
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile, general allylation of unactivated alkyl halides via electrochemically enabled radical-polar crossover†
Haifeng
Chen
 and
Magnus
Rueping
*
and
Magnus
Rueping
*
KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
First published on 12th March 2025
Abstract
Electrochemically driven carbon–carbon formation is receiving considerable interest in organic synthesis. In this study, we present an electrochemically driven method for the formation of C(sp3)–C(sp3) bonds using readily available allylic carbonates, as well as primary, secondary, and tertiary alkyl bromides as electrophiles. This approach offers a highly selective route for synthesizing a broad range of allylic products with excellent functional group tolerance, all without the need for transition metal catalysts. Remarkably, this method also enables the smooth late-stage functionalization of various natural product- and drug-derived substrates, yielding the corresponding complex allylalkanes.
Introduction
The development of new methodologies for constructing C(sp3)–C(sp3) bonds remains a significant challenge in the field of C–C coupling chemistry.1–3 Transition-metal-catalyzed cross-coupling has emerged as a powerful approach for rapid construction and increasing molecular complexity.4–8 However, traditional cross-coupling reactions often require preformed organometallic reagents, which limits their application and substrate scope.9 To address this, reductive cross-coupling using electrophiles as coupling partners has proven to be an efficient strategy,10–13 significantly enhancing molecular complexity by circumventing the need for activated substrates.14–17 Recently, the metallaphotocatalyzed18–21 and metallaelectrocatalyzed22–28 reactions have offered alternative pathways for forming C(sp3)–C(sp3) bonds under mild conditions. Despite these advances,29–33 constructing C(sp3)–C(sp3) bonds remains challenging due to intrinsic obstacles (Scheme 1a). For instance, these methods often rely on expensive metal–ligand catalysts or stoichiometric metal reductants.34,35 Therefore, there is a strong demand for developing new, efficient, mild, metal–ligand-catalyst-free, and environmentally friendly strategies for C(sp3)–C(sp3) bond formation. | ||
| Scheme 1 The development of electrocatalyzed directly formation of the C(sp3) radical. | ||
Electrochemistry has emerged as a versatile and powerful platform for sustainable synthesis, leveraging finely-tuned electron-transfer processes and the use of electrons as traceless redox reagents.36–42 In this context, electroreduction has shown great promise, enabling direct interactions between substrates and electrode surfaces to generate alkyl radicals through the cathodic reduction of alkyl halides.43–47 Elegant studies have explored this concept.48–51 Alkyl halides play a crucial role in organic chemistry due to their diverse reactivity and ease of synthesis.52 As a result, significant progress has been made in the electroreduction of alkyl halides, including applications such as the Giese reaction, cross-electrophile coupling (e-XEC), deuteration, borylation, and bifunctionalization of alkenes, among others.53–57
Allylic moieties are essential substructures in organic molecules and versatile functional groups due to their convertible C–C double bonds and allylic single bonds.58 The allylation of various molecules via the construction of a C–C bond has been thoroughly investigated.3,59 To date, transition-metal catalyzed electrophile allylation of alkyl halides is one of the most important protocols for delivering allylated-alkane compounds (Scheme 1b),60–65 In comparison, the electrochemical allylation of alkyl halides with allylcarbonates, which benefits from transition-metal-free and sustainable conditions, has been relatively underexplored. Thus, direct electrochemical allylation of unactivated alkyl halides presents a practical and attractive alternative for the synthesis of allylated alkane compounds.
In this regard, we present a novel and versatile electroreduction protocol for the formation of allylated alkanes (Scheme 1c). This reaction offers a straightforward and efficient route to allylated products under mild conditions. Moreover, the successful late-stage functionalization of natural products highlights the potential of our methodology. This strategy not only expands the toolkit for C(sp3)–C(sp3) bond formation but also paves the way for future developments in this area.
