
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu-mediated bipolar-type extended π-conjugated microporous polymers for lithium-ion battery cathodes with high energy density and fast-charging capability†
Yitao
Li‡
,
Ju
Duan‡
,
Yuzhu
Wang
,
Likuan
Teng
,
He
Liu
,
Jiaqiang
Li
,
Mengqi
Liu
,
Weisi
He
,
Huawei
Hu
 ,
Lulu
Wang
,
Wei
Lyu
* and
Yaozu
Liao
*
,
Lulu
Wang
,
Wei
Lyu
* and
Yaozu
Liao
*
State Key Laboratory of Advanced Fiber Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: wlyu@dhu.edu.cn; yzliao@dhu.edu.cn
First published on 21st May 2025
Abstract
Engineering lithium-ion battery (LIB) cathode materials with high energy density and fast-charging capability plays a significant role in the development of next-generation lightweight and high-performance storage devices for near space vehicles and electric aircraft. Herein, we propose a post-coordination strategy using Cu2+ to mediate a redox-active bipolar-type conjugated microporous polymer (CMP) incorporating porphyrin and pyrrole active groups (PPCMP). The as-synthesized Cu2+ mediated PPCMP (PPCMP-Cu) not only retains the high surface area (618 m2 g−1) of PPCMP, but also features an extended π-conjugated structure, a narrowed band gap, increased bipolar active sites, and optimized micro/mesopores, maximizing the utilization of active sites and enhancing ion diffusion kinetics. As the LIB cathode, PPCMP-Cu demonstrates an improved ion diffusion rate of 10−9 cm2 s−1 and a higher capacity of 285.1 mA h g−1 at 300 mA g−1, compared to the 10−10 cm2 s−1 and 135.5 mA h g−1 achieved by PPCMP. Moreover, PPCMP-Cu delivers an exceptional energy density of 702 W h kg−1 (based on the cathode) at 300 mA g−1 and an ultra-fast charging capability of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 464 W kg−1 with an ultra-short charging time of just 76 s at 5 A g−1. Besides, PPCMP-Cu shows a stable cycling life, with a 0.010% capacity fading rate per cycle at 2 A g−1 over 5000 cycles. This work paves an avenue for designing high-performance CMP cathode materials for LIBs with high energy density and fast-charging capability.
464 W kg−1 with an ultra-short charging time of just 76 s at 5 A g−1. Besides, PPCMP-Cu shows a stable cycling life, with a 0.010% capacity fading rate per cycle at 2 A g−1 over 5000 cycles. This work paves an avenue for designing high-performance CMP cathode materials for LIBs with high energy density and fast-charging capability.
Introduction
The advancement of near space vehicles and electric aircraft has significantly driven the demand for lightweight and high-performance lithium-ion batteries (LIBs) in the future. As the most critical component of LIBs, the cathode plays a pivotal role in determining the battery's energy storage capability. Inorganic materials, like LiFePO4, LiCoO2, and LiNixCoyMnz, are currently the most widely studied commercial cathode materials due to their easy preparation and stable performance.1–3 However, the limited energy density, poor fast-charging stability, and the shortage of their transition metal resources have greatly limited their further development.4–6 Therefore, it is essential to develop non-polluting, renewable cathode materials that exhibit excellent energy density and fast-charging capability to meet the demands of next-generation energy storage devices for near space electric equipment.7Organic martials are considered as sustainable options for electrodes due to their abundant active sites, low density, environmental friendliness, and sustainable production. Among these, conjugated microporous polymers (CMPs), as a class of porous organic polymers (POPs), have attracted increasing interest as promising electrode materials for LIBs.8,9 This interest primarily stems from the fact that (1) the structural stability of CMPs can prevent dissolution in electrolytes and enhance cycling stability compared to small organic molecules; (2) their intrinsic micropores and high specific surface area facilitate interfacial contact between electrodes and electrolytes, promote ion diffusion and provide abundant active sites for ion storage; (3) the chemical designability of CMPs offers opportunities to develop organic cathode materials with bipolar active sites, i.e., n-type and p-type groups, enabling a broader operating voltage range and high energy density; (4) the intrinsic π-conjugated skeleton facilitates efficient charge transfer.10–15 For example, Zhang et al.16 synthesized CMP (TzPz) using dihydrophenazine and 2,4,6-triphenyl-1,3,5-triazine as monomers via the Buchwald–Hartwig coupling reaction. The obtained high surface area (397 m2 g−1) promotes the contact between the electrolyte and cathode. When served as a cathode for LIBs, a high capacity (202 mA h g−1 at 200 mA g−1) and rate performance (125 mA h g−1 at 5 A g−1) were achieved. Yang et al.17 developed a bipolar-type CMP (TPSZ) using thiazole as an n-type group to store Li+ and triphenylamine as a p-type group to store ClO4−, with a capacity of 237 mA h g−1 at 50 mA g−1 and a wide voltage range from 1.5 to 4.5 V achieved. Although these advancements, challenges remain in optimizing CMP-based cathode materials for achieving LIBs with both high energy density and fast-charging capability, including an optimal volume ratio of micropores and mesopores for improved ion diffusion kinetics, maximized utilization of bipolar active sites for increased capacity, and a narrowed band gap for faster electron transfer.16,18–21
Porphyrin units have been widely utilized in designing organic electrode materials due to their extended π-conjugated structure, multiple electron transfer capabilities, high redox activity, and ability to coordinate with various metals. Specifically, metal-free porphyrin can function as bipolar groups, binding with 2PF6− and 2Li+ through electron acceptance and donation at their macrocyclic core within a single system.15,22,23 Additionally, coordinating metal atoms like Cu can adjust the electronegativity, enabling the binding of 2PF6− and 4Li+ to a single Cu-porphyrin unit. This characteristic makes Cu-porphyrin particularly attractive as bipolar active sites for polymeric cathode materials for LIBs.15 For instance, Liu et al.23 synthesized a Cu-porphyrin-based POP (COP500-CuT2TP) using an electrochemical polymerization method. COP500-CuT2TP showed a specific surface area of 38 m2 g−1, a high capacity of 420 mA h g−1 at 100 mA g−1, and an ion diffusion rate of 10−10 cm2 s−1. It is expected that Cu-porphyrin-based CMPs with higher specific surface area and suitable pore distribution could achieve faster ion diffusion rates, higher energy densities and shorter charging times.
Herein, we propose a Cu2+-mediated strategy to develop a high-performance Cu-porphyrin based CMP. A Cu2+-mediated redox-active CMP, incorporating porphyrin and pyrrole active groups, was synthesized via chemical oxidative polymerization combined with a post-coordination method. Tetrapyrrole phenyl porphyrin (PP), a monomer with bipolar properties, was used to first polymerize into a porphyrin and pyrrole-based CMP (PPCMP). The pyrrole unit was chosen for its typical p-type energy storage sites (binding with one PF6−) and its facilitation of polymerization. Additionally, the incorporation of a benzene unit imparts the CMP with a planar and extended π-conjugated structure, facilitating charge transfer and ensuring a high surface area (968 m2 g−1) due to its rigid skeleton. As shown in Fig. 1, the electron-withdrawing effect of Cu2+ enables its post-coordinating on PPCMP (PPCMP-Cu), which homogenizes the electron distribution within the N4 macrocyclic core and facilitates binding with an additional 2 Li+, similar to a pristine Cu-porphyrin unit. This Cu2+-mediating also increases the conjugation degree of PPCMP, reduces the band gap for promoting electron transfer, and optimizes micropore sizes verified by simulation calculation and experimental tests, improving active site exposure and ion diffusion kinetics. As expected, PPCMP-Cu demonstrates high specific capacities of 285.1 at 300 mA g−1 and 109.2 mA h g−1 at 5 A g−1, respectively, along with exceptional cycling stability, exhibiting a capacity fading rate of 0.010% per cycle at 2 A g−1 over 5000 cycles, with a retained specific capacity of 130.8 mA h g−1. As a result, PPCMP-Cu displays an ultra-high power density of 12464 W kg−1 and energy density of 263 W h kg−1 (based on the cathode) with an ultra-short charging time of just 76 s at 5 A g−1, and a desirable energy density of 702 W h kg−1 with a charging time of 60 min at 300 mA g−1. This study gives insights into synthesizing bipolar-type CMPs with maximum unitization of active sites and facilitated ion diffusion kinetics for high energy density and fast-charging LIB cathodes.
