross-coupling of ene-yne-ketones and difluorocarbene†
Na
Li
a,
Chenghui
Li
a,
Qianying
Zhou
a,
Xin
Zhang
a,
Zhouming
Deng
b,
Zhong-Xing
Jiang
c and
Zhigang
Yang
 *a
*a
aDepartment of Spine Surgery and Musculoskeletal Tumor, Zhongnan Hospital of Wuhan University, School of Pharmaceutical Sciences, Wuhan University, Wuhan, 430071, China. E-mail: zgyang@whu.edu.cn
bDepartment of Spine Surgery and Musculoskeletal Tumor, Zhongnan Hospital of Wuhan University, Wuhan, 430071, China
cState Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, National Center for Magnetic Resonance in Wuhan, Wuhan Institute of Physics and Mathematics, Innovation Academy for Precision Measurement Science and Technology, Chinese Academy of Sciences-Wuhan National Laboratory for Optoelectronics, Wuhan 430071, China
First published on 23rd December 2024
Abstract
Replacement of a carbonyl group with fluorinated bioisostere (e.g., CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) has been adopted as a key tactical strategy in drug design and development, which typically improves potency and modulates lipophilicity while maintaining biological activity. Consequently, new gem-difluoroalkenation reactions have undoubtedly accelerated this shift, and conceptually innovative practices would be of great benefit to medicinal chemists. Here we describe an expeditous protocol for the direct assembly of furan-substituted gem-difluoroalkenes via PFTB-promoted cross-coupling of ene-yne-ketones and difluorocarbene. In this multi-step tandem reaction process, the furan ring and the gem-difluorovinyl group are constructed simultaneously in an efficient manner. These products can serve as bioisosteres of the α-carbonyl furan core, which is an important scaffold present in natural products and drug candidates. The broad generality and practicality of this method for late-stage modification of bioactive molecules, gram-scale synthesis and versatile derivatisation of products has been described. Biological activity evaluation showed that the gem-difluoroalkene skeleton exhibited dramatic antitumor activity.
C) has been adopted as a key tactical strategy in drug design and development, which typically improves potency and modulates lipophilicity while maintaining biological activity. Consequently, new gem-difluoroalkenation reactions have undoubtedly accelerated this shift, and conceptually innovative practices would be of great benefit to medicinal chemists. Here we describe an expeditous protocol for the direct assembly of furan-substituted gem-difluoroalkenes via PFTB-promoted cross-coupling of ene-yne-ketones and difluorocarbene. In this multi-step tandem reaction process, the furan ring and the gem-difluorovinyl group are constructed simultaneously in an efficient manner. These products can serve as bioisosteres of the α-carbonyl furan core, which is an important scaffold present in natural products and drug candidates. The broad generality and practicality of this method for late-stage modification of bioactive molecules, gram-scale synthesis and versatile derivatisation of products has been described. Biological activity evaluation showed that the gem-difluoroalkene skeleton exhibited dramatic antitumor activity.
1 Introduction
Fluorinated compounds are commonly used in a wide range of applications, including pharmaceuticals, materials, agro-chemicals, and 19F MRI agents.1 The fluorine atom contributes to the unique properties of these compounds, including strong electronegativity, a small atomic radius (comparable to that of hydrogen), and a high dissociation energy of the C–F bond.2 Among the fluorinated functionalities, the gem-difluorovinyl group (CF2C) has attracted increasing interest in drug design and discovery, since CF2C can serve as a bioisostere of the carbonyl group, which is highly hydrophilic and electrophilic and susceptible to easy metabolic oxidation (Scheme 1B).3 The direct incorporation of this motif onto organic molecules instead of carbonyl can significantly improve their metabolic stability, bioavailability and modulated lipophilicity compared to their non-fluorinated bioisostere counterpart.4,5 Classically, the gem-difluorovinyl moiety has been acquired via Wittig,6 Julia-Kocienski,7 cross-coupling,8 and dehalogenate functionalization5 reactions (Scheme 1C).