Results and discussion
We initiated the exploration with methyl 2-(4-(tert-butyl) phenyl) allyl ethyl carbonate (1a) and 2-bromo-2-methylpropane (2a) as the coupling partners. As outlined in Table 1, a combination of (+) iron//(−) Ni foam as electrodes, TBAB (tetrabutylammonium bromide) as an electrolyte, and DMA (dimethylacetamide) as solvent with a 4 mA current in an undivided cell delivered the desired product 3a in 65% isolated yield (entry 1). Altering the anode or cathode significantly decreased the reaction efficiency (entries 2–6). Adjusting the current also succeeded in this alkyl-allylation process, albeit with lower efficiency (entries 7–9). Conducting the reaction in alternative solvents, such as DMF or MeCN, did not improve the reaction outcomes (entries 10 & 11). Screening the substrates ratio also led to lower yields (entries 12–14). Applying other electrolytes did not improve the reaction efficiency (entries 15 & 16).|
|
|||||
|---|---|---|---|---|---|
| Optimization of reaction | |||||
| Entry | Variables | Yieldb [%] | Entry | Variables | Yieldb [%] |
| a Reactions were performed with 0.2 mmol of 1a, 0.4 mmol of 2a, 0.2 M of TBAB, 4 mL of DMA in an undivided cell at R. T. and i = 4 mA, 5.2 F mol−1. b GC-FID yield using dodecane as internal standard. c Isolated yield. | |||||
| 1 | Standard | 65c | 10 | DMF as solvent | 50 |
| 2 | RVC(+)//Ni foam(−) w/TMEDA | 8 | 11 | MeCN as solvent | 17 |
| 3 | RVC(+)//RVC(−) w/TMEDA | 12 | 12 | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
51 |
| 4 | (+)Zn//(−)Ni foam | 25 | 13 | 2:1 |
40 |
| 5 | (+)Fe//(−)RVC | 51 | 14 | 1:1 |
36 |
| 6 | (+)Zn//(−)RVC | 27 | 15 | TBACl as electrolyte | 23 |
| 7 | 1 mA | 44 | 16 | TBAI as electrolyte | 45 |
| 8 | 2 mA | 63 | 17 | w/o current | ND |
| 9 | 6 mA | 42 | 18 | w/o TBAB | 32 |
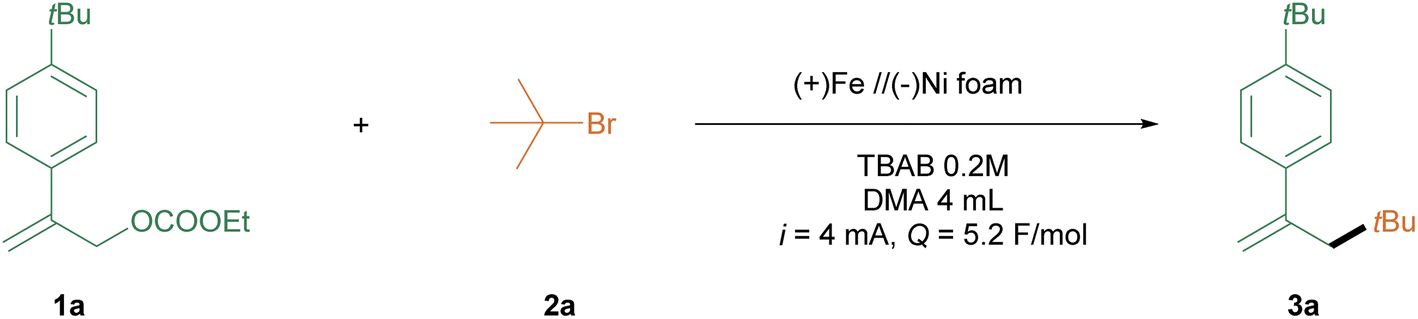
Control experiments revealed that the current and electrolyte were essential for obtaining high reaction results (entries 17 & 18). In order if the iron ions formed at the anode affect the reaction outcome,66 we added different amounts of FeBr2 and FeBr3 as well as additional FeBr2 in combination with different electrodes. However, no reaction enhancement was observed (Fig. S1 & S2†).
With the optimized condition in hand, we moved attention to evaluating the scope of this protocol. As shown in Fig. 1, the installation of electro-donating (3b–f), electron-neutral (3g & h) and electron-withdrawing (3i–m) groups onto the 2-phenylallyl carbonate showed high coupling efficiency and excellent functional group compatibility. Hetero-quinoline substituted substrate also afforded the corresponding product in acceptable yield (3n).
