
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceB(MIDA)-directed site-selective intermolecular halofluoroalkylation of alkenes: synthesis of diversely functionalized building blocks†
Hengbo
Wu‡
ab,
Ruitong
Luo‡
ab,
Jingjing
Peng
ab,
Zijian
Han
ab,
Renjie
Zhang
bc,
Zhijian
Xu
 ab,
Weiliang
Zhu
*ab,
Hong
Liu
*abc and
Chunpu
Li
*abc
ab,
Weiliang
Zhu
*ab,
Hong
Liu
*abc and
Chunpu
Li
*abc
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: wlzhu@simm.ac.cn; hliu@simm.ac.cn; lichunpu@simm.ac.cn
bUniversity of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China
cSchool of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou, 310024, China
First published on 10th January 2025
Abstract
α-Halo borides are generally constructed via Matteson homologation, and the synthesis of both fluorinated and functionalized ambiphilic boronates is challenging and has received inadequate attention. Herein, we describe the N-methyliminodiacetyl boronate [B(MIDA)]-directed halogenation of alkenes via a complementary sequence involving fluoroalkyl radical addition followed by guided radical-to-metal oxidative addition and C–X reductive elimination. The alkali cation and functional groups in B(MIDA) enable coulombic interaction and weak attraction with halogens, which could weaken the Pd–X bond and assist in C–X bond formation and is verified by DFT calculations. As a result, a wide variety of highly functionalized fluorinated α-halo boronates, including drugs and natural products, are obtained in good or moderate yields through the unique catalytic manifold. Notably, the trifunctionalized (F, X, B) building block could be transformed into diverse modified fluorinated products.
Introduction
The α-halo alkylboronic esters containing two reacting functional groups are invaluable synthons for the construction of C–O, C–N, and C–C bonds, which makes them useful for organic synthesis and the total synthesis of natural products.1–5 The α-halo borides, indeed, serve as ambiphilic reagents with both electrophile (C–X) and nucleophile (C–B) centers (Scheme 1A). As an appealing synthesis intermediate, applications in cross-couplings to secondary alkylboronate esters under mild nickel-catalyzed conditions have been disclosed recently.6–8 Moreover, transformations from halogens into amines enable the construction of valuable α-amino boronates, which play a pivotal role in the total synthesis of FDA-approved drugs such as Bortezomib.9 Numerous studies have attempted to exploit effective reactions for the synthesis of α-halo borides (Scheme 1B). Generally, ambiphilic boron reagents are prepared through Matteson homologation,10 a process involving concerted 1,2-metallate rearrangement. However, applying this protocol to the formation of specific boronates is challenging owing to the extremely low temperature and harsh preparation conditions of the organometallic reagent (LiCHCl2). Additionally, in most cases prefunctionalized primary borides, in which simple substitution groups such as linear alkyl, phenyl, and seldom polar units are limited, could be utilized. As a supplement, catalytic hydroboration,11 catalytic hydrohalogenation,12 and photo-induced methodologies13 are alternative approaches, but the substrate scope is limited. The need for moisture or air-sensitive reagents and complicated operations limits the widespread synthetic applications of the abovementioned approaches. Lately, Wang14 and Xu15 reported two elegant strategies involving the radical C–H halogenation of benzyl borides and deoxygenative haloboration from carbonyls. Although some intriguing examples have recently been described, the synthesis of α-halo borides is expected to be very challenging, because these substrates tend to undergo dehalogenation or β-hydride elimination. In addition to some complex functional groups, the concurrent one-step incorporation of fluorine, which has been emphasized in several fields including the pharmaceutical, agrochemical, and material industries,16–18 remains a daunting task. To the best of our knowledge, only a handful of fluorinated iodides, most of which are perfluoroalkyl iodides, could be utilized to form the particular product.19 Thus, developing efficient and facile synthetic patterns for forging diverse fluorine-containing α-halo borides is in high demand. | ||
| Scheme 1 Reaction design. | ||