 | ||
| Fig. 1 Schematic illustration of the advantages of the Cu-mediated bipolar-type CMP with abundant exposed active sites, high conjugation degree, and optimized pore sizes for high-performance LIB cathode materials. | ||
Results and discussion
The schematics for the preparation of PPCMP and PPCMP-Cu samples using the chemical polymerization coupled post-coordination method are shown in Fig. 2a, Schemes S2 and S3.† The PP monomer was synthesized through the Alder–Longo reaction (Scheme S1†). The chemical structures of PP, PPCMP, and PPCMP-Cu samples were first characterized by Fourier Transform Infrared (FT-IR), 1H nuclear magnetic resonance (1H NMR), and solid-state 13C cross-polarization magic-angle-spinning NMR (13C MAS NMR) spectroscopy (Fig. S1–S3†). The disappearance of the stretching vibration bands of the aldehyde at 2730 cm−1 for 4-(1H-pyrrol-1-yl) benzaldehyde (TT) shown in the FT-IR spectrum of PP indicates the successful synthesis of the PP monomer (Fig. S1†).24 The 1H NMR spectrum of PP shown in Fig. S1b† further supports the formation of the PP monomer.24 As shown in Fig. 2b, the decreased intensity of the C–H vibration peak around 720 cm−1 suggested the successful polymerization of PPCMP.25 This is further supported by the observed characteristic peak around 118 ppm, ascribing to the C–C coupling between pyrrole groups,24 in the 13C NMR spectrum of PPCMP (Fig. S2†). Different contents of CuCl2 (i.e., 50, 83, and 91 mol% of CuCl2) were adopted to post-coordinate PPCMP, with samples named PPCMP-Cu-1, PPCMP-Cu, and PPCMP-Cu-10, respectively. The highest Cu element measured from the inductively coupled plasma optical emission spectrometer (ICP-OES) test was obtained for PPCMP-Cu (approximately 80% of the theoretical post-coordination value) (Table S1†). The appearance of a new vibration peak corresponding to the Cu–N bond located at around 1000 cm−1 in the FT-IR spectra of PPCMP-Cu-1, PPCMP-Cu, and PPCMP-Cu-10 suggests that the Cu atom is successfully incorporated into the porphyrin of the polymer (Fig. 2b and S3†).26,27 The powder X-ray diffraction (XRD) patterns of PPCMP and PPCMP-Cu samples exhibit similar broad diffraction peaks without the characteristic peaks of CuCl2 and metal Cu, demonstrating their amorphous nature and the preservation of the polymer skeleton after the introduction of Cu (Fig. S4†). The observed peaks located at 935.1 eV and 954.9 eV in the X-ray Photoelectron Spectroscopy (XPS) spectrum of PPCMP-Cu, corresponding to Cu 2p3/2 and Cu 2p1/2, respectively, demonstrate that Cu is coordinated with the porphyrin ring as the ion state (Fig. 2c).28 Additionally, two characteristic peaks located at 400.3 eV and 398.3 eV corresponding to the amine N (–NH–) and imine N (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) in the porphyrin ring are observed in the N 1s XPS spectra of PPCMP (Fig. 2d and Table S2†).28,29 The observed new peak at 398.8 eV, attributing to the Cu–N bond, and a decreased percentage of pyrrolic N in the N 1s XPS spectra of PPCMP-Cu further indicate that Cu is coordinated by substituting the H of pyrrolic N–H within the porphyrin ring.28,30
C–) in the porphyrin ring are observed in the N 1s XPS spectra of PPCMP (Fig. 2d and Table S2†).28,29 The observed new peak at 398.8 eV, attributing to the Cu–N bond, and a decreased percentage of pyrrolic N in the N 1s XPS spectra of PPCMP-Cu further indicate that Cu is coordinated by substituting the H of pyrrolic N–H within the porphyrin ring.28,30
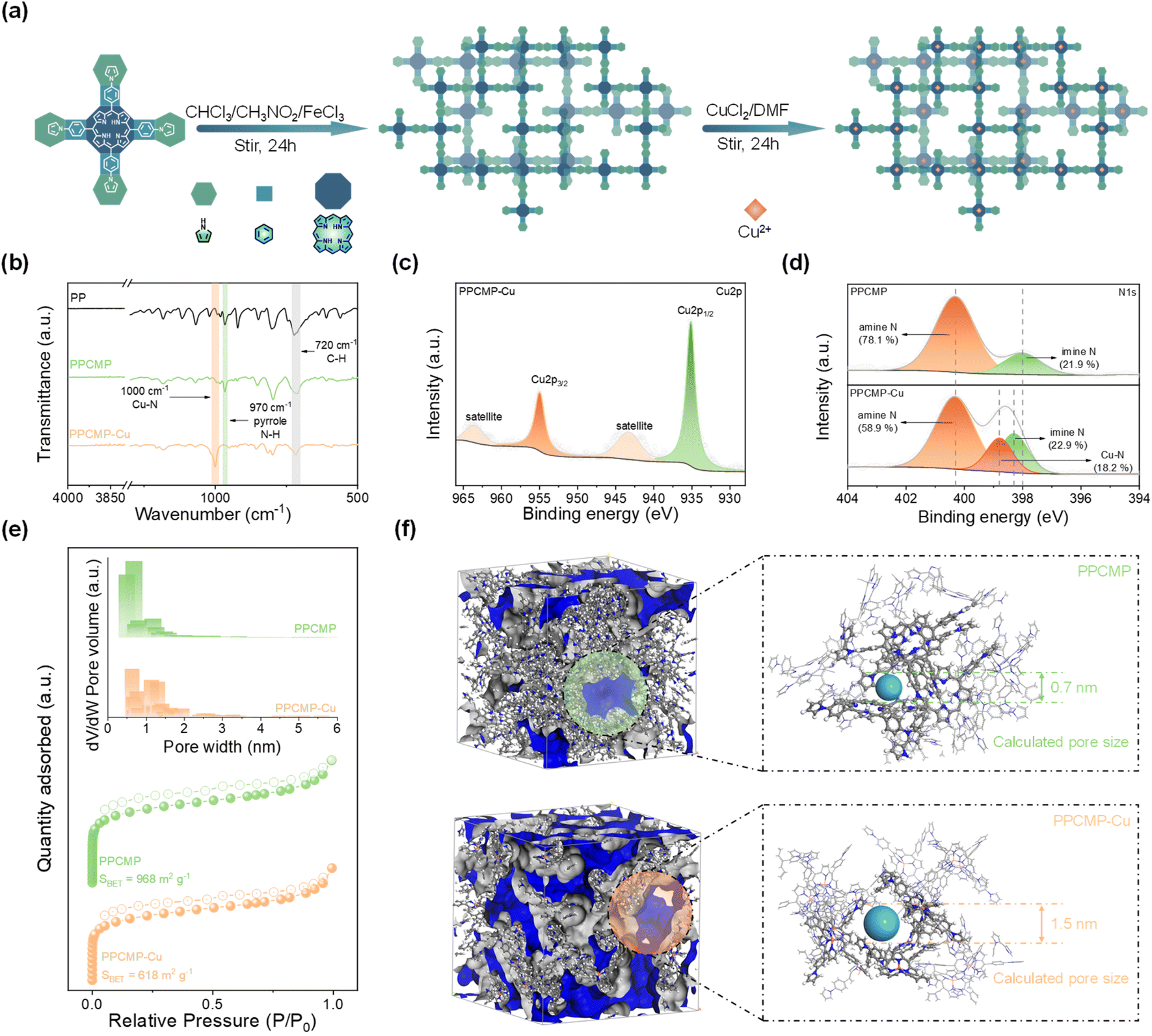 | ||
| Fig. 2 (a) Schematic synthetic procedure of PPCMP-Cu. (b) FT-IR spectra of PP, PPCMP, and PPCMP-Cu. (c) Cu 2p XPS spectra of PPCMP-Cu and (d) N 1s XPS spectrum of PPCMP and PPCMP-Cu. (e) BET specific surface areas and the pore size distribution of PPCMP and PPCMP-Cu. (f) The architecture of pores in the structural models (blue region indicates the void space for N2) and the confined fragmentation with pore sizes of PPCMP and PPCMP-Cu. | ||