 | ||
| Scheme 1 Background and synopsis of work. (A) Value of molecules containing an α-carbonyl furan core in pharmaceutical development. (B) Importance of gem-difluorovinyl moieties. (C) Traditional approaches to the synthesis of gem-difluoroalkenes. (D) This work: potential side reactions and the preparation of furan-substituted gem-difluoroalkenes via a one-pot, multistep process. | ||
The α-carbonyl furan structure is particularly privileged, as it is found in a wide range of natural products and numerous pharmaceuticals (Scheme 1A).9 There are still challenges to overcome to access furan-substituted gem-difluoroalkenes. Firsty, as an electron deficient singlet carbene species, difluorocarbene is more stable, although less reactive than other dihalocarbenes,10 which readily react with carbonyl compounds and alkenes or alkynes and undergo typical carbene transformations such as the Wittig like reaction6 or [2 + 1] cycloaddition.11 Conjugated ene-yne-ketone12b bears ketone, alkenyl and alkynyl motifs, the present cyclized reaction needs to avoid the corresponding three potential side reactions (Scheme 1D). Secondly, most carbene reactions involving alkynes require associated transition metals (e.g., Cu, Pd, Rh, etc.) to form metal carbene intermediates.12 Moreover, the metal difluorocarbene strategy ([M]CF2) has also emerged as a powerful tool for the construction of direct difluoromethylation reactions since Zhang's pioneering work in 2015.13 New reaction designs for the rapid assembly of furan-substituted gem-difluoroalkenes from readily available conjugated ene-yne-ketones with transition-metal-free conditions would be of considerable value in the development of pharmaceuticals and also represent a significant challenge. Continuing our goal of increasing the adoption of fluorinated bioactive compounds in medicinal chemistry,14 we set out our successful efforts to develop an expedient route to furan-substituted gem-difluoroalkenes. Several cyclized adducts were found to have excellent antiproliferative activity against HeLa, 4T1 and HepG2 tumor cell lines.
2 Results and discussion
We commenced studies to optimize the tandem cyclization reaction conditions using conjugated ene-yne-ketone 1 as a model substrate. The reaction was conducted in 1,4-dioxane at 50 °C in the absence of transition metal. Initially, several classes of difluorocarbene reagents, such as TMSCF2Br/TBAF, BrCF2COOEt/Cs2CO3, BrCF2COOK/Cs2CO3, HCF2Cl/Cs2CO3, TMSCF3/NaI and Ph3P+CF2CO2− were investigated, but no any fluorinated adducts were observed, except for the latter, which only gave the expected cyclized product 2 in 45% yield, and by-products 3–5 were not obtained (Table 1, entry 1). Encouraged by this result, common Lewis acids such as AlCl3 or BF3·OEt2 was added individually as a catalyst, found to have no effect on reactivity, and the targeted product 2 was obtained in identical yield (Table 1, entries 2 and 3). For comparison, 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), which acts as an organocatalyst, was also investigated, taking advantage of its low acidity and hydrogen bond donating ability,15 and the yield was slightly improved (Table 1, entry 4). In particular, switching the hydrogen bond donor catalyst from HFIP to perfluoro-tert-butanol (PFTB) gave the desired product 2 in a much better yield (Table 1, entry 5). These results indicate that the hydrogen bond can improve the reaction efficiency, which may be beneficial to facilitate the formation of a zwitterionic intermediate during the intramolecular cyclization process. Subsequently, other ether solvents such as THF, TBME and CPME were then examined, with the latter proving to be the most suitable, giving the expected cyclized adduct 2 in 84% yield (Table 1, entry 8). Finally, the reaction temperature and other Brønsted acids such as H2O, PhOH, PhCOOH, CF3COOH, etc. were also investigated and no superior results were observed (see SI for further optimization trials).|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yield of 2 (%) |
a Reaction conditions: 1 (E/Z = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.1 mmol), Ph3P+CF2CO2− (2.0 equiv.), catalyst (10 mol%) in solvent (1.0 mL) at 50 °C under Ar for 12 h. 19F NMR yields with PhCF3 as internal standard. 3–5 were not found. PFTB = Perfluoro-tert-butanol; CPME = Cyclopentyl methyl ether; HFIP = 1,1,1,3,3,3-Hexafluoroisopropanol. :1, 0.1 mmol), Ph3P+CF2CO2− (2.0 equiv.), catalyst (10 mol%) in solvent (1.0 mL) at 50 °C under Ar for 12 h. 19F NMR yields with PhCF3 as internal standard. 3–5 were not found. PFTB = Perfluoro-tert-butanol; CPME = Cyclopentyl methyl ether; HFIP = 1,1,1,3,3,3-Hexafluoroisopropanol.
|
|||
| 1 | None | 1,4-Dioxane | 45 |
| 2 | AlCl3 | 1,4-Dioxane | 45 |
| 3 | BF3 OEt2 | 1,4-Dioxane | 45 |
| 4 | HFIP | 1,4-Dioxane | 51 |
| 5 | PFTB | 1,4-Dioxane | 63 |
| 6 | PFTB | THF | 51 |
| 7 | PFTB | TBME | 63 |
| 8 | PFTB | CPME | 84 |

After the initial optimization, we investigated the generality of the tandem cyclization reaction by exploring a variety of conjugated ene-yne-ketones (Scheme 2). The influence of the ester group adjacent to the ketone moiety was first investigated. The ester moiety bearing a small hindered group such as methyl and long chain alkyl groups such as n-butyl, n-hexyl can slightly affect the efficiency of the cyclization reaction, the corresponding products 6, 9, 10 were afforded in 60%, 56% and 64% yields respectively. This may be due to the fact that ester groups with little hindrance are more easily hydrolysed in acidic environments. The compatibility of the alkynyl moiety of the conjugated ene-yne-ketone was also further investigated by examining the electronic features and the positional change (para or meta) of the phenyl ring, which provided the expected cyclized adducts 12–22 in 58–99% yields. However, the phenyl ring with electron-deficient substituents or the ortho position of the phenyl ring was substituted and aliphatic alkynyl had a significant effect on the cyclization efficiency, resulting in the failure to form the desired product.