 | ||
| Fig. 1 Allylic carbonate scope. Reactions were performed with 0.2 mmol of 1, 0.4 mmol of 2, 0.2 M of TBAB, and 4 mL of DMA in an undivided cell at R. T. and i = 4 mA, 5.2 F mol−1. | ||
Next, we investigated the scope of the alkyl halides (Fig. 2). To our delight, a series of primary, secondary and tertiary alkyl bromides, in spite of different electronic properties, all could be engaged in the reaction efficiently. For the primary alkyl bromide, alkyl bromides tethered with phenyl and methoxyl substituted phenyl, ether, ester, cyano, benzoate, acetal, trifluoromethyl, N-boc-piperidine and phthalimidyl groups could all be transferred to the cascade products in moderate to excellent yields (4a–k). The use of secondary alkyl bromides with acyclic, cyclic and heteroatoms gave good yields (5a–5l). Unexpectedly, isomerizations (5d & 5i) were detected when bromocyclohexane and 4-bromo-N-boc-piperidine were used. Bicyclic and spiro compounds reacted efficiently and delivered the corresponding products (5j–l) in good yields. In addition, the linear (6a–h) and cyclic tertiary alkyl bromides (6i–j) also reacted efficiently in this system. Significantly, the pentafluoro-substituted substrate could furnish the desired product (6d) in 40% yield. Next, our focus shifted to further demonstrating the practicability and synthetic utility of this protocol, we explored modifications on structurally complex molecules. As displayed in Fig. 3, various primary, secondary, and tertiary natural or pharmaceutical alkyl bromides derived from L-menthol (7a), naproxen (7b), adapalene (7c), indometacin (7d), gemfibrozil (7e), flubiprofen (7f), dehydroabietic acid (7g), acetobromo-a-D-glucose (7h), epiandrosterone (7i), lithocholic acid (7j), tetrahydrolinalool (7k), probenecid (7l),γ-terpineol (7m) and sclareolide (7n) were effectively converted into the corresponding alkyl radical via this protocol, and subsequently delivered a variety of structually complex allylated-alkane derivatives in moderate to good yields.
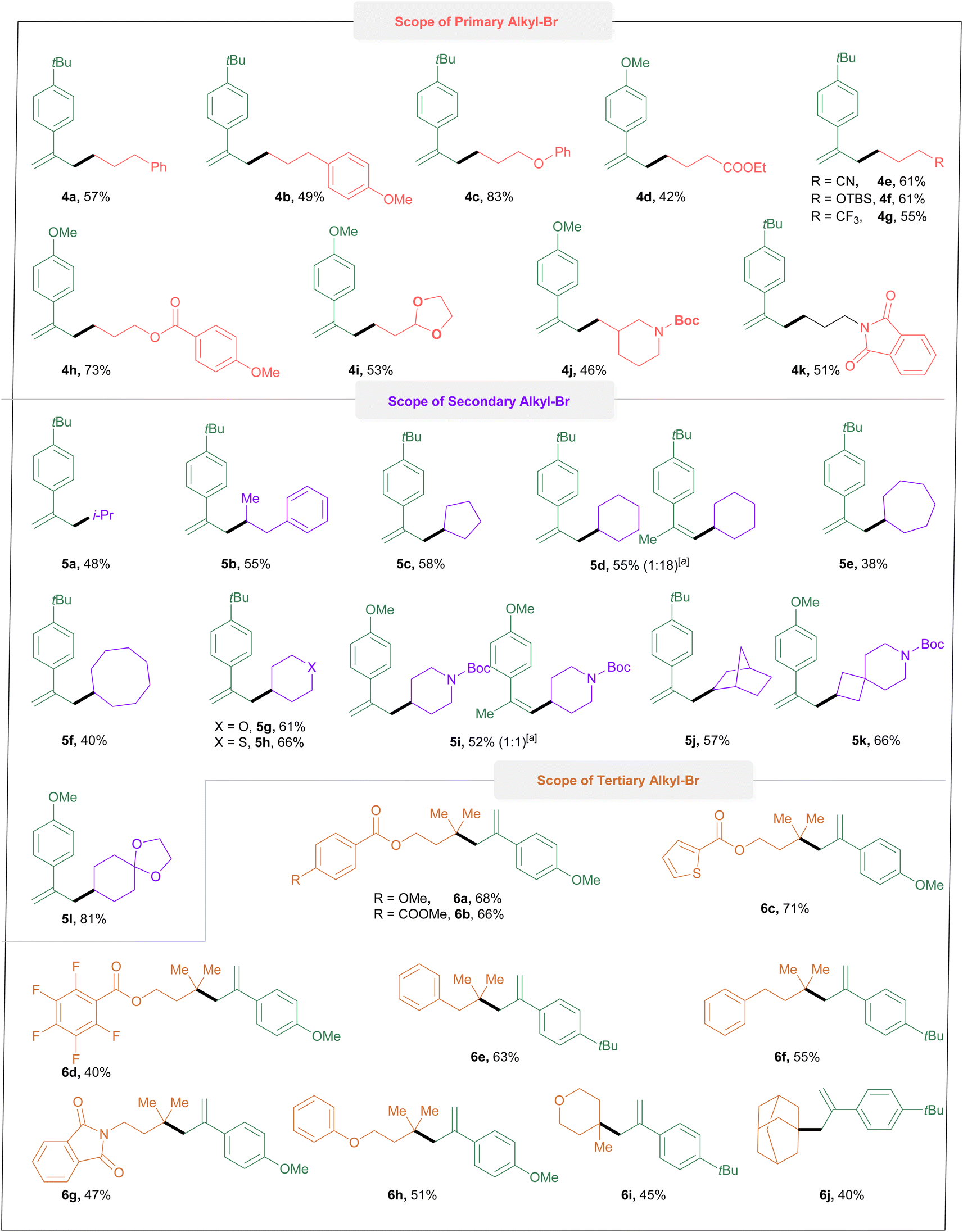 | ||
| Fig. 2 Alkyl-Br substrate scope. Reactions were performed with 0.2 mmol of 1, 0.4 mmol of 2, 0.2 M of TBAB, and 4 mL of DMA in an undivided cell at R. T. and constant current i = 4 mA, 5.2 F mol−1.a Detected by 1H NMR. | ||
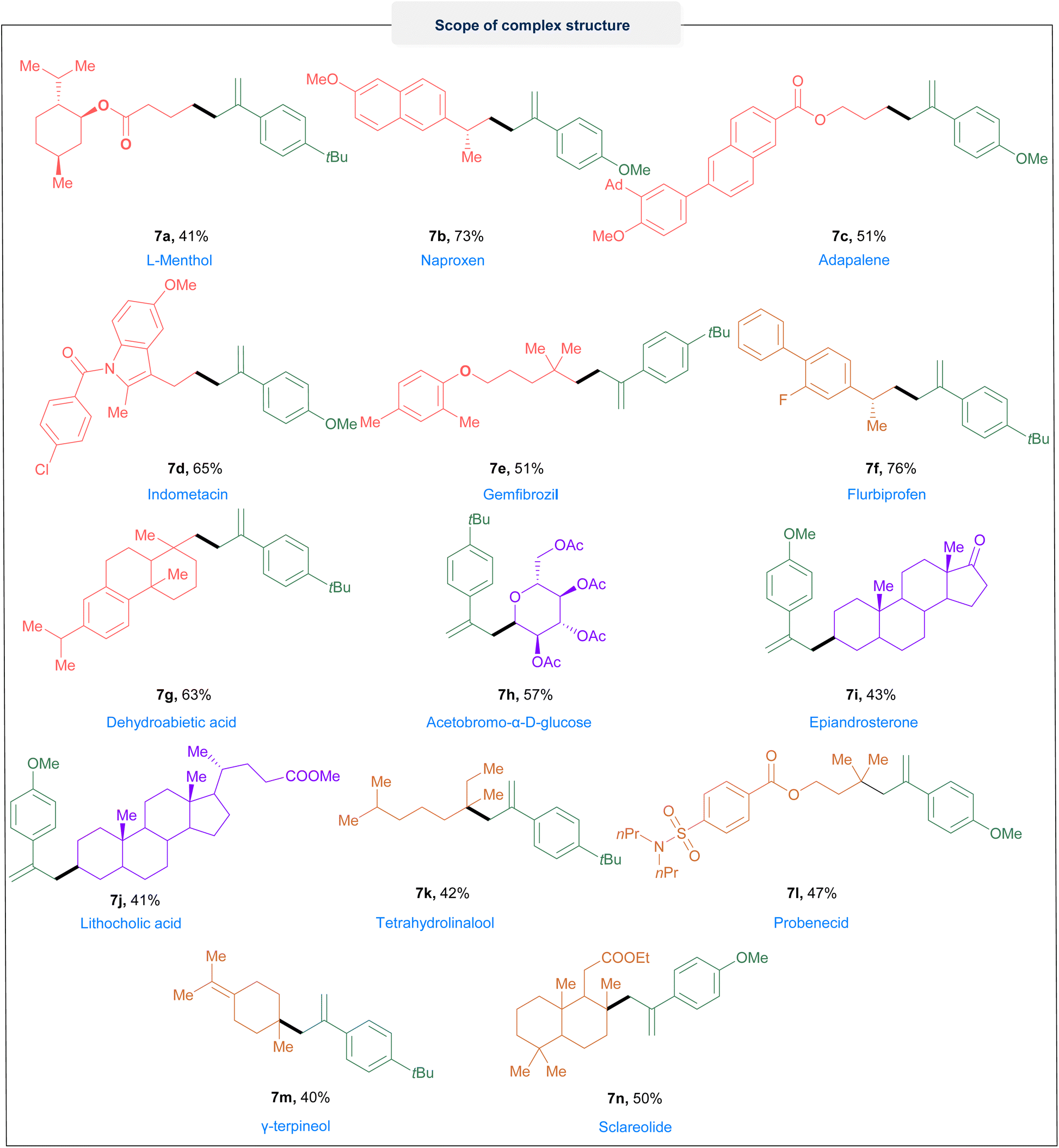 | ||