The porosities of the PPCMP and PPCMP-Cu samples were then investigated by N2 adsorption/desorption measurements at 77.3 K. As displayed in Fig. 2e, both samples exhibit a combination sorption isotherm of type I featuring a sharp uptake in the low-pressure region and type IV featuring a large hysteresis loop in the high-pressure range, indicating the main existence of micropores in PPCMP and PPCMP-Cu.31 The Brunauer–Emmett–Teller (BET) surface area (SBET) of PPCMP-Cu (618 m2 g−1) is lower than that of PPCMP (968 m2 g−1), which may be due to the enlarged micropore size after coordinating with Cu2+. As the pore size distribution curves shown in Fig. 2e, the pore size proportion of 1–2 nm increases from 21.9% to 47.1% and the proportion of 0–1 nm decreases from 69.8% to 37.5% upon increasing the coordinating Cu2+ ratio from 0 (PPCMP) to 80% coordination ratio (PPCMP-Cu) (Fig. S5†). Such a trend confirms that the Cu2+ coordination can regulate the pore size of PPCMP. Pore size distribution simulations were conducted to help understanding the possible reason. Dimers of PPCMP and PPCMP-Cu were developed as models for LAMMPS calculation (Fig. S6 and 2f, for details see the ESI†). It can be seen that the Cu2+ coordination makes the connected spatial angle of dimers reduce from 128.0° (PPCMP) to 80.3° (PPCMP-Cu) (Fig. S6a and b†). The possible reason is that the formation of the Cu–N bond leads to the extended π-conjugated skeleton of the porphyrin ring and lowers the distortion degree, which could make the intrinsic micropore size larger.27,28 As shown in Fig. 2f, S6c and d,† the simulated PPCMP-Cu displays a larger average pore size of 1.5 nm, compared with the average pore size of 0.7 nm for simulated PPCMP. The simulated specific surface area of PPCMP-Cu (772 m2 g−1) and PPCMP (952 m2 g−1) are close to those obtained from the experimental results, indicating the rationality of the simulated structures. These obtained larger pore sizes and high surface area are expected to be more favorable for ion insertion and faster diffusion with the large ion radius as PF6− for the cathode material, which could promote the activation of the redox reaction and ion storage.15,21 Therefore, it is expected that PPCMP-Cu samples are more favorable for cathode materials than PPCMP. The morphologies of samples were then investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. S7,† all samples exhibit close-packed nanosphere-like morphology. TEM images (Fig. S8†) further demonstrate the close-packed spherical particles for PPCMP and PPCMP-Cu. The similar morphologies of PPCMP and PPCMP-Cu indicate that the polymer retains the skeleton after the coordination of Cu2+. The absence of dark aggregated dots of metal cluster in the TEM images (Fig. S8†) of PPCMP-Cu suggests the homogeneous distribution of coordinated Cu2+. It can be further supported by the observed homogeneous distribution of C, N, and Cu in the element energy dispersive spectroscopy (EDS) mapping of PPCMP-Cu (Fig. S9†). The high thermal stability of PPCMP and PPCMP-Cu was finally studied by thermogravimetric analysis (TGA) with an initial decomposition temperature at around 350 °C in a nitrogen (N2) atmosphere. The PPCMP displayed a 63% mass retention upon increasing the temperature to 900 °C. In comparison, PPCMP-Cu exhibits better thermal stability, with a minimal weight loss occurring until 500 °C and a 73% mass retention at 900 °C, further indicating the possible enhanced intermolecular π-stacking of PPCMP-Cu (Fig. S10†).32
To further elucidate the structural characteristics of PPCMP-Cu, X-ray absorption near-edge structure (XANES) spectroscopy was employed to probe the oxidation state and coordination environment of Cu atoms (Fig. 3a). The Cu K-edge XANES spectrum of PPCMP-Cu (Fig. 3a) displays a position nearly identical to that of copper phthalocyanine (CuPc), indicating that the Cu ions in PPCMP-Cu predominantly exist in the +2 state. Additionally, in the k3-weighted Fourier transform (FT) extended X-ray absorption fine structure (EXAFS) spectra (Fig. 3b), both PPCMP-Cu and CuPc exhibit a prominent peak at approximately 1.53 Å, which is notably different from the characteristic Cu–Cu peak at 2.24 Å observed in metallic Cu foil. This clearly suggests that the dominant coordination environment in PPCMP-Cu is Cu–N bonding. The coordination number of the Cu ions was determined by fitting of the EXAFS data (Fig. 3c, S11, and Table S3†), the coordination environment in PPCMP-Cu gives mainly priority to Cu–N coordination. Wavelet transform (WT) contour plots were also employed to investigate the local environment of the Cu ions (Fig. 3d–f). Compared to the Cu–Cu bond peak located at 8 Å−1 observed in Cu foil, the peak maximum of PPCMP-Cu appears at 6.5 Å−1 corresponding to the Cu–N bonds, which are also observed in CuPc. These results collectively confirm that Cu atoms in PPCMP-Cu are predominantly coordinated by nitrogen atoms rather than forming metallic clusters, reinforcing the successful integration of Cu2+ into the porphyrin framework via Cu–N bonds.33