 | ||
| Scheme 2 Substrate scope.a Standard reaction conditions: conjugated ene-yne-ketone (E/Z = 1:1, 0.5 mmol), Ph3P+CF2CO2− (2.0 equiv.), and PFTB (10 mol%) in CPME (5.0 mL) at 50 °C under Ar for 12 h. Isolated yields are shown. | ||
Fortunately, when the β-naphthylacetylene-derived substrate was subjected to this transformation, the reaction proceeded smoothly and delivered the desired product 23 in good yield. Subsequently, our attention was directed towards the evaluation of the scope of different substituents on the ketone skeleton. The synthesis of conjugated ene-yne-ketones can be achieved through the condensation of alkyne aldehydes and β-keto ethyl esters, which are commercially available in numerous candidate structures. Therefore, ethyl was chosen as the ester group for our next investigation. In general, the cyclization reactions of a wide range of aliphatic and aromatic ketone-containing substrates with Ph3P+CF2CO2− proceeded smoothly, enabling the furyl-difluoroalkenes with good yields (products 24–45). For example, ketone moieties containing aliphatic groups such as ethyl, cyclopropyl, cyclobutyl and cyclohexyl were found to be well compatible and provided the desired products 24–27 in 63–78% yields. For the aromatic ketones, the steric hindrance (ortho) and the different electron properties at the para and meta positions had little effect on the reaction efficiency under the present conditions, giving the desired adducts 28–38 in good yields. Incorporating multiple substituents such as 2,5-dimethyl, 3,4-dimethoxy, 3,4,5-trimethoxy into the benzene ring could slightly improve the reaction efficiency (products 39–41). Gratifyingly, the present protocol has also been successfully applied to medically relevant heteroaromatic ring (e.g., dihydrobenzofuryl, furyl, and thienyl) and fused ring (2-naphthyl) substrates with no loss of efficiency (products 42–45). Finally, the structure of 33 was unambiguously confirmed by X-ray crystallography analysis (CCDC 2356931).
The excellent functional group compatibility mentioned above further encourages us to apply this approach to the late-stage modification of bioactive molecules, natural products and therapeutic agents. To our delight, furyl-difluoroalkene analogues of polyethylene glycol and 2-adamantanol were successfully synthesized in satisfactory yields (products 46–47). Conjugated ene-yne-ketones derived from small molecules such as sesamol, L-menthol and (+)-fenchol were suitable substrates, although the former gave a relatively low yield (products 48–50). Similarly, substrates derived from glucose and dibenzosuberone were readily converted to the corresponding adducts 51 and 52 in identical yields (61%). Geraniol and citronellol,16 which have similar structures, were isolated from plants of the genus Geranium and derivatised on the substrates, the former delivering the corresponding product 53 in excellent yields, while the latter was moderate (54). Vitamin E, a plant-based antioxidant essential for human health,17 and cholesterol are both crucial for cell membrane structure, signal transduction, and overall human health.18 Conjugated ene-yne ketones derived from them as complex natural products could also be readily subjected to cyclized gem-difluorovinylation reactions despite unprecedentedly high molecular weights (products 55–56). All these results show that this current protocol opens a new door for the direct modification of biologically active molecules and drugs without the need for multi-step parallel synthesis, which could facilitate new drug design in the future (Scheme 3).
 | ||
| Scheme 3 Late-stage functionalization of bioactive molecules.a Standard reaction conditions: conjugated ene-yne-ketone (E/Z = 1:1, 0.3 mmol), Ph3P+CF2CO2− (2.0 equiv.), and PFTB (10 mol%) in CPME (3.0 mL) at 50 °C under Ar for 12 h. Isolated yields are shown. | ||
To demonstrate the applicability of the cyclized gem-difluorovinylation reaction, a gram-scale reaction was carried out with 1.01 g of conjugated ene-yne-ketone and Ph3P+CF2CO2− (2.0 equiv.), delivering the corresponding adduct 18 (0.92 g) in 80% isolated yield, with a slight decrease in efficiency compared to the small-scale reaction (Scheme 4A). Furans and their derivatives are important scaffolds and are routinely used in drug discovery, while the gem-difluorovinyl group, which serves as a fluorinated synthon, has recently attracted considerable attention from chemists, such as its use for nucleophilic additions,19 defluorinative functionalization,5eetc. Therefore, we expected the resulting furan-substituted gem-difluoroalkenes to undergo various synthetic transformations. As shown in Scheme 4A, the treatment of product 18 with benzoyl hydrazine in DMSO at 80 °C resulted in the observation of a disubstituted 1,3,4-oxadiazole 57 in a yield of 61% by a cyclization process in the presence of an excess amount of Cs2CO3. Interestingly, the nucleophilic addition reactions of gem-difluoroalkenation were observed when heteroatomic nucleophiles, such as 4-methylbenzenethiol and imidazole, were employed, resulting in the corresponding products 58 and 60 with satisfactory yields. As anticipated, the gem-difluorovinyl moiety was also readily hydrogenated in THF in the presence of hydrogen (balloon) and catalytic palladium on activated carbon (Pd/C), resulting in the formation of CF2H 59 in a moderate yield. Defluorinating substitutions can be achieved by treating product 18 with strong nucleophiles such as indole or TMSCN at room temperature in the presence of Cs2CO3, and the trisubstituted α-monofluoroalkenes 61–62 were obtained in good yields.