| Fig. 3 Modification of structurally complex molecules. Reactions were performed with 0.2 mmol of 1, 0.4 mmol of 2, 0.2 M of TBAB, and 4 mL of DMA in an undivided cell at R. T. and constant current i = 4 mA, 5.2 F mol−1. | ||
To further elucidate the possible mechanism of this electrochemically driven formation of allylated-alkanes, a range of control experiments were performed. Firstly, the use of stoichiometric amounts of radical scavengers (1, 1- diphenyethylene and TEMPO) under the standard conditions was applied. Both desired products and alkyl radical captured products could be observed (Fig. 4a). In principle, the allylic carbonate might also be reduced by the cathode to release the allyl radical. However, we did not observe the formation. Alkyl radical clock experiments were performed by using the cyclopropyl tethered (bromomethyl) cyclopropane (2d) and ethyl 2-bromo-2-cyclopropylacetate (2e). Here, we obtained the ring-opening product 14 in 55%, although the secondary alkyl substrate only furnished the desired product 15 in trace yield and by-product 15a was formed in 45%, which indicates that the secondary alkyl radical is involved in the reaction (Fig. 4b). Thus, radical cyclization experiments were also performed. The cyclization products 16 and 17 were obtained in 67% and 31%, respectively (Fig. 4c). These observations suggest the involvement of alkyl radicals in this protocol. To further investigate, a competition reaction was performed (Fig. 4d), yielding products 3a, 4a, and 5a with yields of 59%, 8%, and 17%, respectively. These results demonstrated that the alkyl reactivity followed the sequence: tertiary > secondary > primary. Additionally, reaction monitoring indicated that the allylation reaction was completed within 7 hours (Fig. 4e). To explore the electrochemical behavior of the reactants, cyclic voltammetry (CV) measurements were conducted. As shown in Fig. 4f, the reduction peaks for 3-phenylpropyl bromide, 2-bromopropane, and tert-butyl bromide were observed at −2.70 V, −2.66 V, and −2.52 V versus the saturated calomel electrode (SCE), and the onset values were −2.4 V, −2.3 V and −2.2 V, respectively. For 2-(4-(tert-butyl)phenyl) allyl ethyl carbonate, two onset reduction peaks were recorded at −2.74 V and −2.40 V versus SCE. These findings indicate that alkyl halides can be reduced to generate the corresponding alkyl radicals under the conditions of this protocol.
 | ||