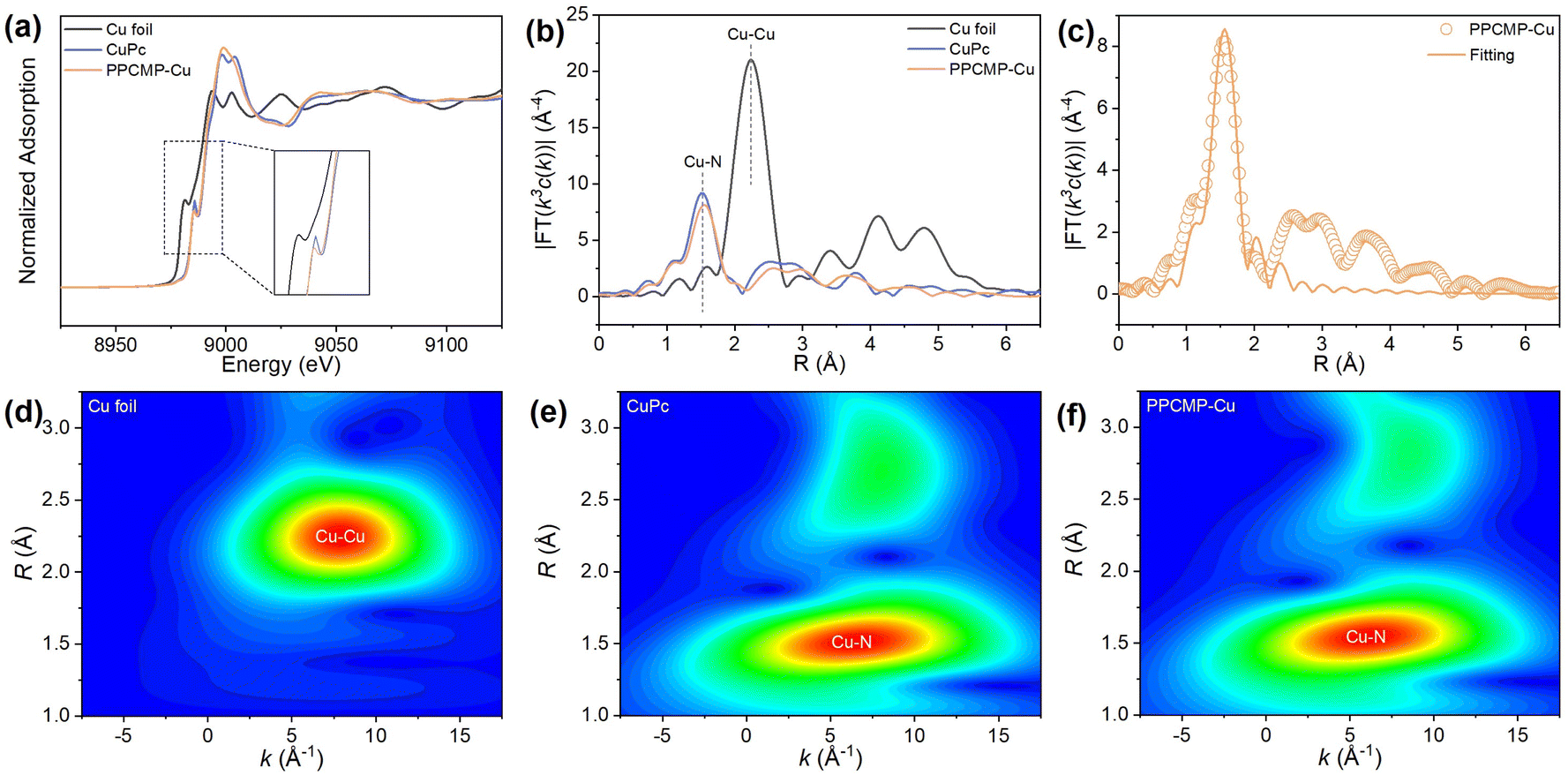 | ||
| Fig. 3 (a) Normalized XANES spectra and (b) FT-EXAFS spectra at the Cu K-edge of PPCMP-Cu, copper phthalocyanine, and Cu foil. (c) EXFAS fitting curves for PPCMP-Cu in R-space. WT-EXAFS spectra of discriminating radial distance and k-space resolution of (d) Cu foil, (e) copper phthalocyanine, and (f) PPCMP-Cu. | ||
Density functional theory (DFT) calculations were first performed to understand the Cu2+ post-coordinate effect on the band gap and charge distribution of PPCMP and PPCMP-Cu (calculation details are presented in the ESI†). The repeating units of PPCMP (PP) and PPCMP (Cu-PP) were adopted as models (Fig. S12†). As shown in Fig. 4a, the band gap reduces from 2.67 eV for PP to 2.49 eV for Cu-PP, with the highest occupied molecular orbital and lowest unoccupied molecular orbital (HOMO/LUMO) energy of −5.09, −5.12, and −2.42, −2.63 eV calculated for PP and Cu-PP, respectively. Meanwhile, the electrical conductivity of PPCMP-Cu was 8.20 × 10−15 S cm−1, which is higher than that of PPCMP (1.08 × 10−15 S cm−1) in Fig. S13.† This improvement can be attributed to the enhanced charge delocalization resulting from the π–d conjugation effect introduced by Cu2+ coordination.34,35 The obtained results further support the enhanced conjugation degree after coordinating Cu2+, which is beneficial for faster electron transfer and redox kinetics.15,18,36 From the molecular electrostatic potential (MESP) map shown in Fig. 4b, the inner region of the porphyrin ring tends to be electrically neutral for Cu-PP in contrast to that in PP. It indicates that Cu2+ can regulate the electric field distribution of N4 at its macrocyclic core to combine additional 2 cations than PP, imparting Cu-PP with a bipolar characteristic of 4 cations and 2 anions storage.23 Combined with the one anion storage for per pyrrole, the theoretical capacity of PPCMP-Cu is calculated as high as 286.2 mA h g−1 (calculation details based on eqn (S1) are displayed in the ESI†). Benefitted by its high porosity and larger micropore sizes, it is expected that PPCMP-Cu is an ideal cathode material with high energy density and fast-charging performance.
 | ||
| Fig. 4 (a) HOMO/LUMO energy diagrams with the corresponding configuration and (b) the electrostatic potential map of the PP and Cu-PP (red indicates the positive charge and blue indicates the negative charge). (c) CV curves at a scan rate of 0.2 mV s−1 of PPCMP-Cu. (d) Cycling performances at a current density of 300 mA g−1, (e) rate performances at different current densities, and (f) long-term cycling stability at a current density of 2 A g−1 of PPCMP and PPCMP-Cu. (g) Comparison of the electrochemical properties of PPCMP-Cu to other cathodes in LIBs.17,29–31 | ||
The electrochemical performance of PPCMP and PPCMP-Cu was then tested. From the cyclic voltammetry (CV) curves shown in Fig. S14† and 4c, four distinct couples of redox peaks (around 3.85/3.84, 3.09/3.03, 2.56/2.58, and 1.65/1.59 V vs. Li+/Li), corresponding to the two combination steps of 6 PF6− and two combination steps of 4 Li+ can be observed for PPCMP-Cu, while only two obvious couples of redox peaks located at 2.31/3.17 and 2.75/3.90 V vs. Li+/Li are noted for PPCMP.15 The observed peaks in the low-voltage range and high-voltage range belonging to the n-type and p-type active sites, respectively, observed in the CV cures of PPCMP-Cu demonstrate the typical bipolarization redox characteristics.37 This result is in accordance with the different active sites analysis in the above DFT calculation, the porphyrin center can be combined with Li+ and PF6− and the pyrrole ring can be combine with PF6−. The larger area of CV curves of PPCMP-Cu, compared with PPCMP at the same scan rate, also indicates its increased capacity. As shown in Fig. 4c, the CV curves of PPCMP-Cu are well over-lapped and the redox peaks remain stable in the subsequent cycling, revealing its remarkable stability.