 | ||
| Scheme 4 Gram-scale synthesis, synthetic transformations, and mechanistic studies. | ||
In order to elucidate the possible reaction mechanism, a series of control experiments were conducted (Scheme 4B). The addition of TEMPO, a free radical inhibitor, under standard conditions resulted in a slight effect on the reaction efficiency, indicating that the free radical pathway could be excluded. As a common carbene scavenger, 1,1-diphenylethylene was then subjected to the cyclized gem-difluorovinylation reaction, the reaction was significantly blocked and accompanied by the formation of a gem-difluorinated cyclopropane 63 in 30% yield. Furthermore, the reactions were almost completely suppressed when phenol and 3-azaindole were added individually to the reaction system. The accompanying difluorocarbene-trapped products 64 and 65 were identified by 19F NMR in 21% and 72% yields, respectively. The above results indicated that the present cyclized reaction proceeded through a difluorocarbene route.
Based on the results of the above control experiments and related literature studies,10 a hypothetical reaction pathway is illustrated as shown in Scheme 4C. Initially, the conjugated ene-yne ketone 1 was activated by the (CF3)3COH (PFTB), which can take advantage of its low acidity and hydrogen bonding ability, similar to (CF3)2CHOH (HFIP),15 followed by an intramolecular nucleophilic attack of carbonyl oxygen, forming the zwitterionic intermediate A, which could be further stabilized by PFTB. Subsequently, the vinyl anion of intermediate A rapidly captured a difluorocarbene species (:CF2) generated by the thermal dissociation of Ph3P+CF2CO2− to form a new zwitterionic intermediate B/C. Finally, the expected furan-substituted gem-difluoroalkene 2 was obtained via the tautomerism equilibrium of intermediate B/C.
Given the prevalence of the α-carbonyl furan core in medicinal design, we are particularly interested in exploring the antitumor activities of the synthesized furan-substituted gem-difluoroalkene, which serve as its bioisostere. After initial evaluation in HeLa human cervical cancer cells using a CCK-8 assay, compounds 11, 17, 38, 41, 42, and 44 were found to exhibit high activities (see SI for details). These compounds were then selected for further testing against cancer cell lines using the well-known fluorinated anticancer drug 5-fluorouracil (5-FU) as a positive control. Besides HeLa cells, the other two cancer cell lines are murine breast cancer cells (4T1) and human hepatoma cells (HepG2). As shown in Fig. 1A, the results of the structure–activity relationship (SAR) studies indicated that compounds bearing three methoxy groups at the 3,4,5 positions of the phenyl ring in vicinity to the ester moiety, or one methoxy at the para position of the phenyl ring in vicinity to the gem-difluorovinyl moiety, exhibited enhanced activity. The compounds 17 and 41 exhibited potent inhibitory potency in the 4T1 cell line, with IC50 values of 21.01 and 24.27 μM, respectively.