We anticipated that a site-oriented α-halogenation could be achieved using the borides. Pioneered by Burke and co-workers, N-methyliminodiacetyl boronates [B(MIDA)s] have been identified as a purifiable, stable, and compatible surrogate for traditional boronates20 and the use of B(MIDA)s has been helpful in overcoming various obstacles in synthesizing challenging organoboron compounds.21–23 Conceptually, inspired by a N-tetranuclear organoboron catalyst which was similar to the B–N-containing BMIDA group,24–26 we envisioned that potential interactions between boron/nitrogen/oxygen in BMIDA and halogen atoms might be helpful for orienting halogens. Fluoroalkyl halides (Rf–X), an ideal functionalized fluorine source, could be initiated by palladium to generate fluoroalkyl radicals.27,28 Building on the above principles, we employed olefin B(MIDA) derivatives that could undergo a free fluorine radical recombination after the Pd(0) activation, to generate the key Pd(I)–X species and reactive α-borylalkyl radicals.29,30 Consequently, colligation of the mentioned species would result in a C(sp3)–Pd(II)–X system where direct reductive elimination is thermodynamically disfavored.31 Computational studies have proved that X–Pd(II) dissociation was a critical step in the C(sp3)–Pd(II)–X RE process,32 so we supposed that additional interactions resulting from functional groups in BMIDA could reduce the Pd-center electron density and therefore facilitate heterolytic bond dissociation. Presumably, the potential X–B attraction and alkali metal cation aided33 coulombic interaction were thought to suppress the kinetic hindrance and then lead to direct reductive elimination. Finally, α-halo boronates characterized by useful trifunctionalized building blocks may serve as a platform for further decorations (Scheme 1C). Herein, we report the B(MIDA)-dictated palladium-catalyzed halofluoroalkylation of alkenes with fluoroalkyl halides (Rf–X) and the detailed mechanism was revealed by density functional theory (DFT) calculations. In contrast to the existing methods that rely on sophisticated manipulations to prepare starting organoboron precursors, this unique technique is characterized by high efficiency, synthesis simplicity, the commercial availability of reagents, and a remarkably broad substrate scope, thus allowing for straightforward access to fluorinated α-halo boronates even for the late-stage modification of biologically relevant systems, which may be widely applied to medicinal chemistry and chemical biology.
Results and discussion
Considering the significant role of difluoromethylene (CF2) in improving the physicochemical and biological properties of drugs and natural products,34 we conducted our investigation using the commercially available bromodifluoroacetate, which has been intensively developed,35 and vinyl B(MIDA) for the construction of fluorinated α-halo boronates (Table 1).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb |
| a Reaction conditions: B-1 (0.27 mmol, 1.0 equiv.), S-1 (2.0 equiv.), PdCl2(dppf)·DCM (10 mol%), BINAP (10 mol%), K2CO3 (2.0 equiv.), EtOAc (2 mL), 85 °C, argon, 12 h, if otherwise noted. b Determined by 19F NMR using fluorobenzene as an internal standard, and the number in parentheses is the isolated yield. | ||
| 1 | None | 90 (89) |
| 2 | Air | 66 |
| 3 | PdCl2(MeCN)2 instead of PdCl2(dppf)·DCM | 59 |
| 4 | Dppe instead of BINAP | 15 |
| 5 | t BuOK instead of K2CO3 | 23 |
| 6 | Dioxane instead of EtOAc | 61 |
| 7 | S-1 (4 equiv.) | 70 |
| 8 | Without K2CO3 | <5 |

To test our hypothesis, a variety of palladium catalysts, ligands, bases, and solvents were applied for the model reaction. Remarkably, a combination of PdCl2(dppf)·DCM (10 mol%) with an electron-rich racemic BINAP ligand in the presence of a Brønsted base K2CO3 in ethyl acetate at 85 °C could generate the desired product in a 90% yield, as determined via19F NMR (entry 1, for detailed optimization studies, see Tables S1–S4, ESI†). Even the standard condition was similar to that employed in Heck coupling, no Heck-type product was detected and 1 was formed in a moderate yield (entry 2) regardless of the presence of an argon atmosphere. Among the palladium catalysts, PdCl2(dppf)·DCM exerted the best catalytic activity, which surpassed that of PdCl2(MeCN)2 (entry 3). Specifically, variation from BINAP to Dppe resulted in a decreased yield (15%), which suggests that maintaining the steric hindrance of the ligand was essential for the reaction (entry 4). Moreover, a relatively low yield was achieved viatBuOK utilization (entry 5). Replacing ethyl acetate with dioxane still provided 1 in a moderate yield, indicating that the reaction was relatively insensitive to the change of solvent (entry 6). Likewise, a good yield of 1 was achieved at a high S-1 amount of 4 equiv. (entry 7). The lack of K2CO3 significantly reduced the reaction yield (entry 8).