| Fig. 4 Mechanistic studies. aGC-FID yield using dodecane as internal standard. bIsolated yield. (a) Radical scavengers; (b) fragmentation cyclopropyl substrates; (c) radical cyclization experiment; (d) competition reaction; (e) reaction monitor; (f) CV measurement; (g) propososed mechanism. | ||
Mechanism
Based on the above analysis results and literature report,53,65 we propose a plausible mechanism for the new electrochemical C(sp3)–C(sp3) bond formation (Fig. 4g). Initially, the allylic carbonate reacts with the cathode-activated alkyl radicals, forming a stable tertiary benzyl radical. This radical is subsequently reduced at the cathode to generate a carbanion. The carbanion intermediate can then undergo β-elimination, leading to the formation of the desired products.Conclusions
In conclusion, we have successfully developed an efficient electrochemically-driven radical polar crossover method for the formation of allylated alkanes using allylic carbonate and unactivated alkyl bromides. This protocol operates under mild conditions, enabling the synthesis of a diverse range of synthetically valuable allylated alkanes. Given the commercial availability of the reactants and the properties of allyl and alkyl compounds, we believe that this electroreduction protocol will have broad applicability, expand the library of allylated alkanes, and inspire further exploration in the field of C(sp3)–C(sp3) coupling reactions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. C., and M. R. conceived the idea of this work. H. C. carried out the reaction optimization and substrate scope. H. C. and M. R. co-wrote the manuscript. M. R. directed the entire research.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the King Abdullah University of Science and Technology (KAUST), Saudi Arabia, Office of Sponsored Research. This publication is based upon work supported by King Abdullah University of Science and Technology (KAUST) under Award No. RFS-OFP2023-5517 and URF/1/5033.Notes and references
- E. J. C. Corey and X.-M. Cheng, The Logic Of Chemical Synthesis, Wiley, New York, 1995 Search PubMed.
- H. M. Huang, P. Bellotti and F. Glorius, Transition metal-catalysed allylic functionalization reactions involving radicals, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC.
- L. Susse and B. M. Stoltz, Enantioselective Formation of Quaternary Centers by Allylic Alkylation with First-Row Transition-Metal Catalysts, Chem. Rev., 2021, 121, 4084–4099 CrossRef PubMed.