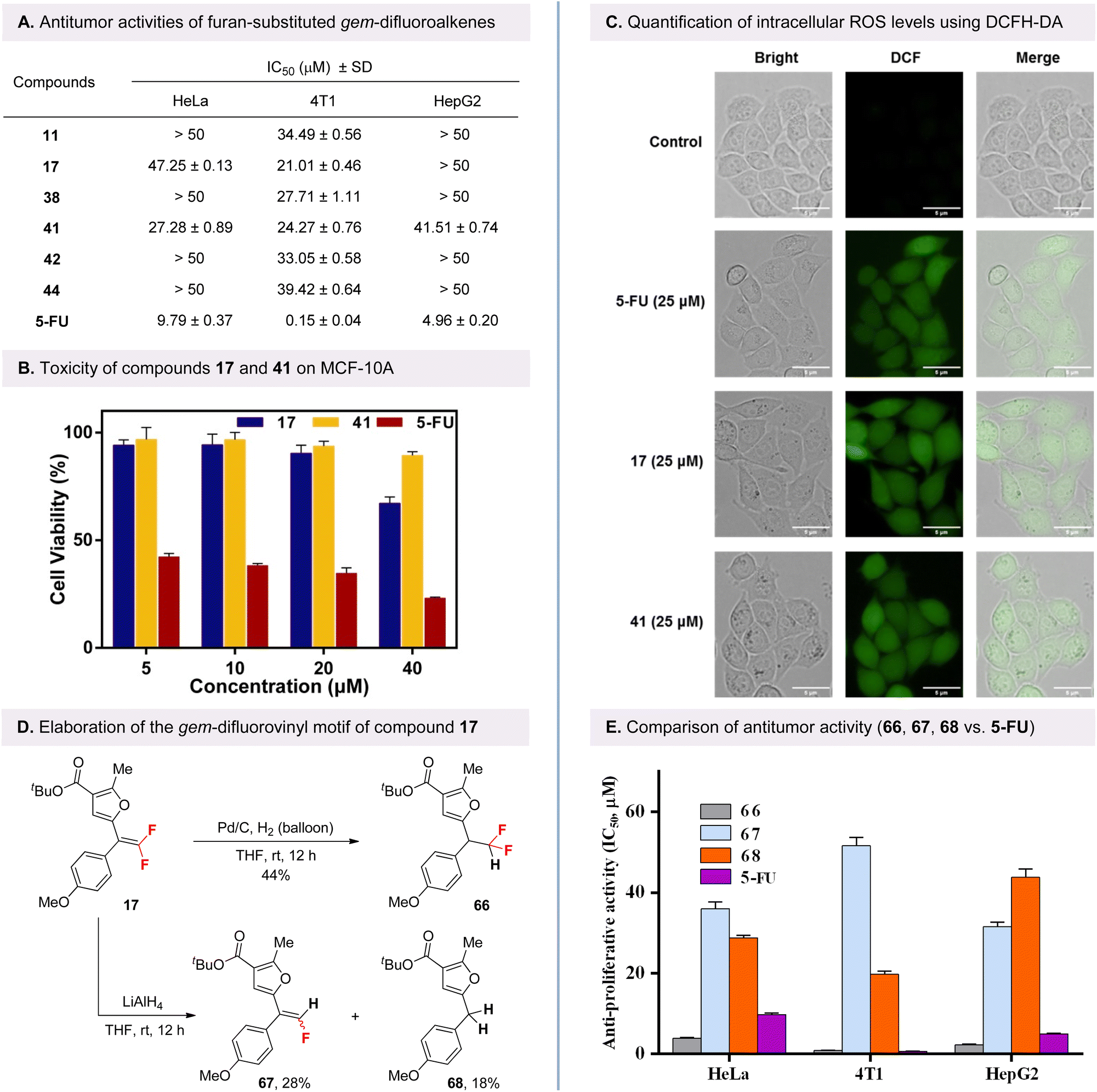 | ||
| Fig. 1 Biological activity evaluation. (A) Cells were incubated with compounds for 48 h and cell viability was measured by CCK-8 assay; IC50 is the concentration of compound required to inhibit cell growth by 50%; SD (standard deviation) of three independent experiments. (B) Toxicity of compounds 17, 41 and 5-FU on normal cell (MCF-10A). (C) ROS levels in HeLa cells were detected by DCFH-DA staining after incubation with compounds 17, 41 and 5-FU at 25 μM for 24 h. (D) Diversification of gem-difluorovinyl-containing product 17. (E) Anti-proliferative activities on HeLa, 4T1 and HepG2 cell lines for compound 17 and its derivatives (IC50, μM). | ||
Subsequently, the cytotoxicity of gem-difluoroalkene analogs 17 and 41 was evaluated in human normal mammary epithelial cells (MCF-10A) to ascertain their potential as therapeutic agents, with 5-FU serving as the positive control. As illustrated in Fig. 1B, at concentrations of 5, 10, 20 and 40 μM, the cytotoxicity of compounds 17 and 41 to MCF-10A cells was found to be significantly lower than that of 5-FU. The results indicate that furan-substituted gem-difluoroalkenes 17 and 41 may be relatively safe in vitro and feasible as effective agents for cancer therapy, which deserves further focus in the future.
Typically, the level of intracellular reactive oxygen species (ROS) is often associated with apoptosis of tumor cells.20 DCFH-DA is an oxidation-sensitive fluorescent probe that can penetrate cell membranes and be enzymatically hydrolyzed into free DCFH, which is then oxidized by intracellular ROS to form DCF with green fluorescence. As shown in Fig. 1C, after 24 hours of incubation of 4T1 cells with 25 μM of 17 and 41, the green fluorescence was significantly increased compared to the control group as observed by fluorescence microscopy. These results suggest that both compounds are capable of inducing ROS production in 4T1 cells, thereby increasing intracellular ROS levels.