To gain some insights into the reaction's compatibility with different functional groups, under the optimized reaction conditions, we first explored the scope of bromodifluoro esters and amides. Accordingly, the methyl ester derivate (2) was well applicable to the reaction, with a yield of 82%. As demonstrated in Scheme 2, the protocol was found to be general and characterized by remarkable tolerance in the presence of amides with commonly encountered fragments. The steric hindrance of amines with tert-butyl (3) and cyclohexyl (4) could provide the corresponding products, with yields of ∼70%. Aniline and benzylamine derivatives containing electron-withdrawing difluoro, chlorine, and trifluoromethyl were smoothly processed (5–7, 9, 11), which provides good opportunities for downstream transformations. The benzylamines containing isopropyl (8) and naphthalene (10) underwent a successful reaction, although the yields were moderate. Versatile synthetic handles, such as electron-donating methylenedioxy, heterocyclic benzofuran, and thiophene were all compatible under the standard conditions (12–14). Encouraged by the abovementioned results, we sought to examine other bromodifluoro sources including phenyl sulfone, diethyl phosphonate, and benzo[d]oxazole, and the direct substitution of benzene (15–22). In the cases of sulfone (15) and phosphonate (16), the reaction was less efficient than the reactions involving the above substrates. Nevertheless, the oxazole ring (17) and other aryl halides (18–22) exhibited promising tolerance, attributable to reactive benzyl radicals generated by palladium catalysts. In addition to the universality of bromo, we questioned whether this strategy could enable the creation of iodofluorinated products. Surprisingly, α-iodo B(MIDA) boronates were also readily generated using the corresponding iodofluorides. The ester (23), alkyl amines (24, 25), and aniline (26) could be successfully converted into the corresponding products, with similarly high levels of yields. The structure of 23 was identified via X-ray crystallography. Considering the two electron-withdrawing groups, OCF3-substituted aniline (28) maintained a good yield, while the F-substituted one (27) showed a moderate yield. Regarding the benzylamines (29–33), the method was also applicable, regardless of the electrical properties, and was more efficient than the bromo products. Of note, product 33 could be conveniently achieved with no difficulties when argon was displaced by air. In terms of methylenedioxy (34) and thiophene (35), the required products were obtained with moderate efficiency. Moreover, other types of iodofluorides, for instance, phenyl sulfone (36), were applicable under standard conditions, and particularly, perfluoroalkyl iodide (37) and perfluoroalkyl bromide (37-2) were installed at the right position, with excellent yields, owing to the convenient initiation from palladium catalysts.27c,28
 | ||
| Scheme 2 Substrate scope of fluorides. Reaction conditions: B(MIDA) (0.27 mmol, 1.0 equiv.), fluorides (2.0 equiv.), PdCl2(dppf)·DCM (10 mol%), BINAP (10 mol%), K2CO3 (2.0 equiv.), EtOAc (2 mL), 85 °C, argon, 12 h. aThe reaction was conducted in air. bThe reaction was conducted for 5 h. | ||
Then, we modified the bioactive molecules with complex motifs. To our delight, a chiral-derived compound 38 was generated in a 75% yield. For the decorations of drugs, mexiletine, a drug against ventricular arrhythmia, and rimantadine, a drug with great influence on the influenza virus, were modified to provide 39 in a 70% yield and 40 in a 54% yield. As a critical area in biology research, a series of amino acid substrates from alanine, glutamate, and phenylalanine were tested to evaluate the generality of the palladium-catalyzed system, where all products were furnished smoothly (41–43). Further exploration using L-menthol (44) and α-D-galactopyranose (45) from carbohydrates was highly efficient, as proved by the good yields. Prominently, transformations proceeded excellently in the presence of steroids (46–48) and pentacyclic triterpene (49) under standard conditions. The assembly of 46 and 48, which possess internal alkenes, yielded predominantly addition products neighbored by boron atoms with good site-selectivity, suggesting a directing effect of the B(MIDA) moiety on the adjacent alkene. The above results demonstrated that the current method was applicable to biologically relevant systems, thereby expanding the utilization scope of the reaction for convenient late-stage modifications. Moreover, iodofluorides from mexiletine (50) and amino acids (51–53) could produce expected products, with yields of 67%, 75%, 76%, and 69%, respectively.
Next, we concluded that the reaction could be extended to even substituted olefins with B(MIDA). As illustrated in Scheme 3, the majority of substrates suffered from relatively low yields largely because of the stabilized hyperconjugation effect on the substituted radicals. Alkyls (54) and halogens such as chlorine (55) were amenable to the general method. An attractive rearrangement product (56a) bearing cyclopropyl as the starting material was unexpectedly generated, which broadened the vision of constructing α-difluoromethylene (CF2)-containing B(MIDA) boronates and indirectly verified the radical process occurring in this reaction. The ratio of 56a to 56b was 6.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 according to the 19F NMR analysis. In addition to the above compounds, functionalized side chains possessing benzoyl (57) and phthalimide (58) groups were tolerant to the established reaction. Consistent with the non-substituted examples, iodine could be installed properly when substituted olefins were employed. The rearrangement iodinated product 59a was afforded in a 31% yield, including 10% 59b. Benzoyl (60) and phthalimide (61) substrates could be transformed into the corresponding iodine-containing boronates, in which preliminary diastereoisomer ratio values were observed: 6.7:1 for 60 and 2.5:1 for 61, respectively.