- R. Jana, T. P. Pathak and M. S. Sigman, Advances in transition metal (Pd, Ni, Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners, Chem. Rev., 2011, 111, 1417–1492 CrossRef PubMed.
- L. J. Cheng and N. P. Mankad, C–C and C–X coupling reactions of unactivated alkyl electrophiles using copper catalysis, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC.
- M. J. Goldfogel, L. Huang and D. J. Weix, in Nickel Catalysis In Organic Synthesis: Methods And Reactions, ed. S. Ogoshi, Wiley-VCH, Weinheim, 2020, pp. 183–222, DOI:10.1002/9783527813827.ch9.
- W. Xue, X. Jia, X. Wang, X. Tao, Z. Yin and H. Gong, Nickel-catalyzed formation of quaternary carbon centers using tertiary alkyl electrophiles, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC.
- Y. Wei, L. Q. H. Lin, B. C. Lee and M. J. Koh, Recent Advances in First-Row Transition Metal-Catalyzed Reductive Coupling Reactions for π-Bond Functionalization and C-Glycosylation, Acc. Chem. Res., 2023, 56, 3292–3312 CrossRef.
- W. Shi, C. Liu and A. Lei, Transition-metal catalyzed oxidative cross-coupling reactions to form C–C bonds involving organometallic reagents as nucleophiles, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC.
- X. Wang, Y. Dai and H. Gong, Nickel-Catalyzed Reductive Couplings, Top. Curr. Chem., 2016, 374, 43 CrossRef.
- K. E. Poremba, S. E. Dibrell and S. E. Reisman, Nickel-Catalyzed Enantioselective Reductive Cross-Coupling Reactions, ACS Catal., 2020, 10, 8237–8246 CrossRef PubMed.
- L. M. Chen and S. E. Reisman, Enantioselective C(sp2)–C(sp3) Bond Construction by Ni Catalysis, Acc. Chem. Res., 2024, 57, 751–762 CrossRef.
- Y. Gong, J. Hu, C. Qiu and H. Gong, Insights into Recent Nickel-Catalyzed Reductive and Redox C–C Coupling of Electrophiles, C(sp3)-H Bonds and Alkenes, Acc. Chem. Res., 2024, 57, 1149–1162 CrossRef.
- H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Nickel-Catalyzed Reductive Allylation of Tertiary Alkyl Halides with Allylic Carbonates, Angew. Chem., Int. Ed., 2017, 56, 13103–13106 CrossRef.
- Y. Ye, H. Chen, J. L. Sessler and H. Gong, Zn-Mediated Fragmentation of Tertiary Alkyl Oxalates Enabling Formation of Alkylated and Arylated Quaternary Carbon Centers, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef.
- C. Dorval, M. Tricoire, J.-M. Begouin, V. Gandon and C. Gosmini, Cobalt-Catalyzed C(sp2)–CN Bond Activation: Cross-Electrophile Coupling for Biaryl Formation and Mechanistic Insight, ACS Catal., 2020, 10, 12819–12827 CrossRef.
- H. Chen, H. Yue, C. Zhu and M. Rueping, Reactivity in Nickel-Catalyzed Multi-component Sequential Reductive Cross-Coupling Reactions, Angew. Chem., Int. Ed., 2022, 61, e202204144 CrossRef PubMed.
- C. Zhu, H. Yue, L. Chu and M. Rueping, Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity, Chem. Sci., 2020, 11, 4051–4064 RSC.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Carboxylic Acids as Adaptive Functional Groups in Metallaphotoredox Catalysis, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef PubMed.
- J. Zhang and M. Rueping, Metallaphotoredox catalysis for sp3 C–H functionalizations through single-electron transfer, Nat. Catal., 2024, 7, 963–976 CrossRef.
- P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, 3d metallaelectrocatalysis for resource economical syntheses, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC.
- J.-S. Zhong, Y. Yu, Z. Shi and K.-Y. Ye, An electrochemical perspective on the roles of ligands in the merger of transition-metal catalysis and electrochemistry, Org. Chem. Front., 2021, 8, 1315–1328 RSC.
- A. L. Xu Cheng, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis, CCS Chem., 2022, 4, 1120–1152 CrossRef.
- H. Chen, C. Zhu, H. Yue and M. Rueping, Group 14 Elements Hetero-Difunctionalizations via Nickel-Catalyzed Electroreductive Cross-Coupling, Angew. Chem., Int. Ed., 2023, 62, e202306498 CrossRef CAS.
- C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Advances on the Merger of Electrochemistry and Transition Metal Catalysis for Organic Synthesis, Chem. Rev., 2022, 122, 3180–3218 CrossRef CAS PubMed.