Finally, to determine whether the fluorine-containing group on the product is beneficial to its antitumor activity, several transformations were performed focusing on the gem-difluorovinyl motif of compound 17 (Fig. 1D). The gem-difluoroalkene 17 is readily converted to the CF2H-containing product 66 in the presence of Pd/C and hydrogen. In addition, the monofluoroalkene 67 was readily accessible by further hydrodefluorination of the gem-difluorovinyl motif with LiAlH4 as reductant, accompanied by the formation of the defluorinated product 68, which is difficult to obtain by conventional strategies. With these derivatives of gem-difluoroalkene 17, we then evaluated their biological activity in cells, focusing on their fluorinated units for comparison. As shown in Fig. 1E, fluorinated compounds 66 and 67 as well as non-fluorinated furan core compound 68 showed good anticancer activities against all the above three cancer cell lines. In particular, compound 66, in which the gem-difluorovinyl group is hydrogenated, was found to exhibit the most excellent inhibitory activity against HeLa and HepG2 cancer cell lines, significantly exceeding that of the classical fluorine-containing anticancer drug 5-FU, with IC50 values of 3.92 and 2.31 μM, respectively. These results clearly demonstrated the potential of the fluorine-containing scaffold as a favorable motif for enhancing the biological properties of drug candidates containing a furan core.
3 Conclusions
In conclusion, we have developed a general PFTB-promoted cyclized gem-difluorovinylation approach by using conjugated ene-yne-ketones as the furan source and Ph3P+CF2CO2− as an efficient :CF2 agent to access furan-substituted gem-difluoroalkenes that serve as bioisosteres of the α-carbonyl furan core. Three potential side reactions based on the ketone, alkenyl, and alkynyl moieties of conjugated ene-yne-ketones with difluorocarbene were not been identified. The late-stage functionalization of naturally occurring biomolecules and drug-derived complex molecules, as well as the synthetic conversion of the resulting furyl-difluoroalkenes to high-value fluorinated compounds, were demonstrated to highlight the synthetic value of this protocol. Many products showed dramatic antiproliferative activity in HeLa, 4T1 and HepG2 tumor cell lines. Among them, the derivative with hydrogenated gem-difluorovinyl moiety (66) showed the most potent effect against the HeLa and HepG2 cells with IC50 values of 3.92 and 2.31 μM, respectively, exceeding that of the positive control 5-fluorouracil (5-FU), opening new opportunities in cancer therapy.Data availability
Details on experimental procedures and characterization data, as well as X-ray data of 33, are available in the ESI.†Author contributions
Z. Yang conceived the idea, designed the research project, and drafted the manuscript. N. Li performed the chemical experiments, conducted cell-based assays and was responsible for compiling the ESI.† C. Li, Q. Zhou and X. Zhang helped with the chemical experiments. Z. Deng and Z.-X. Jiang assisted in the design of the research project. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for financial support from the National Key Research and Development Program of China (grant no. 2018YFA0704000), the National Natural Science Foundation of China (grant no. 22077098), and the Natural Science Foundation of Hubei Province (2022CFB204). We also thank Ran Zhang from the Core Facility of Wuhan University for his assistance with NMR and X-ray crystallographic analysis.Notes and references
- For selected reviews and book, see: (a) K. Uneyama, Organofluorine Chemistry, Wiley-Blackwell, New Delhi, 2006 CrossRef; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (c) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (d) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS; (e) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef PubMed; (f) J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718–10801 CrossRef PubMed; (g) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef PubMed; (h) P. T. Lowe and D. O'Hagan, Chem. Soc. Rev., 2023, 52, 248–276 RSC.
- (a) J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef PubMed; (b) T. Wu, A. Li, K. Chen, X. Peng, J. Zhang, M. Jiang, S. Chen, X. Zheng, X. Zhou and Z.-X. Jiang, Chem. Commun., 2021, 57, 7743–7757 RSC.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef PubMed; (b) H. Yanai and T. Taguchi, Eur. J. Org Chem., 2011, 2011, 5939–5954 CrossRef; (c) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef PubMed.
- (a) C. Leriche, X. He, C.-W. T. Chang and H.-W. Liu, J. Am. Chem. Soc., 2003, 125, 6348–6349 CrossRef PubMed; (b) G. Magueur, B. Crousse, M. Ourévitch, D. Bonnet-Delpon and J.-P. Bégué, J. Fluorine Chem., 2006, 127, 637–642 CrossRef.
- For selected reviews, see: (a) G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 CrossRef; (b) M. Decostanzi, J.-M. Campagne and E. Leclerc, Org. Biomol. Chem., 2015, 13, 7351–7380 RSC; (c) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef; (d) X.-J. Zhang, Y.-M. Cheng, X.-W. Zhao, Z.-Y. Cao, X. Xiao and Y. Xu, Org. Chem. Front., 2021, 8, 2315–2327 RSC; (e) P. H. S. Paioti, S. A. Gonsales, S. Xu, A. Nikbakht, D. C. Fager, Q. Liu and A. H. Hoveyda, Angew. Chem., Int. Ed., 2022, 61, e202208742 CrossRef; (f) K. Wang and W. Kong, ACS Catal., 2023, 13, 12238–12268 CrossRef.