:1 according to the 19F NMR analysis. In addition to the above compounds, functionalized side chains possessing benzoyl (57) and phthalimide (58) groups were tolerant to the established reaction. Consistent with the non-substituted examples, iodine could be installed properly when substituted olefins were employed. The rearrangement iodinated product 59a was afforded in a 31% yield, including 10% 59b. Benzoyl (60) and phthalimide (61) substrates could be transformed into the corresponding iodine-containing boronates, in which preliminary diastereoisomer ratio values were observed: 6.7:1 for 60 and 2.5:1 for 61, respectively.
 | ||
| Scheme 3 Substrate scope of substituted olefin B(MIDA)s. Reaction conditions: B(MIDA) (0.27 mmol, 1.0 equiv.), fluorides (2.0 equiv.), PdCl2(dppf)·DCM (10 mol%), BINAP (10 mol%), K2CO3 (2.0 equiv.), EtOAc (2 mL), 85 °C, argon, 12 h. | ||
In view of the potential synthetic building blocks of fluorinated α-halo boronates, the advantage of this strategy has also been exemplified by the efficient gram-scale synthesis of product 23, and 81% isolated yield was obtained under air conditions (Scheme 4A). The presence of different esters and amides provided valuable opportunities for diverse organic applications as privileged trifunctionalized moieties. As shown in Scheme 4B, starting from 23, the iodine could be easily substituted by nucleophilic benzenethiol derivatives in good yields (62 and 63). This special iodine was not only selectively dehalogenated but also eliminated readily to furnish fluorinated vinyl and alkyl B(MIDA) boronates (64 and 66) in moderate yields. Treating 23 with KHF2 could afford its BF3K analog (65). We also examined the prospect of using bromine atoms. Treating 5 with sodium azide could yield a C–N bond containing derivative 68. Moreover, treating 5 with pinacol under an alkaline condition could generate difluoropyrrolidine pinacol boronates (67) for the first time in high yield. The coupling nature of B(MIDA) boronates was leveraged during the mixing of eliminated-5 with benzene halides to produce styrene-Heck-type compounds 69 and 70. Furthermore, an amide group-bearing compound 29 could smoothly yield benzylmercaptan together with benzoate substituted products, which constructed C–O (73) and C–S (74) bonds directly. The dehalogenation intermediate (71) of 29 was oxidized, during which the B(MIDA) motif was converted into the hydroxyl group (72) for further transformation. Fortunately, treating 12via a Bischler–Napieralski reaction led to the generation of an unprecedented fluorinated crispine A analog (75) in an acceptable yield, which offers a new approach to tetrahydroisoquinoline engineering (Scheme 4C).
 | ||
| Scheme 4 Gram-scale preparation and synthetic utilities. Reaction conditions: for the gram scale reaction: B(MIDA) (2.73 mmol, 1.0 equiv.), ICF2COOEt (2.0 equiv.), PdCl2(dppf)·DCM (10 mol%), BINAP (10 mol%), K2CO3 (2.0 equiv.), EtOAc (10 mL), 85 °C, air, 12 h. aPhSNa, DMF, 85 °C; bsodium benzo[d]thiazole-2-thiolate, DMF, 85 °C; cTTMSS, PhMe, 85 °C; dKHF2, MeOH, 70 °C; eBnNH2, DMF, 85 °C; fpinacol, K3PO4, DCM/THF/H2O, 60 °C; gNaN3, DMF, 85 °C; h(1) BnNH2, DMF, 85 °C; (2) ArI, Pd(OAc)2, SPhos, K3PO4, THF/H2O, 60 °C; iTTMSS, DME, 85 °C; jNaOH, 30% H2O2, THF, 0 °C to rt; kPhCOONa, DMF, 85 °C; lBnSH, Et3N, DMF, 85 °C; mPOCl3, MeCN, 110 °C. For more details, see the ESI.† | ||
Furthermore, we investigated the mechanistic aspects of the palladium catalytic cycle and highlighted the role of the B(MIDA) moiety. To identify whether a difluoroalkyl free radical was involved in the process, a mixture of vinyl B(MIDA) and ICF2COOEt was treated with the radical scavenger TEMPO. As depicted in Scheme 5A, no desired product was detected, and a TEMPO–CF2COOEt complex 76 was validated via high-resolution mass spectrometry. A radical clock experiment was also performed using α-cyclopropylstyrene, which is considered to participate in radical rearrangement,36,37 as a probe (Scheme 5B). A ring-open difunctionalized product 77 was obtained in a 78% yield