- L. Zou, X. Wang, S. Xiang, W. Zheng and Q. Lu, Paired Oxidative and Reductive Catalysis: Breaking the Potential Barrier of Electrochemical C(sp3)-H Alkenylation, Angew. Chem., Int. Ed., 2023, 62, e202301026 CrossRef CAS.
- N. Li, R. Sitdikov, A. P. Kale, J. Steverlynck, B. Li and M. Rueping, A review of recent advances in electrochemical and photoelectrochemical late-stage functionalization classified by anodic oxidation, cathodic reduction, and paired electrolysis, Beilstein J. Org. Chem., 2024, 20, 2500–2566 CrossRef CAS.
- D. Brosamlen and M. Oestreich, Regioselective Hydroalkylation of Vinyl and Allylsilanes as Well as Vinylgermanes under Ni–H Catalysis, Org. Lett., 2023, 25, 5319–5323 CrossRef.
- A. Hossain, R. L. Anderson, C. S. Zhang, P. J. Chen and G. C. Fu, Nickel-Catalyzed Enantioconvergent and Diastereoselective Allenylation of Alkyl Electrophiles: Simultaneous Control of Central and Axial Chirality, J. Am. Chem. Soc., 2024, 146(11), 7173–7177 CrossRef CAS PubMed.
- R. Chen, N. E. Intermaggio, J. Xie, J. A. Rossi-Ashton, C. A. Gould, R. T. Martin, J. Alcazar and D. W. C. MacMillan, Alcohol-alcohol cross-coupling enabled by S(H)2 radical sorting, Science, 2024, 383, 1350–1357 CrossRef.
- X. C. Gan, B. Zhang, N. Dao, C. Bi, M. Pokle, L. Kan, M. R. Collins, C. C. Tyrol, P. N. Bolduc, M. Nicastri, Y. Kawamata, P. S. Baran and R. Shenvi, Carbon quaternization of redox active esters and olefins by decarboxylative coupling, Science, 2024, 384, 113–118 CrossRef.
- P. Li, Z. Zhu, C. Guo, G. Kou, S. Wang, P. Xie, D. Ma, T. Feng, Y. Wang and Y. Qiu, Nickel-electrocatalysed C(sp3)–C(sp3) cross-coupling of unactivated alkyl halides, Nat. Catal., 2024, 7, 412–421 CrossRef.
- C. L. Sun and Z. J. Shi, Transition-metal-free coupling reactions, Chem. Rev., 2014, 114, 9219–9280 CrossRef.
- J. Xie, H. Jin and A. S. K. Hashmi, The recent achievements of redox-neutral radical C–C cross-coupling enabled by visible-light, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC.
- A. Jutand, Contribution of electrochemistry to organometallic catalysis, Chem. Rev., 2008, 108, 2300–2347 CrossRef PubMed.
- G. S. Sauer and S. Lin, An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes, ACS Catal., 2018, 8, 5175–5187 CrossRef.
- T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Resource Economy by Metallaelectrocatalysis: Merging Electrochemistry and C H Activation, Trends Chem., 2019, 1, 63–76 CrossRef.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, Organic Electrochemistry: Molecular Syntheses with Potential, ACS Cent. Sci., 2021, 7, 415–431 CrossRef PubMed.
- Y. Liu, P. Li, Y. Wang and Y. Qiu, Electroreductive Cross-Electrophile Coupling (eXEC) Reactions, Angew. Chem., Int. Ed., 2023, 62, e202306679 CrossRef.
- C. Zhu, H. Chen, H. Yue and M. Rueping, Electrochemical chemo- and regioselective arylalkylation, dialkylation and hydro(deutero)alkylation of 1,3-enynes, Nat. Synth., 2023, 2, 1068–1081 CrossRef.
- H. Chen, C. Zhai, C. Zhu and M. Rueping, Allylgermane synthesis via facile and general nickela-electrocatalyzed electrophile coupling, Chem Catal., 2025 DOI:10.1016/j.checat.2024.101257.
- J. B. Sperry and D. L. Wright, The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Modern strategies in electroorganic synthesis, Chem. Rev., 2008, 108, 2265–2299 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef.
- S. Mohle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef.
- W. Zhang, W. Guan, J. I. Martinez Alvarado, L. F. T. Novaes and S. Lin, Deep Electroreductive Chemistry: Harnessing Carbon- and Silicon-Based Reactive Intermediates in Organic Synthesis, ACS Catal., 2023, 13, 8038–8048 CrossRef.
- A. Claraz and G. Masson, Recent Advances in C(sp3)–C(sp3) and C(sp3)–C(sp2) Bond Formation through Cathodic Reactions: Reductive and Convergent Paired Electrolyses, ACS Org. Inorg. Au, 2022, 2, 126–147 CrossRef.