Herein, we utilized PPCMP and PPCMP-Cu as the active materials for the fabrication of electrodes. The tap densities for the electrodes with low mass loading as around 0.3–0.35 mg cm−2 were measured to be 0.211 g cm−3, which increased to 0.300 g cm−3 with a high mass loading of 1.0 mg cm−2 (calculation details based on eqn (S2)†). The tap density of PPCMP-Cu is significantly lower than that of conventional inorganic materials, which can be attributed to the higher proportion of conductive additives and binders, as well as the inherently lightweight feature of CMPs. The electrolyte uptake for the electrodes with low mass loading (around 0.3–0.35 mg cm−2) were measured as 280% and 360% for PPCMP and PPCMP-Cu (calculation details based on eqn (S3)†), respectively. With a high mass loading of 1.0 mg cm−2, the electrolyte uptake of PPCMP-Cu reached 390%. The high electrolyte uptake facilitates sufficient accessibility to active sites during cycling.38
As expected, PPCMP-Cu exhibits an outstanding initial capacity of 301.7 mA h g−1 at a current density of 300 mA g−1 (Fig. 4d), which stabilizes at 285.1 mA h g−1 in the second cycle. This value is consistent with theoretical expectations, with an almost 100% utilization of active sites. After 200 cycles, an excellent specific capacity of 277.3 mA h g−1 remained for PPCMP-Cu, while only 135.5 mA h g−1 was kept for PPCMP. It should be mentioned that the capacity of PPCMP-Cu increased slightly after several cycles, which may be ascribed to the gradual activation of the electrode material.39 The rate performance of PPCMP and PPCMP-Cu was then evaluated at various current densities. As shown in Fig. 4e and S15,† PPCMP-Cu delivers specific capacities of 295.0, 238.2, 191.1, 148.3, and 109.2 mA h g−1 at current densities of 0.3, 0.5, 1, 2, and 5 A g−1, respectively. In particular, during the return process from 5 to 0.3 A g−1, the capacities recover to their initial values, indicating the excellent reversible redox kinetics for PPCMP-Cu.
Fig. 4f shows the long-term cycling performance of both samples tested at a current density of 2 A g−1. It can be seen that PPCMP-Cu shows an ultra-long stable cycling performance, with a capacity fading rate of only 0.010%, approximately 100% of coulombic efficiency, and a specific capacity of 130.8 mA h g−1 remained after 5000 cycles. Conversely, PPCMP exhibits a lower specific capacity of only 75.4 mA h g−1 after 5000 cycles. Furthermore, PPCMP-Cu delivers a high specific capacity of 207.1 mA h g−1 and an energy density of 457 W h kg−1 at 300 mA g−1 with the mass loading as ∼1.0 mg cm−2 (Fig. S16a†). When the current density is increased to 5000 mA g−1, PPCMP-Cu retains a specific capacity of 72.5 mA h g−1 with a power density of 12725 W kg−1 within 58 s. When the current density recovered to 300 mA g−1, a reversible capacity of 180.3 mA h g−1 is retained. Long-term cycling at a higher mass loading (1.0 mg cm−2) of PPCMP-Cu was also evaluated at 2000 mA g−1 (Fig. S16b†), where PPCMP-Cu retains a capacity of 85.1 mA h g−1 even after 2000 cycles. The capacity and high-rate performance of PPCMP-Cu are superior to those of recently reported CMPs and the porphyrin-based cathode (Fig. 4g), which possesses higher capacity (285.1 mA h g−1) at higher current density (300 mA g−1) with high active site utilization (∼100%) and exhibits 130 mA h g−1 after 5000 cycles at 2 A g−1. For example, TAPBA-NTCDA@MWCNTs displays only 97.6 mA h g−1 at 100 mA g−1 and 51.4 mA h g−1 after 10000 cycles at 2 A g−1.40 Meanwhile, Ni-TABQ displays 139 mA h g−1 (at 1 A g−1) after only 1000 cycles with limited active site utilization (65%).41 The superior electrochemical performance of PPCMP-Cu may be because the coordination of Cu2+ increases the exposed bipolar active sites of porphyrin and provides more pores in the scale of 1–2 nm and a smaller band gap of 2.49 eV.
Electrochemical reaction kinetics of the PPCMP and PPCMP-Cu cathodes were further analyzed. Fig. S17a† and 5a show the CV curves of PPCMP and PPCMP-Cu cathodes measured at various scan rates from 0.2 to 1 mV s−1. It can be seen that, when the scan rate increases, the CV curves of both PPCMP and PPCMP-Cu show similar shapes and the redox peaks shift slightly. These results indicated that both samples have good rate performance.42 We used Dunn's method43 to explore the contribution of the capacitive process for PPCMP and PPCMP-Cu (eqn (S4) and (S5)†). The peak currents in CV curves follow the linear relationship with the scan rate, indicating the characteristic of capacitive behavior and the related b values of both samples are calculated (Fig. S17b, 5b, and Table S4†). The b values of redox peaks in the high voltage range marked in Fig. S17b† and 5b are 0.78, 0.99, 0.62, and 0.99 for PPCMP (O1 and R1) and PPCMP-Cu (O2 and R2), respectively. It demonstrates that the ion storage for both samples is mainly controlled by the surface-dominated pseudocapacitive characteristic.43 It indicates that the easily accessible and exposed active sites existed in both samples. Trasatti analysis was then performed to quantitatively separate the diffusion-controlled and capacitive-controlled contributions. As shown in Fig. 5c and S18†, the capacitive-controlled contribution of PPCMP-Cu is about 51% at 0.2 mV s−1, which increases to 83% at 1 mV s−1. However for PPCMP, the capacitive-controlled contribution increased from 67% (0.2 mV s−1) to 88% (1 mV s−1). The less capacitive-controlled contribution and high diffusion-controlled contribution of PPCMP-Cu indicate that it exhibits an optimized pore distribution for ion diffusion.44
 | ||
| Fig. 5 (a) CV curves of PPCMP-Cu at different scan rates. (b) The b values of different peaks of PPCMP-Cu. (c) The capacitive-controlled contribution ratios at different rates of PPCMP and PPCMP-Cu. (d) GITT curve and corresponding ion diffusion coefficient for PPCMP-Cu. (e) The Ea was calculated from the EIS at different temperatures for PPCMP and PPCMP-Cu. (f) The concept image of ion transport variation between PPCMP and PPCMP-Cu. (g) Ragone plots compiling energy density versus power density (p-DPPZ,47 PT-BTA,48 TAPT-CNTCDA@CNTq,49 YPTPA,50 TPA-co-DDP-PROXYL,51 COP500-CuT2TP23). | ||