along with a cis–trans isomerization ratio of 5:1, and a trace amount of 23 was observed. Both results imply that a single-electron transfer pathway drove the generation of the difluoroalkyl radical, and the radical addition had priority over Pd(I)–X oxidative addition. To confirm the guiding role of B(MIDA), two comparative experiments in which vinyl Bpin and styrene were used to displace B(MIDA) as starting materials were conducted (Scheme 5C), and no target products were formed, which offered some evidence for capturing Pd(I)–X species by B(MIDA). As illustrated in Scheme 5D, no reaction occurred in the absence of the palladium catalyst, suggesting that the palladium catalyst was the key factor for fluoroalkyl-radical initiation. We chose another path involving a nickel catalyst and a bpy-ligand to observe whether the reaction would proceed given that nickel could also generate difluoroalkyl free radicals.38 However, nearly no product was obtained, which was in sharp contrast to the model reaction. Besides, it was also unable to carry out the standard reaction thermally initiated by AIBN. Therefore, we added TEMPO trying to capture TEMPO-CF2COOEt. Unfortunately, neither 76 nor 1 was detected by MS or 19F NMR, which means that Ni(II) and AIBN are not a suitable initiator to induce the difluoroalkyl radical in our reaction system. Finally, deficiency of the base led to a remarkable decrease in the yield, which was <5%, as detected via19F NMR, further substantiating the idea that the alkali cation might be critical in inducing C–X formation.
 | ||
| Scheme 5 Mechanistic studies. | ||
Since we observed the unclear role of bases in our reaction system, to provide more insight into the role of the BMIDA group and observed selectivity, density functional theory (DFT) calculations were carried out with the Gaussian 16 package.39 As shown in Fig. 1, the reactant S-1 was selected as the zero-point of the potential energy surface (0.0 kcal mol−1). The direct generation of free radical INT1 from S-1 without the catalyst Pd-BINAP (C1) required a very high free energy barrier of 51.9 kcal mol−1 (Fig. S1†), indicating that the Pd catalyst was essential to initiate the radical process and then form INT1 and C2. As a reversible regio-selectivity was observed in compound 56, two addition pathways were compared in both our model reaction and the formation of 56 (for 56, see ESI Fig. S4†). In the model reaction, INT1 underwent a radical-addition process with B-1 to produce the key intermediate P1-INT2via transition state P1-TS1 in the main reaction (pathway1) with a free energy barrier of 8.2 kcal mol−1, which is more favorable than the side reaction (pathway2) viaP2-TS1 (11.8 kcal mol−1). P1-INT2 or P2-INT2 further reacted with C2 to form the C(sp3)–Pd(II)–X system P1-INT3 or P2-INT3, respectively, which was then stabilized by alkali metal cations (e.g. K+) via its potential coulombic interaction with Br and carbonyl-O of BMIDA (P1-INT4 and P2-INT4). Subsequently, Br was transferred to the α-C of the boron atom through direct RE to form the final product 1via the transition state P1-TS2 with a free energy barrier of 17.9 kcal mol−1, which was lower than that viaP2-TS2 (20.9 kcal mol−1). Notably, it was found that the free energy barrier of the transition state P1-TSNO without alkali metal cations (e.g. K+) was 33.3 kcal mol−1, indicating the critical role of the BMIDA-chelated alkali metal ion which enabled the sp3-reductive-elimination to proceed successfully (Fig. S2†). In addition, a weak attraction between B and Br was recognized by IGM40,41 analysis using Multiwfn,42 which could further stabilize the alkali metal ion complex and might be helpful for heterolytic Pd–X bond dissociation (Fig. S3†). Based on the DFT calculations, it could be concluded that C1, BMIDA and alkali metal carbonates (e.g. K2CO3) were important to the reaction of S-1 and B-1 to form the product 1; moreover, generally observed thermodynamic and dynamic stability made pathway1 feasible while regio-reversible pathway2 was not.