- B. Huang, Z. Sun and G. Sun, Recent progress in cathodic reduction-enabled organic electrosynthesis: Trends, challenges, and opportunities, eScience, 2022, 2, 243–277 CrossRef.
- T. B. Hamby, M. J. LaLama and C. S. Sevov, Controlling Ni redox states by dynamic ligand exchange for electroreductive Csp3–Csp2 coupling, Science, 2022, 376, 410–416 Search PubMed.
- H. Xiang, J. He, W. Qian, M. Qiu, H. Xu, W. Duan, Y. Ouyang, Y. Wang and C. Zhu, Electroreductively Induced Radicals for Organic Synthesis, Molecules, 2023, 28, 857–879 CrossRef PubMed.
- X. Sun and K. Zheng, Electrochemical halogen-atom transfer alkylation via alpha-aminoalkyl radical activation of alkyl iodides, Nat. Commun., 2023, 14, 6825 CrossRef PubMed.
- W. Zhang and S. Lin, Electroreductive Carbofunctionalization of Alkenes with Alkyl Bromides via a Radical-Polar Crossover Mechanism, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef.
- B. Wang, P. Peng, W. Ma, Z. Liu, C. Huang, Y. Cao, P. Hu, X. Qi and Q. Lu, Electrochemical Borylation of Alkyl Halides: Fast, Scalable Access to Alkyl Boronic Esters, J. Am. Chem. Soc., 2021, 143, 12985–12991 CrossRef PubMed.
- P. Li, C. Guo, S. Wang, D. Ma, T. Feng, Y. Wang and Y. Qiu, Facile and general electrochemical deuteration of unactivated alkyl halides, Nat. Commun., 2022, 13, 3774 CrossRef.
- W. Zhang, L. Lu, W. Zhang, Y. Wang, S. D. Ware, J. Mondragon, J. Rein, N. Strotman, D. Lehnherr, K. A. See and S. Lin, Electrochemically driven cross-electrophile coupling of alkyl halides, Nature, 2022, 604, 292–297 CrossRef.
- L. Lu, Y. Wang, W. Zhang, W. Zhang, K. A. See and S. Lin, Three-Component Cross-Electrophile Coupling: Regioselective Electrochemical Dialkylation of Alkenes, J. Am. Chem. Soc., 2023, 145, 22298–22304 CrossRef PubMed.
- J. F. Han, P. Guo, X. G. Zhang, J. B. Liao and K. Y. Ye, Recent advances in cobalt-catalyzed allylic functionalization, Org. Biomol. Chem., 2020, 18, 7740–7750 RSC.
- S. Dutta, T. Bhattacharya, D. B. Werz and D. Maiti, Transition-metal-catalyzed C–H allylation reactions, Chem, 2021, 7, 555–605 Search PubMed.
- Y. Dai, F. Wu, Z. Zang, H. You and H. Gong, Ni-catalyzed reductive allylation of unactivated alkyl halides with allylic carbonates, Chem.–Eur. J., 2012, 18, 808–812 CrossRef PubMed.
- X. Qian, A. Auffrant, A. Felouat and C. Gosmini, Cobalt-catalyzed reductive allylation of alkyl halides with allylic acetates or carbonates, Angew. Chem., Int. Ed., 2011, 50, 10402–10405 CrossRef PubMed.
- Y. Yu, H. Chen, Q. Qian, K. Yao and H. Gong, An extension of nickel-catalyzed reductive coupling between tertiary alkyl halides with allylic carbonates, Tetrahedron, 2018, 74, 5651–5658 CrossRef.
- H. Chen, Y. Ye, W. Tong, J. Fang and H. Gong, Formation of allylated quaternary carbon centers via C–O/C–O bond fragmentation of oxalates and allyl carbonates, Chem. Commun., 2020, 56, 454–457 RSC.
- Y. Xu, M. Zhang and M. Oestreich, Photochemical, Nickel-Catalyzed C(sp3)–C(sp3) Reductive Cross-Coupling of α-Silylated Alkyl Electrophiles and Allylic Sulfones, ACS Catal., 2022, 12, 10546–10550 CrossRef.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Julia and D. Leonori, Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides, Science, 2020, 367, 1021–1026 CrossRef PubMed.
- G. S. Kumar, A. Peshkov, A. Brzozowska, P. Nikolaienko, C. Zhu and M. Rueping, Nickel-Catalyzed Chain-Walking Cross-Electrophile Coupling of Alkyl and Aryl Halides and Olefin Hydroarylation Enabled by Electrochemical Reduction, Angew. Chem., Int. Ed., 2020, 59, 6513–6519 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07923j |
| This journal is © The Royal Society of Chemistry 2025 |