Galvanostatic intermittent titration technique (GITT) measurements were conducted to reveal the ions diffusion characteristics of both samples during the charging and discharging processes (eqn (S6)†). As shown in Fig. S19† and 5d, the average ion diffusion coefficient based on the GITT results is 2.98 × 10−12 cm2 s−1 for PPCMP, while that of PPCMP-Cu is 1.14 × 10−10 cm2 s−1.45 Obviously, the ion diffusion coefficient of PPCMP-Cu is significantly higher than that of PPCMP. These results are mainly because of the larger pore size structure with more accessible active sites of PPCMP-Cu. Molecular dynamics (MD) simulations were conducted using models of PPCMP and PPCMP-Cu in 1 M LiPF6 excluding solvent components of the electrolyte (Fig. S20a†).46 The Li+ diffusion in PPCMP-Cu is significantly faster than that in PPCMP over 250 ps from the time evolution of the mean square displacement (MSD) (Fig. S20b†), consistent with the experimentally observed enhancement in Li+ transport for PPCMP-Cu. Moreover, according to the electrochemical impedance spectroscopy (EIS) at different temperatures, the activation energy (Ea) of both samples as cathodes in the half-battery system was calculated (Fig. 5e and eqn (S7)†). The Ea of PPCMP-Cu is 0.46 eV, which is significantly lower than that of PPCMP (0.68 eV), further indicating that the facilitated ion transport in PPCMP-Cu resulted from its narrowed band gap (Fig. 5f). As a result, PPCMP-Cu delivers an ultra-high energy density of 702 W h kg−1 and a power density of 702 W kg−1 with lithium metal as the anode (charging time of 60 min) at a current density of 300 mA g−1 calculated from eqn (S8) and (S9)†. Moreover, upon increasing the current density to 5 A g−1, PPCMP-Cu displays an energy density of 263 W h kg−1 and an excellent power density of 12464 W g−1, with a charging time of just 76 s. The above results indicate that PPCMP-Cu exhibits a competitive higher energy density and fast-charging performance with an ultra-fast charging time, among POP-based cathode materials23,47–51 (Fig. 5g and Table S5†). For comparison, PPCMP only presents an energy density of 272 W h kg−1 with a power density of 680 W kg−1 (charging time of 24 min) at 300 mA g−1 and an energy density of 130 W h kg−1 with a power density of 12000 W kg−1 at 5 A g−1 (charging time of 39 s), respectively (Table S5†). In addition, a full-battery was also assembled by using PPCMP-Cu as the cathode and pre-lithiated graphite as the anode, demonstrating good electrochemical performance, including high energy density (216 W h kg−1 at 0.3 A g−1) and fast-charge capability (6684 W kg−1 at 5 A g−1) with a charging time of around 1 min (Fig. S21†). The increased energy density and power density, as well as the short charging time, indicates that the coordination of Cu2+ on PPCMP effectively increases the active sites and promotes electron transfer. To the best of our knowledge, PPCMP-Cu exhibits high performance of energy density and fast charging capability which ranks among organic cathode materials and also is the best of CMP-based cathode materials. In addition, PPCMP-Cu displays superior electrochemical performance, including higher energy density and enhanced fast-charging capability, compared to the recently reported conventional inorganic cathodes in lithium metal battery (LMB) and LIB systems (Table S6†).
Fig. S22† presents the ex situ FT-IR spectra of PPCMP-Cu at selected voltages (1.2–4.4 V) during the charge–discharge process to understand the ion storage mechanism. Notably, compared with the pristine FT-IR spectra, the characteristic peak intensity of P–F at 846 cm−1 gradually increased at voltages higher than 3.3 V during the charging process (p-doping reaction),19 which declined during the discharging process. It indicates the insertion and deintercalation of PF6−.52 Meanwhile, when the voltage was under 3.5 V during discharging (n-doping reaction), the peak intensity of CN (around 1645 cm−1) decreased, with an increased peak intensity of C–N (at 1330 cm−1) observed. This variation of peaks can be attributed to the occurrence of the insertion of Li+ on the CN reaction sites and the formation of Li–C–N.53 These phenomena demonstrate the bipolar reaction characteristics of PPCMP-Cu, which matches the bipolar characteristic analysis derived from the CV curves shown in Fig. 4c.
The same results can also be observed in ex situ XPS spectra. As shown by the N 1s XPS spectra of as-prepared pristine PPCMP-Cu in Fig. 6a, the peaks at 400.3, 398.8, and 398.3 eV, ascribed to pyrrolic N (C–N), Cu–N, and imine N (CN), respectively,30,54,55 are observed. After discharging to 1.2 V from the pristine state, the decreased proportion of the CN peak (from 20.1% to 9.8%) indicates Li+ insertion on the CN active sites from porphyrin during the discharge.55,56 Further charging to 4.4 V with Li+ desertion, the peak proportion of CN rose back to 20.0%. Notably, when charging or discharging to the voltage around 3 V, the peaks corresponding to Cu–N and CN are consistent with the position of the pristine state. In addition, a new peak corresponding to Cu+ appears at 932.8 eV when discharging to 1.2 V, in which the percentage of the Cu2+ peak (395.3 eV) decreased from 78.2% (4.4 V) to 69.9% (1.2 V).23 Upon subsequent charging to 4.4 V, the proportion for the Cu+ peak decreases from 30.1% to 21.8% (Fig. S23†). The valence changes of Cu in Cu–N bonds suggest that the coordinated Cu2+ can occur the redox reaction during cycling. Meanwhile, upon coordination of Cu2+ within the porphyrin ring, the resulting Cu–N bonds in PPCMP-Cu offer more active sites for Li ion storage than PPCMP. This result is also evidenced by the electric field distribution optimization in the macrocyclic core of porphyrin as discussed above in Fig. 4b.23 Notably, the peak area proportions of Cu–N remain stable at about 15% during cycling (Fig. 6a), confirming the structural robustness of the Cu–N coordination bond.
 | ||
| Fig. 6 (a) N 1s and (b) F 1s of ex situ XPS spectra for PPCMP-Cu at different charge states. (c) The structure evolution of the repeating unit of PPCMP-Cu during the charge/discharge process. (d) The discharge pathway was obtained from simulations. (e) The optimized different active sites were obtained via the MESP method. | ||
The same trend can be observed in the subsequent cycle process, proving the reversible bipolar ion storage capability of the porphyrin ring. In Fig. 6b, the observed characteristic peak at 688.0 eV in the F 1s XPS spectra for the as-prepared PPCMP-Cu was attributed to the polyvinylidene difluoride (PVDF) binder in the electrode slurry.57 After discharging to 1.2 V, a new peak at 685.4 eV, corresponding to the PF6− from the residual electrolyte, appeared.53,57 When charging to 4.4 V, the percentage of the P–F peak originating from PF6− increased from 9.1% to 18.0%. The variation of P–F peak intensity shows a consistent trend during the subsequent charging and discharging processes, indicating the occurrence of doping and de-doping of PF6− on PPCMP-Cu from the electrolyte. The above results confirm that PPCMP-Cu serves as both active storage sites of Li+ and PF6− during the charging and discharging processes, therefore endowing the cathode with high energy density.