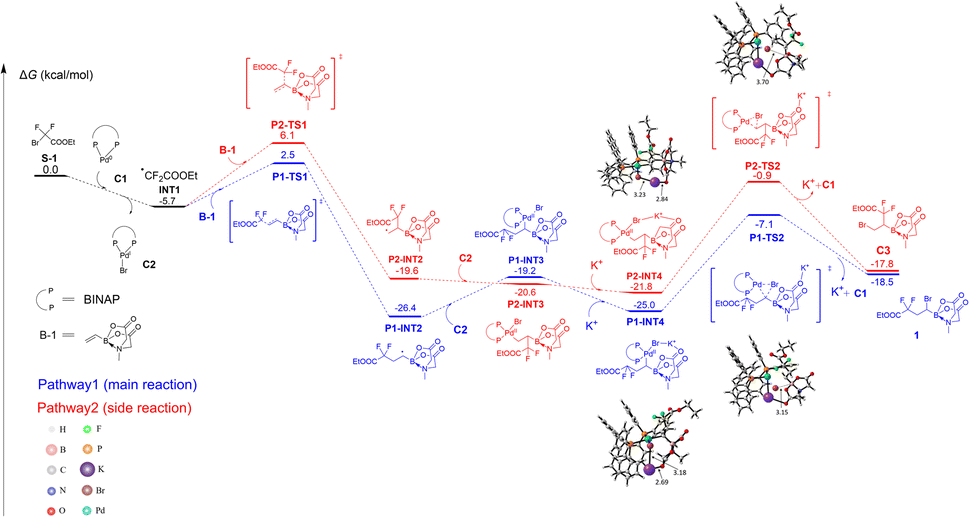 | ||
| Fig. 1 Free-energy profile for the formation of 1 and C3, calculated at the M06-D3/6-311++G(d,p) SDD//B3LYP-D3(BJ)/6-31G(d) SDD level of theory. The SDD basis set was used for Pd. Free energies are given in kcal mol−1. | ||
However, it should be mentioned that XAT (halogen atom transfer) could also be another possible pathway which occurred in our reaction system.35g Once the fluoroalkyl-radical is initiated by Pd(0), halogens of the other difluoroalkyl halides might be abstracted and therefore form the product as well as create a new difluoroalkyl radical via the chain reaction mechanism. The Pd(I) species could also provide the halogens for the fluoroalkyl-radical and be reduced to Pd(0) at the same time. Research trying to find the correct mechanism is still ongoing.
Conclusions
In summary, a high-efficiency and facile synthesis of fluorinated α-halo boronates was established in this study. The anticipated functional groups of B(MIDA) and the alkali cation aided the direct reductive elimination of the C(sp3)–Pd(II)–X system, which was fully characterized by DFT calculations. A series of useful trifunctionalized (F, X, B) building blocks were furnished through the designed plan. This catalytic strategy provides extensive access to esters, amides, drugs, and even natural products modified with fluorinated α-halo boronates, and demonstrates a high level of functional group tolerance, which is unusual for these types of products. Additionally, the palladium-catalyzed process overcomes the challenges associated with substituted olefin boronates, thereby improving the possibilities for diverse functionalization. Follow-up decoration of the products further expands the variety and verifies the practicality of this method. This unique protocol has immense potential for application in the drug industry and chemical biology and will pave a new path for the synthesis of organoboron compounds.Data availability
All experimental, DFT calculation and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compound 23 have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2236620.Author contributions
W. Z., C. L. and H. L. conceived this project. H. W., R. L., W. Z., C. L., and H. L. designed the experiments. H. W. performed the experiments. J. P. and R. Z. prepared some starting materials. R. L., Z. H. and Z. X. performed DFT calculations. H. W., R. L., W. Z., C. L., and H. L. analyzed and interpreted the results. H. W., R. L., W. Z., C. L., and H. L. wrote and revised this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Key R&D Program of China (No. 2023YFF1205104 and 2022YFA1302900), the National Natural Science Foundation of China (No. 82273766 and 82130105), and the Youth Innovation Promotion Association CAS (2020282). We thank LetPub (https://www.letpub.com/) for its linguistic assistance during the preparation of this manuscript.Notes and references
- O. Andler and U. Kazmaier, Org. Lett., 2021, 23, 8439 CrossRef CAS PubMed.
- O. Andler and U. Kazmaier, Org. Biomol. Chem., 2021, 19, 4866 RSC.
- J. Gorges and U. Kazmaier, Org. Lett., 2018, 20, 2033 CrossRef CAS PubMed.
- Y. Lou, J. Qiu, K. Yang, F. Zhang, C. Wang and Q. Song, Org. Lett., 2021, 23, 4564 CrossRef CAS PubMed.
- D. S. Matteson, H.-W. Man and O. C. Ho, J. Am. Chem. Soc., 1996, 118, 4560 CrossRef CAS.
- S. Z. Sun, M. Borjesson, R. Martin-Montero and R. Martin, J. Am. Chem. Soc., 2018, 140, 12765 CrossRef CAS PubMed.
- S. Z. Sun and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 3622 CrossRef CAS PubMed.
- J. Schmidt, J. Choi, A. T. Liu, M. Slusarczyk and G. C. Fu, Science, 2016, 354, 1265 CrossRef CAS PubMed.