- (a) D. G. Cox, N. Gurusamy and D. J. Burton, J. Am. Chem. Soc., 1985, 107, 2811–2812 CrossRef; (b) I. Nowak and M. J. Robins, Org. Lett., 2005, 7, 721–724 CrossRef; (c) J. Zheng, J. Cai, J.-H. Lin, Y. Guo and J.-C. Xiao, Chem. Commun., 2013, 49, 7513–7515 RSC; (d) J. Zheng, J.-H. Lin, J. Cai and J.-C. Xiao, Chem.–Eur. J., 2013, 19, 15261–15266 CrossRef; (e) K. Aikawa, W. Toya, Y. Nakamura and K. Mikami, Org. Lett., 2015, 17, 4996–4999 CrossRef.
- (a) Y. Zhao, W. Huang, L. Zhu and J. Hu, Org. Lett., 2010, 12, 1444–1447 CrossRef; (b) B. Gao, Y. Zhao, M. Hu, C. Ni and J. Hu, Chem.–Eur. J., 2014, 20, 7803–7810 CrossRef PubMed; (c) X.-P. Wang, J.-H. Lin, J.-C. Xiao and X. Zheng, Eur. J. Org Chem., 2014, 2014, 928–932 CrossRef; (d) Q. Luo, X. Wang and J. Hu, Tetrahedron, 2022, 113, 132694 CrossRef.
- (a) M. Hu, C. Ni, L. Li, Y. Han and J. Hu, J. Am. Chem. Soc., 2015, 137, 14496–14501 CrossRef PubMed; (b) Z. Zhang, W. Yu, C. Wu, C. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 273–277 CrossRef; (c) R.-Y. Yang, H. Wang and B. Xu, Chem. Commun., 2021, 57, 4831–4834 RSC; (d) G. Wang, W. Li, T. Liu, Y. Zhang, B. Wang, F. Xue, W. Jin, C. Ma, Y. Xia and C. Liu, Chem. Sci., 2022, 13, 11594–11599 RSC.
- (a) H.-P. Fiedler, C. Bruntner, J. Riedlinger, A. T. Bull, G. Knutsen, M. Goodfellow, A. Jones, L. Maldonado, W. Pathom-aree, W. Beil, K. Schneider, S. Keller and R. D. Sussmuth, J. Antibiot., 2008, 61, 158–163 CrossRef PubMed; (b) C. C. C. Wang, Y.-M. Chiang, M. B. Praseuth, P.-L. Kuo, H.-L. Liang and Y.-L. Hsu, Basic Clin. Pharmacol. Toxicol., 2010, 107, 583–589 CrossRef; (c) S. K. Manna, S. Giri, S. Mondal, R. N. Sana, A. K. Samal and A. Mandal, ChemistrySelect, 2023, 8, e202203150 CrossRef; (d) C. Besley, D. P. Rhinehart, T. Ammons, B. C. Goess and J. S. Rawlings, Bioorg. Med. Chem. Lett., 2017, 27, 3087–3091 CrossRef PubMed.
- For selected reviews, see: (a) P. J. Brothers and W. R. Roper, Chem. Rev., 1988, 88, 1293–1326 CrossRef; (b) C. Ni and J. Hu, Synthesis, 2014, 46, 842–863 CrossRef; (c) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef; (d) J. W. Lee, K. N. Lee and M.-Y. Ngai, Angew. Chem., Int. Ed., 2019, 58, 11171–11181 CrossRef PubMed; (e) J.-H. Lin and J.-C. Xiao, Acc. Chem. Res., 2020, 53, 1498–1510 CrossRef PubMed; (f) J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214–8247 RSC; (g) F.-L. Qing, X.-Y. Liu, J.-A. Ma, Q. Shen, Q. Song and P. Tang, CCS Chem., 2022, 4, 2518–2549 CrossRef; (h) X. Ma, J. Su and Q. Song, Acc. Chem. Res., 2023, 56, 592–607 CrossRef PubMed.
- (a) F. Wang, T. Luo, J. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153–7157 CrossRef PubMed; (b) L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2013, 52, 12390–12394 CrossRef PubMed; (c) X.-Y. Deng, J.-H. Lin, J. Zheng and J.-C. Xiao, Chem. Commun., 2015, 51, 8805–8808 RSC; (d) A. García-Domínguez, T. H. West, J. J. Primozic, K. M. Grant, C. P. Johnston, G. G. Cumming, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2020, 142, 14649–14663 CrossRef PubMed.