DFT theory calculations were finally conducted to help understand the sequential structural evolution of PPCMP-Cu during successive cationization and anionization (calculation details are shown in the ESI†). Combined with FT-IR, XPS, and MESP of Cu-PP analysis above, the simulated charging and discharging processes of Cu-PP are shown in Fig. 6c.58 Upon charging, porphyrin and pyrrolic N act as p-type active sites for accepting electrons and binding with 2PF6− and 4PF6−, respectively. As shown in Fig. 6d, the Gibbs free energies of Cu-PP with 3 and 6PF6− (i.e., Cu-PP-3PF6 and Cu-PP-6PF6) are −28.46 and −30.66 eV, respectively, confirming the strong binding ability between these p-type active sites and PF6−. Fig. 6e shows that binding three PF6− (Cu-PP-3PF6) were located in the porphyrin ring with one PF6− and two pyrroles with two PF6−. The MESP maps of unbound pyrrole in Cu-PP-3PF6 exhibit red positive charge, in which PF6− prefers to interact with in further charging process. As more PF6− anions bind to the porphyrin ring and pyrrole units, the MESP map of Cu-PP-6PF6 shows a tendency toward a neutral electrostatic state. It should be mentioned that the increased distortion in the spatial structure of the Cu-PP monomer with higher PF6− binding is attributed to the large size of PF6−. Upon discharging, the Cu-porphyrin unit allows for the binding of 4Li+ by donating electrons (Fig. 6c). The calculated Gibbs free energies of Cu-PP with 2 and 4Li+ (i.e., Cu-PP-2Li and Cu-PP-4Li) are −6.17 and −14.30 eV, respectively (Fig. 6d). From the MESP maps of Cu-PP-2Li and Cu-PP-4Li, we can see that the N atoms in the porphyrin ring begin to saturate when gradually binding 4Li+. This phenomenon further explains the role of the Cu–N bond in Li+ storage. According to the simulation results, Cu-PP undergoes a total of 10 electron transfer during the charging and discharging process, and the theoretical capacity calculated based on this conclusion is 286.2 mA h g−1, which is comparable to the actual reversible capacity (285.1 mA h g−1). The excess capacity in Fig. 4d and e may be attributed to the valence change of Cu ions as discussed above (Fig. S23†) and the formation of the cathode-electrolyte interphase (CEI) film.59
Conclusions
In this work, a Cu2+-mediated bipolar-type PPCMP-Cu incorporating porphyrin and pyrrole groups, as a promising organic cathode material for LIBs with high energy density and fast-charging capability, is synthesized through a simple oxidation polymerization combined with post-coordination method. DFT theoretical calculations, ex situ FI-TR, and XPS spectra analysis verify the bipolar ion storage mechanism, with Cu2+ coordination increasing active sites from seven to a total of ten ion storage and extending π–π conjugation with a narrowed band gap for the facilitated charge transfer. N2 adsorption/desorption isotherm analysis, simulation calculation, and kinetic studies show that Cu2+ coordination regulates the pore structures with an increased micropore size for facilitated ion diffusion and maximum active site utilization, thereby endowing an organic cathode with high capacity and excellent cycling stability. As expected, PPCMP-Cu displays a high energy density of 702 W h kg−1 (based on the cathode) and retains a high capacity of 277.3 mA h g−1 after 200 cycles at 300 mA g−1. Moreover, PPCMP-Cu delivers an ultra-fast charging capability of 12464 W kg−1 with an exceptionally short charging time of 76 s at 5 A g−1. This work not only introduces a high-performance, easily synthesized bipolar cathode material for LIBs, but also presents a novel strategy for the design and modulation of the band gap and porous structure of metal coordination porphyrin-based porous polymer electrodes.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. T. L. and J. D. contributed equally to this paper. W. L. and Y. Z. L. conceived the project. Y. Z. W. and J. Q. L. synthesized the samples, performed the structural characterization, and implemented structural simulations. J. D., L. K. T., and H. L. participated in the electrochemical tests. W. S. H., H. W. H., and L. L. W. performed the formatting of the manuscript. All authors analyzed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (52373172 and 52473055), the National Key Research and Development Program of China (2022YFB3807100 and 2022YFB3807102), the Program of Shanghai Academic Research Leader (21XD1420200), the Chang Jiang Scholar Program (T2023082), the Natural Science Foundation of Shanghai (23ZR1401100), and the State Key Laboratory for Modification of Chemical Fibers and Polymer Materials (KF2204, KF2306, and KF2316).References
- X. G. Yang, T. Liu and C. Y. Wang, Nat. Energy, 2021, 6, 176–185 CrossRef CAS.
- J. Y. Li, C. Lin, M. Y. Weng, Y. Qiu, P. H. Chen, K. Yang, W. Y. Huang, Y. X. Hong, J. Li, M. J. Zhang, C. Dong, W. G. Zhao, Z. Xu, X. Wang, K. Xu, J. L. Sun and F. Pan, Nat. Nanotechnol., 2021, 16, 599–605 CrossRef CAS PubMed.
- L. Liu, M. Li, L. Chu, B. Jiang, R. Lin, X. Zhu and G. Cao, Prog. Mater. Sci., 2020, 111, 100655 CrossRef CAS.
- Z.-C. Jian, J.-X. Guo, Y.-F. Liu, Y.-F. Zhu, J. Q. Wang and Y. Xiao, Chem. Sci., 2024, 15, 19698–19728 RSC.
- X. H. Wang, C. H. Li, S. Y. Liu and Y. M. Sun, Chem. Sci., 2024, 15, 17979–17987 RSC.
- W. D. Li, B. H. Song and A. Manthiram, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC.
- Y.-X. Yao, L. Xu, C. Yan and Q. Zhang, EES Batteries, 2025, 1, 9–22 RSC.
- J. S. M. Lee and A. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- K. Amin, N. Ashraf, L. Mao, C. F. J. Faul and Z. Wei, Nano Energy, 2021, 85, 105958 CrossRef CAS.
- L. G. Yue, X. Y. Wang, L. Chen, D. J. Shen, Z. H. Shao, H. Wu, S. F. Xiao, W. Q. Liang, Y. J. Yu and Y. Y. Li, Energy Environ. Sci., 2024, 17, 1117–1131 RSC.
- Q. S. Bai, J. L. Huang, K. H. Tang, Y. L. Zhu and D. C. Wu, Adv. Mater., 2025, 37, 2416661 CrossRef CAS PubMed.
- K. Amin, B. C. Baker, L. Pan, W. Mehmood, Z. Hao, R. Nawaz, Z. Wei and C. F. Faul, Adv. Mater., 2025, 37, 2410262 CrossRef CAS PubMed.
- H. Zhang, X. Ma, D. Guo, M. Jiao, Y. Liu, Y. Fang, M. Wu and Z. Zhou, Adv. Funct. Mater., 2025, 2419764 CrossRef CAS.
- Z. Sun, M. Shu, J. Li, B. Liu, H. Yao, S. Guan and Z. Sun, J. Energy Chem., 2023, 78, 30–36 CrossRef CAS.
- X. Wu, X. Feng, J. Yuan, X. Yang, H. Shu, C. Yang, Z. liu, J. Peng, E. Liu, S. Tan and P. Gao, Energy Storage Mater., 2022, 46, 252–258 CrossRef.
- W. Y. Ma, L. W. Luo, P. H. Dong, P. Y. Zheng, X. H. Huang, C. Zhang, J. X. Jiang and Y. Cao, Adv. Funct. Mater., 2021, 31, 2105027 CrossRef CAS.
- Y. Yang, J. Cai, Y. Zuo, K. Zhang, C. Gao, L. Zhou, Z. Chen, W. Chu and D. Xia, Energy Storage Mater., 2024, 71, 103587 CrossRef.
- J. Min Park, J. H. Lee and W.-D. Jang, Coord. Chem. Rev., 2020, 407, 213157 CrossRef.
- Q. M. Xu, Z. X. Liu, Y. C. Jin, X. Y. Yang, T. T. Sun, T. Y. Zheng, N. Li, Y. H. Wang, T. X. Li, K. Wang and J. Z. Jiang, Energy Environ. Sci., 2024, 17, 5451–5460 RSC.
- X. Y. Yang, Y. M. Hu, N. Dunlap, X. B. Wang, S. F. Huang, Z. P. Su, S. Sharma, Y. H. Jin, F. Huang, X. H. Wang, S. H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20385–20389 CrossRef CAS PubMed.
- A. Q. Wang, C. Breakwell, F. Foglia, R. Tan, L. Lovell, X. C. Wei, T. Wong, N. Q. Meng, H. D. Li, A. Seel, M. Sarter, K. Smith, A. Alvarez-Fernandez, M. Furedi, S. Guldin, M. M. Britton, N. B. McKeown, K. E. Jelfs and Q. L. Song, Nature, 2024, 635, 353–358 CrossRef CAS PubMed.
- Y. Sun, F. He, X. Huang, B. Ren, J. Peng, D. Chen, X. Hu, X. Sun and P. Gao, Chem. Eng. J., 2023, 451, 138734 CrossRef CAS.
- X. Wu, W. Zhou, C. Ye, J. H. Zhang, Z. Y. Liu, C. K. Yang, J. F. Peng, J. L. Liu and P. Gao, Angew. Chem., Int. Ed., 2024, 63, e202317135 CrossRef CAS PubMed.
- Y. Z. Wang, Z. H. Cheng, W. J. Li, X. J. Zhao, S. Li, C. Cheng, A. Thomas and Y. Z. Liao, Adv. Sustainable Syst., 2024, 8, 2400142 CrossRef CAS.
- H. Yang, S. L. Zhang, L. H. Han, Z. Zhang, Z. Xue, J. Gao, Y. J. Li, C. S. Huang, Y. P. Yi, H. B. Liu and Y. L. Li, ACS Appl. Mater. Inter., 2016, 8, 5366–5375 CrossRef CAS PubMed.
- S. C. Qi, Y. J. Zhao, X. J. Lu, Y. L. Liu, Z. Sun, X. Q. Liu and L. B. Sun, Chem. Sci., 2024, 15, 7285–7292 RSC.
- J. Han, Z. B. Zhu, N. J. Li, D. Y. Chen, Q. F. Xu, H. Li, J. H. He and J. M. Lu, Appl. Catal., B, 2021, 291, 120108 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- S. Yuan, L. L. Cui, Z. Y. Dou, X. Ge, X. Q. He, W. Zhang and T. Asefa, Small, 2020, 16, 2000742 CrossRef CAS PubMed.
- R. R. Liang, Z. S. Han, P. Y. Cai, Y. H. Yang, J. Rushlow, Z. Y. Liu, K. Y. Wang and H. C. Zhou, J. Am. Chem. Soc., 2024, 146, 14174–14181 CrossRef CAS PubMed.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS.
- K. M. Barkigia, L. Chantranupong, K. M. Smith and J. Fajer, J. Am. Chem. Soc., 1988, 110, 7566–7567 CrossRef CAS.
- Q. He, H. Li, Z. Hu, L. Lei, D. Wang and T. T. Li, Angew. Chem., Int. Ed., 2024, 63, e202407090 CrossRef CAS PubMed.
- J. Chu, Z. Liu, J. Yu, H.-G. Wang, F. Cui and G. Zhu, Nat. Commun., 2025, 16, 3549 CrossRef CAS PubMed.
- Y. Wu, J. Shen, Z. Sun, Y. Yang, F. Li, S. Ji, M. Zhu and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202215864 CrossRef CAS PubMed.
- L. F. Zhong, Z. S. Fang, C. H. Shu, C. S. Mo, X. C. Chen and D. S. Yu, Angew. Chem., Int. Ed., 2021, 60, 10164–10171 CrossRef CAS PubMed.
- L. Cheng, X. Yan, J. Yu, X. Zhang, H.-G. Wang, F. Cui and Y. Wang, Adv. Mater., 2024, 2411625 Search PubMed.
- S. Zheng, Y. Fu, S. Bi, X. Yang, X. Xu, X. Li, Q. Xu and G. Zeng, Angew. Chem., Int. Ed., 2025, 64, e202417973 CrossRef CAS PubMed.
- X. Y. Xu, S. Q. Zhang, K. Xu, H. Z. Chen, X. L. Fan and N. Huang, J. Am. Chem. Soc., 2023, 145, 1022–1030 CrossRef CAS PubMed.
- L. Chen, Y. Li, X. Wang, J. Wu, Y. Ding, S.-B. Ren, L. Zhang, Z. Xu, B. Chen, D.-M. Han and Y.-P. Wu, Chem. Eng. J., 2023, 464, 142658 CrossRef CAS.
- K. Li, J. Yu, Z. Si, B. Gao, H.-G. Wang and Y. Wang, Chem. Eng. J., 2022, 450, 138052 CrossRef CAS.
- X. L. Fan and C. S. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. C. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- X. L. Chen, M. Xie, Z. L. Zheng, X. Luo, H. C. Jin, Y. F. Chen, G. Z. Yang, D. S. Bin and D. Li, J. Am. Chem. Soc., 2023, 145, 5105–5113 CrossRef CAS PubMed.
- W. Li, S. Han, C. Xiao, J. Yan, B. Wu, P. Wen, J. Lin, M. Chen and X. Lin, Angew. Chem., Int. Ed., 2024, 63, e202410392 CAS.
- W. Y. Yuan, J. Y. Weng, M. H. Ding, H. M. Jiang, Z. G. Fan, Z. J. Zhao, P. J. Zhang, L. P. Xu and P. F. Zhou, Energy Storage Mater., 2024, 65, 103142 CrossRef.
- K. Li, Q. Li, Y. N. Wang, H. G. Wang, Y. H. Li and Z. J. Si, Mater. Chem. Front., 2020, 4, 2697–2703 RSC.
- K. Li, Y. Wang, B. Gao, X. Lv, Z. Si and H.-G. Wang, J. Colloid Interface Sci., 2021, 601, 446–453 CrossRef CAS PubMed.
- C. Zhang, X. Yang, W. Ren, Y. Wang, F. Su and J.-X. Jiang, J. Power Sources, 2016, 317, 49–56 CrossRef CAS.
- Y. Ou, Y. F. Xiong, Z. F. Hu, Y. Zhang and L. J. Dong, J. Mater. Chem. A, 2022, 10, 10373–10382 RSC.
- J. Duan, K. Wang, L. Teng, H. Liu, L. Xu, Q. Huang, Y. Li, M. Liu, H. Hu, X. Chen, J. Wang, W. Yan, W. Lyu and Y. Liao, ACS Nano, 2024, 18, 29189–29202 CrossRef CAS PubMed.
- Z. X. Liu, X. Y. Yang, K. Wang, B. W. Li, W. B. Liu, X. Chen, D. D. Qi, L. Gong and J. Z. Jiang, Energy Storage Mater., 2024, 68, 103337 CrossRef.
- X. L. Liu, Y. C. Jin, H. L. Wang, X. Y. Yang, P. P. Zhang, K. Wang and J. Z. Jiang, Adv. Mater., 2022, 34, 2203605 CrossRef CAS PubMed.
- X. Y. Yang, L. Gong, X. L. Liu, P. P. Zhang, B. W. Li, D. D. Qi, K. Wang, F. He and J. Z. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202207043 CrossRef CAS PubMed.
- Z. D. Lei, Q. S. Yang, Y. Xu, S. Y. Guo, W. W. Sun, H. Liu, L. P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- Y. L. Lin, H. L. Cui, C. Liu, R. Li, S. P. Wang, G. M. Qu, Z. Q. Wei, Y. H. Yang, Y. X. Wang, Z. J. Tang, H. F. Li, H. Y. Zhang, C. Y. Zhi and H. M. Lv, Angew. Chem., Int. Ed., 2023, 62, 14329–14333 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Xiao, N. Adelstein, Y. Bi, W. Bian, J. Cabana, C. L. Cobb, Y. Cui, S. J. Dillon, M. M. Doeff and S. M. Islam, Nat. Energy, 2024, 9, 1463–1473 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08244c |
| ‡ Y. T. Li and J. Duan contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2025 |