- (a) R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156 CrossRef CAS PubMed; (b) S. Touchet, F. Carreaux, B. Carboni, A. Bouillon and J. L. Boucher, Chem. Soc. Rev., 2011, 40, 3895 RSC; (c) D. B. Diaz and A. K. Yudin, Nat. Chem., 2017, 9, 731 CrossRef CAS PubMed; (d) V. M. Dembitsky and M. Srebnik, Tetrahedron, 2003, 59, 579 CrossRef CAS; (e) E. S. Priestley and C. P. Decicco, Org. Lett., 2000, 2, 3095 CrossRef CAS PubMed.
- (a) D. S. Matteson, J. Org. Chem., 2013, 78, 10009 CrossRef CAS PubMed; (b) D. S. Matteson, Chem. Rev., 1989, 89, 1535 CrossRef CAS; (c) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588 CrossRef CAS; (d) D. S. Matteson and D. Majumdar, Organometallics, 1983, 2, 1529 CrossRef CAS.
- (a) S. Elgendy, G. Patel, V. V. Kakkar, G. Claeson, D. Green, E. Skordalakes, J. A. Baban and J. Deadman, Tetrahedron Lett., 1994, 35, 2435 CrossRef CAS; (b) S. Elgendy, G. Claeson, V. V. Kakkar, D. Green, G. Patel, C. A. Goodwin, J. A. Baban, M. F. Scully and J. Deadman, Tetrahedron, 1994, 50, 3803 CrossRef CAS.
- (a) B. Zheng and M. Srebnik, Tetrahedron Lett., 1993, 34, 4133 CrossRef CAS; (b) B. Zheng and M. Srebnik, Tetrahedron Lett., 1994, 35, 1145 CrossRef CAS.
- (a) D. J. Pasto, J. Chow and S. K. Arora, Tetrahedron, 1969, 25, 1557 CrossRef CAS; (b) H. C. Brown, N. R. De Lue, Y. Yamamoto and K. Maruyama, J. Org. Chem., 1977, 42, 3252 CrossRef CAS; (c) D. S. Matteson and D. Fernando, J. Organomet. Chem., 2003, 680, 100 CrossRef CAS; (d) H. C. Brown and Y. Yamamoto, J. Chem. Soc. D, 1971, 1535 RSC.
- L. Yang, D. H. Tan, W. X. Fan, X. G. Liu, J. Q. Wu, Z. S. Huang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 3454 CrossRef CAS PubMed.
- D. Wang, J. Zhou, Z. Hu and T. Xu, J. Am. Chem. Soc., 2022, 144, 22870 CrossRef CAS PubMed.
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (b) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- X. Chen, K. Fan, Y. Liu, Y. Li, X. Liu, W. Feng and X. Wang, Adv. Mater., 2022, 34, e2101665 CrossRef PubMed.
- (a) K. Marcin, D. Marek, D. Krzysztof, K. Tomasz, S. Janusz and W. Krzysztof, Tetrahedron Lett., 2017, 58, 2162 CrossRef; (b) M. Ueda, Y. Kato, N. Taniguchi and T. Morisaki, Org. Lett., 2020, 22, 6234 CrossRef CAS PubMed; (c) T. Fang, J. Qiu, K. Yang and Q. Song, Org. Chem. Front., 2021, 8, 1991 RSC.
- (a) J. Li and M. D. Burke, J. Am. Chem. Soc., 2011, 133, 13774 CrossRef CAS PubMed; (b) D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961 CrossRef CAS PubMed; (c) E. M. Woerly, A. H. Cherney, E. K. Davis and M. D. Burke, J. Am. Chem. Soc., 2010, 132, 6941 CrossRef CAS PubMed; (d) G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667 CrossRef CAS PubMed; (e) E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484 CrossRef CAS PubMed; (f) J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221 CrossRef CAS PubMed; (g) A. M. Kelly, P.-J. Chen, J. Klubnick, D. J. Blair and M. D. Burke, Org. Lett., 2020, 22, 9408 CrossRef CAS PubMed; (h) D. J. Blair, S. Chitti, M. Trobe, D. M. Kostyra, H. M. S. Haley, R. L. Hansen, S. G. Ballmer, T. J. Woods, W. Wang, V. Mubayi, M. J. Schmidt, R. W. Pipal, G. F. Morehouse, A. M. E. Palazzolo Ray, D. L. Gray, A. L. Gill and M. D. Burke, Nature, 2022, 604, 92 CrossRef CAS PubMed.