- For selected reviews on metal carbenes and conjugated ene-yne-ketones, see: (a) L. D. Shirtcliff, S. P. McClintock and M. M. Haley, Chem. Soc. Rev., 2008, 37, 343–364 RSC; (b) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef PubMed; (c) S. Teng and J. Zhou, Chem. Soc. Rev., 2022, 51, 1592–1607 RSC . For selected recent examples on conjugated ene-yne-ketones, see:; (d) S. Mao, L. Tang, C. Wu, X. Tu, Q. Gao and G. Deng, Org. Lett., 2019, 21, 2416–2420 CrossRef PubMed; (e) T. Chen, W. Wu and Z. Weng, Tetrahedron, 2019, 75, 130751 CrossRef; (f) J. Huang, F. Li, L. Cui, S. Su, X. Jia and J. Li, Chem. Commun., 2020, 56, 4555–4558 RSC; (g) H. Peng, Y. Wan, Y. Zhang and G. Deng, Chem. Commun., 2020, 56, 1417–1420 RSC; (h) L. Tang, Y. Zhang and G. Deng, J. Org. Chem., 2021, 86, 13245–13251 CrossRef PubMed; (i) Y. Wan, Y. Zhu, H. Peng and G. Deng, J. Org. Chem., 2022, 87, 281–293 CrossRef PubMed; (j) Q. Jiang, A. Li, X. Liu, Y. Yu, B. Zhu and H. Cao, J. Org. Chem., 2022, 87, 7056–7063 CrossRef PubMed; (k) H.-J. Cho, Y. L. Kim and J. H. Kim, J. Org. Chem., 2022, 87, 16424–16435 CrossRef PubMed; (l) M. Zhang, M. Liu, Y. Qiu, M. Peng, Z. Zhang, S. Song, X.-B. Chen and F. Yu, Adv. Synth. Catal., 2024, 366, 2363–2369 CrossRef; (m) G. Zhang, X. Cai, J. Jia, B. Feng, K. Yang and Q. Song, ACS Catal., 2023, 13, 9502–9508 CrossRef; (n) H. Peng, Y. Zhang and G. Deng, J. Org. Chem., 2023, 88, 7038–7045 CrossRef PubMed; (o) J. Wang, Z. Li, S. Bai, Q. Zhou, T. Wu, Z. Hu and X. Xu, Chin. Chem. Lett., 2023, 34, 107823 CrossRef; (p) S.-Y. Zou, F. Yang, X. Zhao, X.-G. Ren, Z.-S. Chen and K. Ji, Org. Chem. Front., 2023, 10, 767–773 RSC.
- (a) Z. Feng, Q.-Q. Min and X. Zhang, Org. Lett., 2016, 18, 44–47 CrossRef; (b) X. Zeng, Y. Li, Q.-Q. Min, X.-S. Xue and X. Zhang, Nat. Chem., 2023, 15, 1064–1073 CrossRef PubMed.
- (a) H. Zhang, X. Li, Q. Shi, Y. Li, G. Xia, L. Chen, Z. Yang and Z.-X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3763–3767 CrossRef PubMed; (b) X. Li, N. Li, L. Yang, R. Jiang, Y. He, J. Shi, Z.-X. Jiang and Z. Yang, ACS Catal., 2023, 13, 12755–12765 CrossRef; (c) Y. Mao, N. Li, J. Liu, Z.-X. Jiang and Z. Yang, Org. Lett., 2023, 25, 7567–7572 CrossRef; (d) Q. Zhou, M. Huang, Y. Shen, Z. Chen, L. Xu and Z. Yang, Org. Lett., 2024, 26, 1212–1217 CrossRef.
- H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aubé, Chem. Rev., 2022, 122, 12544–12747 CrossRef.
- M. E. Bergman, M. Bhardwaj and M. A. Phillips, Plant J., 2021, 107, 493–510 CrossRef.
- L. M. Ulatowski and D. Manor, Neurobiol. Dis., 2015, 84, 78–83 CrossRef PubMed.
- E. J. Westover and D. F. Covey, J. Membr. Biol., 2004, 202, 61–72 CrossRef PubMed.
- (a) P. Tian, C.-Q. Wang, S.-H. Cai, S. Song, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2016, 138, 15869–15872 CrossRef PubMed; (b) H.-J. Tang, L.-Z. Lin, C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2017, 56, 9872–9876 CrossRef; (c) W.-J. Yoo, J. Kondo, J. A. Rodríguez-Santamaría, T. V. Q. Nguyen and S. Kobayashi, Angew. Chem., Int. Ed., 2019, 58, 6772–6775 CrossRef; (d) X. Yu, A. Maity and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202310288 CrossRef.
- (a) K. Sinha, J. Das, P. B. Pal and P. C. Sil, Arch. Toxicol., 2013, 87, 1157–1180 CrossRef; (b) H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental procedures, screening of reaction conditions, characterization data, and copies of NMR. CCDC 2356931. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08247h |
| This journal is © The Royal Society of Chemistry 2025 |