- (a) S. Adachi, A. B. Cognetta, M. J. Niphakis, Z. He, A. Zajdlik, J. D. St Denis, C. C. Scully, B. F. Cravatt and A. K. Yudin, Chem. Commun., 2015, 51, 3608 RSC; (b) A. Holownia, C. H. Tien, D. B. Diaz, R. T. Larson and A. K. Yudin, Angew. Chem., Int. Ed., 2019, 58, 15148 CrossRef CAS PubMed; (c) S. J. Kaldas, C. H. Tien, G. D. P. Gomes, S. Meyer, M. Sirvinskas, H. Foy, T. Dudding and A. K. Yudin, Org. Lett., 2021, 23, 324 CrossRef CAS PubMed.
- (a) M. L. Lepage, S. Lai, N. Peressin, R. Hadjerci, B. O. Patrick and D. M. Perrin, Angew. Chem., Int. Ed., 2017, 56, 15257 CrossRef CAS PubMed; (b) Y. M. Ivon, I. V. Mazurenko, Y. O. Kuchkovska, Z. V. Voitenko and O. O. Grygorenko, Angew. Chem., Int. Ed., 2020, 59, 18016 CrossRef CAS PubMed.
- (a) W. X. Lv, Y. F. Zeng, Q. Li, Y. Chen, D. H. Tan, L. Yang and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 10069 CrossRef CAS PubMed; (b) Y. F. Zeng, W. W. Ji, W. X. Lv, Y. Chen, D. H. Tan, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 14707 CrossRef CAS PubMed; (c) W. X. Lv, Q. Li, J. L. Li, Z. Li, E. Lin, D. H. Tan, Y. H. Cai, W. X. Fan and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 16544 CrossRef CAS PubMed; (d) D. H. Tan, Y. H. Cai, Y. F. Zeng, W. X. Lv, L. Yang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 13784 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, J. Am. Chem. Soc., 2020, 142, 12245 CrossRef CAS PubMed.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, X.-F. Zhu and G.-P. Wu, J. Am. Chem. Soc., 2021, 143, 3455 CrossRef CAS PubMed.
- Y.-Y. Zhang, G.-W. Yang, R. Xie, X.-F. Zhu and G.-P. Wu, J. Am. Chem. Soc., 2022, 144, 19896 CrossRef CAS PubMed.
- (a) D. T. Rosevear and F. G. A. Stone, J. Chem. Soc. A, 1968, 164 RSC; (b) H. D. Empsall, M. Green and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1972, 96 RSC; (c) Q.-Y. Chen, Z.-Y. Yang, C.-X. Zhao and Z.-M. Qiu, J. Chem. Soc., Perkin Trans. 1, 1988, 563 RSC.
- Z. Feng, Q. Q. Min, H. Y. Zhao, J. W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270 Search PubMed.
- B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 2015, 137, 6762 CrossRef CAS PubMed.
- N. Kumar, R. R. Reddy, N. Eghbarieh and A. Masarwa, Chem. Commun., 2019, 56, 13 RSC.
- X. Chen, J. Zhao, M. Dong, N. Yang, J. Wang, Y. Zhang, K. Liu and X. Tong, J. Am. Chem. Soc., 2021, 143, 1924 CrossRef CAS PubMed.
- Y. Lan, P. Liu, S. G. Newman, M. Lautens and K. N. Houk, Chem. Sci., 2012, 3, 1987 RSC.
- J. Z. Essman and E. N. Jacobsen, J. Am. Chem. Soc., 2024, 146, 7165 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed.
- (a) C. Xu, R. Cheng, Y. C. Luo, M. K. Wang and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18741 CrossRef CAS PubMed; (b) L. An, Y. L. Xiao, S. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6921 CrossRef CAS PubMed; (c) H. Y. Zhao, Z. Feng, Z. Luo and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10401 CrossRef CAS PubMed; (d) L. An, F. F. Tong, S. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 11884 CrossRef CAS PubMed; (e) X.-P. Fu, X.-S. Xue, X.-Y. Zhang, Y.-L. Xiao, S. Zhang, Y.-L. Guo, X. Leng, K. N. Houk and X. Zhang, Nat. Chem., 2019, 11, 948 CrossRef CAS PubMed; (f) C. Xu, Z.-F. Yang, L. An and X. Zhang, ACS Catal., 2019, 9, 8224 CrossRef CAS; (g) W.-H. Guo, H.-Y. Zhao, Z. J. Luo, S. Zhang and X. Zhang, ACS Catal., 2019, 9, 38 CrossRef CAS.
- J. E. Baldwin, Chem. Rev., 2003, 103, 1197 CrossRef CAS PubMed.
- E. Shirakawa, X. Zhang and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 4671 CrossRef CAS PubMed.
- J. W. Gu, Q. Q. Min, L. C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Gaussian. Inc., Wallingford CT, 2016 Search PubMed.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928 RSC.
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization data, and original 1H and 13C NMR spectra. CCDC 2236620 (for 23). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07900k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |