
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnisotropy-dependent chirality transfer from cellulose nanocrystals to β-FeOOH nanowhiskers†
Jinyu
Tang
,
Shouhua
Feng
 and
Ming
Yang
*
and
Ming
Yang
*
The State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun, 130012, Jilin, China. E-mail: mingyang@jlu.edu.cn
First published on 13th March 2025
Abstract
Chiral iron oxides and hydroxides have garnered considerable interest owing to the unique combination of chirality and magnetism. However, improving their g-factor, which is critical for optimizing the chiral magneto-optical response, remains elusive. We demonstrated that the g-factor of β-FeOOH could be boosted by enhancing the anisotropy of nanostructures during a biomimetic mineralization process. Cellulose nanocrystals were used as both mineralization templates and chiral ligands, driving oriented attachment of β-FeOOH nanoparticles and inducing the formation of highly aligned chiral nanowhiskers. Circular dichroism spectra and time-dependent density-functional theory proved that chirality transfer was induced from cellulose nanocrystals to β-FeOOH through ligand–metal charge transfer. Interestingly, chirality transfer was significantly enhanced during the elongation of nanowhiskers. A nearly 34-fold increase in the g-factor was observed when the aspect ratio of nanowhiskers increased from 2.6 to 4.4, reaching a g-factor of 5.7 × 10−3, superior to existing dispersions of chiral iron oxides and hydroxides. Semi-empirical quantum calculations revealed that such a remarkable improvement in the g-factor could be attributed to enhanced dipolar interactions. Cellulose nanocrystals exert vicinal actions on highly anisotropic β-FeOOH with a large dipole moment, increasing structural distortions in the coordination geometry. This mechanism aligns with the static coupling principle of one-electron theory, highlighting the strong interaction potential of supramolecular templates. Furthermore, paramagnetic β-FeOOH nanowhiskers alter the magnetic anisotropy of cellulose nanocrystals, leading to a reversed response of helical photonic films to magnetic fields, promising for real-time optical modulation.
Introduction
Chiral inorganic nanomaterials have captured the attention of researchers due to their interesting optical properties and diverse applications.1–3 Imposing symmetry breaking of organic matter on inorganic nanomaterials is the most effective means for inducing chirality transfer.4–6 The fundamental mechanism is related to interfacial electronic coupling, which has allowed the emergence of chiroptical properties that are otherwise hard to achieve in traditional chiral molecules.4,6 Among various chiroptical inorganic nanoparticles (NPs), iron oxides stand out as an intriguing platform owing to their unique spin and magnetic characteristics.7–9 Despite the progress made in the synthesis of chiral iron oxides using amino acids as chiral agents,2,8,10 challenges persist in (1) understanding the relationship between structural parameters and the g-factor, (2) extending to supramolecular templates for the potential increase of asymmetry, and (3) exploring novel anisotropy-dependent chiromagnetic properties. In particular, while different strategies have been proposed to improve the g-factor, such as the engineering of transition dipoles,6 exciton coupling,11 and long-range order,3 none have been proven effective for iron oxides and hydroxides.Polysaccharides are intricately linked with biogenic iron oxyhydroxides,12 particularly in the form of aligned goethite13 and magnetite nanowhiskers (NWs).14 Primarily associated with hard biomineralized tissues, the potential for chirality transfer from polysaccharides to iron oxides remains elusive. This connection between polysaccharides and iron oxides offers a great opportunity to combine chirality with magnetism through a biomimetic concept for generating chiromagnetic effects.7,10 Combining two earth-abundant materials in a hybrid chiral system also mirrors the elegant strategies employed by living organisms to maximize the use of limited resources, highlighting the sustainable advantages of such applications. In contrast to small chiral molecules such as amino acids, structural polysaccharides may not only provide chiral interfaces for facilitating chirality transfer but also act as supramolecular templates, offering significant potential for strengthened interactions.15,16 This aspect, often overlooked in theories focusing on excited state coupling,17 could be better understood through one-electron theory based on the static coupling mechanism.18
Supramolecular templates with multiscale architecture can induce helical organization to manipulate chirality,3,19 with cellulose nanocrystals (CNCs) serving as one such example. As a renewable resource, CNCs, assembled from β-1,4-linked anhydro-D-glucose chains with asymmetric carbon atoms, were proven to be twisted.20 A chiral nematic phase can be produced through evaporation-induced self-assembly (EISA) of CNCs at room temperature.21 This hierarchical chiral template has been widely used for the synthesis of hybrid chiral photonic films,22–25 chiral inorganic films,26–29 and helical assembly of inorganic NPs.30–32 It is worth noting that in these previous efforts, chirality transfer from CNCs to inorganic NPs occurred at the micro- and nano-scales. The asymmetric packing of inorganic NPs is the main reason for the observation of chiroptical properties.
In this work, oriented attachment of β-FeOOH NPs onto CNCs was demonstrated to form highly aligned chiral NWs. Chirality transfer from CNCs to β-FeOOH occurs at the molecular level, and the induced optical activity of β-FeOOH was confirmed by circular dichroism (CD) spectra, in agreement with time-dependent density-functional theory (TD-DFT). An enhancement of the g-factor of β-FeOOH by up to 34-fold during its oriented growth was observed, which was elaborated by one-electron theory. Dipole moment (DM) calculations showed that CNCs introduce field perturbations through dipolar interactions with the crystal lattice of β-FeOOH, resulting in structural distortions of β-FeOOH, beneficial to chiroptical properties. Orderly arranged paramagnetic β-FeOOH NWs modify CNCs' magnetic anisotropy, leading to a unique magneto-optical response of helical photonic films.
Results and discussion
CNCs were directly employed to induce oriented mineralization of iron oxides under mild conditions. In a typical synthesis, certain amounts of CNC solutions were added dropwise to a diluted Fe3+ solution. After a 36-hour room-temperature reaction, high-contrast NWs were observed, exhibiting a strong orientation along the axis of CNCs (Fig. 1a). Low-magnification TEM images confirmed the alignment, displaying two substances (CNCs and β-FeOOH) with distinctly different contrasts simultaneously (Fig. S1†). Concurrently, small NPs were also present (Fig. 1b and S2a–d†). Their sizes closely match the diameters of NWs (Fig. S2e and f†), aligning well with the elementary nanofibrils in CNCs.21 A high-resolution transmission electron microscopy (HRTEM) image revealed inter-planar spacing distances of approximately 0.330 nm and 0.255 nm, corresponding to the lattice planes of (30–1) and (21–1) for β-FeOOH, respectively (Fig. 1c, inset). Fast Fourier transform (FFT) analysis confirmed the growth direction of NWs aligned along the b-axis (Fig. 1c, inset).33,34 Further examination through high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDXS) mapping revealed the homogeneous dispersion of β-FeOOH on CNCs (Fig. 1d). In a solution lacking CNCs, oval NPs with diameters of approximately 15–30 nm were obtained (Fig. S3†), implying the critical role of CNCs in the confined growth of β-FeOOH. The template effect of CNCs is likely favoured by structural matching35 because the Fe–Fe distance of 3.03 Å along the b-axis of β-FeOOH perfectly matches the O–O distance of 2.99 Å in cellulose (Fig. 1e). This geometric alignment is accompanied by the strong binding of O in the D-glucose units with Fe3+ (Fig. S4†). The binding of Fe3+ with sulfate half-ester groups, primarily located at C6, is also possible, although they constitute only a minor percentage.36 The alignment of β-FeOOH with CNCs suggests a level of structural control akin to that exerted by chitin over iron oxides during mineralization,37,38 although the precise pathways may differ. | ||
| Fig. 1 (a and b) TEM images of β-FeOOH adhering to the surface of cellulose nanocrystals (CNCs). The white and yellow arrows indicate the presence of β-FeOOH NWs and NPs, respectively. (c) An HRTEM image with the upper and lower insets showing the lattice fringes and FFT analysis of the central square part, respectively. The yellow line is perpendicular to (020) planes, parallel to the b-axis and the growth direction of NWs. (d) An HAADF-STEM image and EDXS mappings. (e) Structural matching between cellulose and β-FeOOH. | ||
Time-dependent TEM observations acquired insight into the mechanism of oriented mineralization. To visualize the growth of β-FeOOH clearly, high-magnification TEM images were taken. Under such imaging conditions, CNCs could hardly be seen due to their extremely low contrast. However, because the size of β-FeOOH NWs is much smaller than that of CNCs, it is practically feasible to differentiate β-FeOOH NWs from CNCs. After 6 hours of reaction, the resultant material was labelled as CNC-Fe-6, with similar abbreviations used for other composites obtained at different reaction times. The surface of CNCs exhibited scattered small NPs in CNC-Fe-6 (Fig. 2a and S5a†), which aligned along the long axis and subsequently transitioned into NWs at a later stage (CNC-Fe-12) (Fig. 2b and S5b†). Additionally, a few NPs free from CNCs were also observed (Fig. S5b and S5c†). With prolonged reaction time, the coverage of NWs increased, but NPs persisted (Fig. 2c–e and S5d†), continuing to align linearly (Fig. S6†). The aspect ratios (ARs) of β-FeOOH exhibited continuous growth before experiencing a slight drop at 48 h (Fig. S7†). X-ray diffraction (XRD) patterns confirmed that both NPs and NWs could be assigned to β-FeOOH (JCPDS 42-1315) (Fig. 2f). These results indicated that oriented attachment of β-FeOOH to CNCs constitutes a fundamental mechanism in the formation of aligned NWs. Fe3+ interacts with functional groups on CNCs, promoting the nucleation of small NPs, which is favoured by the electrostatic attraction between Fe3+ and CNCs, as confirmed by the sharp decline of electrokinetic potential after the addition of Fe3+ (Table S1†). Small NPs may also form directly in the solution but tend to assemble with those already anchored to CNCs, creating linear chains that eventually grow into NWs (Fig. 2a–e, inset, and S8†).
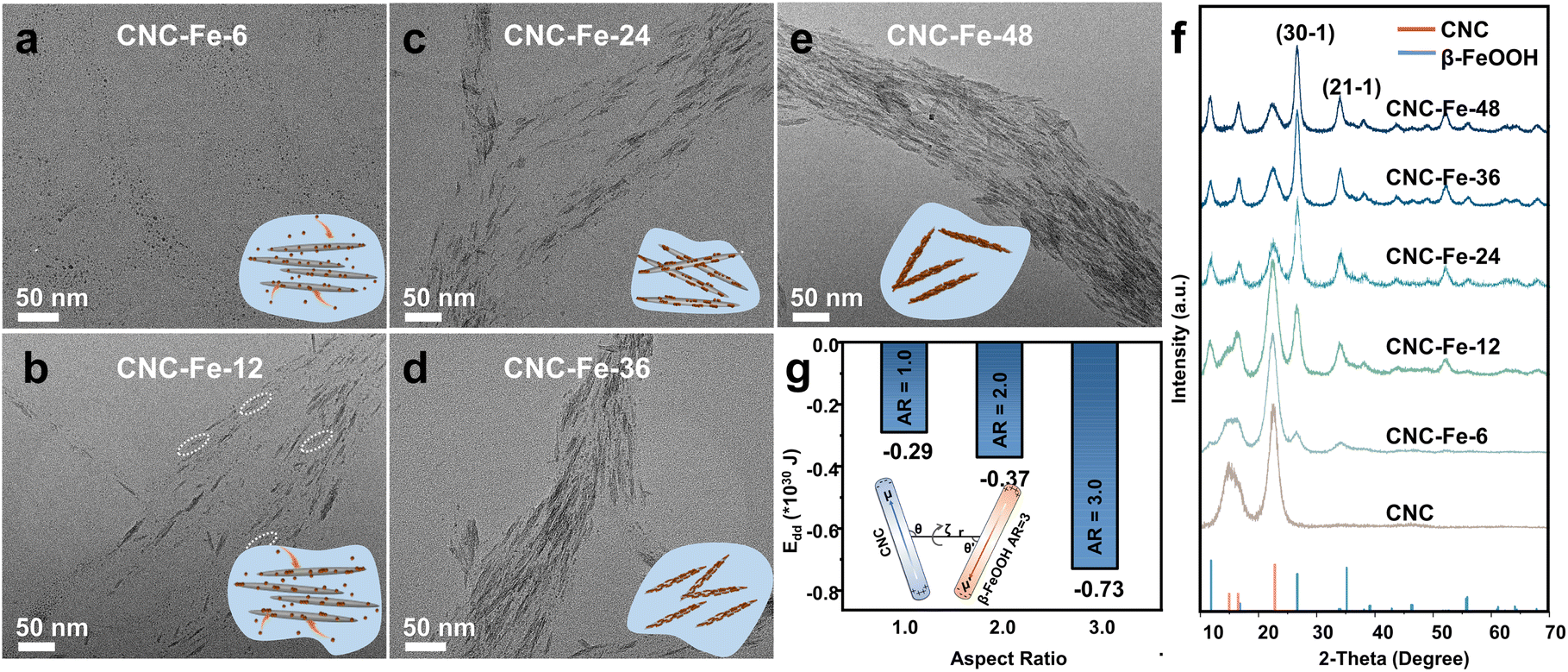 | ||
| Fig. 2 (a–e) TEM images of mineralized CNCs obtained at different reaction times (6–48 h) and (f) the corresponding XRD patterns. The inset shows the progression of oriented attachment. (g) The calculated dipole–dipole interaction potential (Edd) between CNCs and β-FeOOH with different aspect ratios (ARs). The inset displays the geometry used for the calculation of Edd when AR = 3. | ||
Quantum mechanical calculations based on semi-empirical PM6 theory revealed that the DM of β-FeOOH increases as ARs become larger (Fig. S9†). The dipole–dipole interaction potential (Edd)39 between CNCs and β-FeOOH is strengthened according to the variations of DMs (μ′) and angles (θ′) formed between the vectors of DMs and the line connecting the centre (Fig. 2g and S10†), encouraging oriented attachment of β-FeOOH on CNCs. However, dipolar interactions between CNCs and β-FeOOH could limit particle mobility, which explains the presence of small NPs even at the end of reactions, differing from oriented attachment in the solution.40–42
The UV-vis spectra of CNC dispersions exhibited a distinct UV absorption profile43 (Fig. 3a). The appearance of new absorption peaks during the reaction implied the formation of β-FeOOH (Fig. 3a). Specifically, the peaks centred at 216, 268, and 350 nm are attributed to O 2p → Fe 3d ligand–metal charge transfer (LMCT) transitions.44 The peak at 450 nm results from the double excitation of the Fe–Fe pair transition, while the peak at 538 nm is ascribed to a single ion transition.45,46 Additionally, the broad peak beyond 600 nm may be related to other forbidden d–d transitions. The redshift in the absorption edge, which occurred as NWs grew, can be attributed to changes in the energy levels of β-FeOOH with varying ARs.47
 | ||
| Fig. 3 (a) UV-vis and (b) CD spectra of dispersions of CNCs and CNC-Fe-x (x = 6–48). (c) Atomistic models used for TD-DFT calculations. (d) Simulated CD and absorption spectra of Fe3O6-3Glu. | ||
In the CD spectra, CNC dispersions exhibited a conspicuous positive signal (Fig. 3b). However, this signal significantly diminished after a 6-hour reaction (CNC-Fe-6) (Fig. 3b). With the reaction's extension, negative peaks began to emerge, gradually increasing in intensity and shifting bathochromically from 12 to 48 hours (Fig. 3b). Compared with time-dependent TEM observations (Fig. 2a–e), the emergence of NWs appeared to be critical for an apparent negative CD signal. In contrast, the dispersion of β-FeOOH NPs measuring 15–30 nm exhibited a bare CD signal, albeit with similar absorption spectra (Fig. S11†).
The above results indicated the chirality transfer from CNCs to β-FeOOH. To elucidate the mechanism of chirality transfer, we conducted calculations using the TD-DFT method.48 The geometry of a D-glucopyranose unit extracted from the structure of CNCs has a considerable effect on the simulated CD spectra, primarily due to the different orientations of –OH groups (Fig. S12†). However, the calculation of two D-glucopyranose units linked by β-(1–4) glycosidic bonds extracted from different positions showed similar CD spectra, indicating that two glucose monomers may represent cellulose quite well (Fig. S13†). Thus, to simplify the search for excited states, we substituted the complex cellulose molecules with two glucose monomers linked by β-(1–4) glycosidic bonds, denoted as Glu (Fig. 3c). The calculated absorption peak of Glu is in the UV-vis range and the apparent Cotton effect can be observed because of the σ to σ* and n to σ* transitions according to the hole–electron distribution and charge differential density plot (Fig. S14†).
We then established Fe3O6, an elementary Fe–O structure in the akaganéite unit cell, bonded with three Glu units through C3–O, denoted as Fe3O6-3Glu (Fig. 3c). The formation of Fe–O bonds between CNCs and β-FeOOH was confirmed by both FT-IR and XPS spectra. The broad peaks between 3500 and 3350 cm−1 are attributed to the stretching vibrations of –OH,49 which exhibited an obvious redshift in CNC-Fe-36 compared with CNCs and β-FeOOH (Fig. S15†), implying hydrogen bonding interactions between CNCs and β-FeOOH. The FT-IR bands at 703 cm−1 for β-FeOOH corresponding to Fe–O bonds50 shifted to 696 cm−1 in CNC-Fe-36 (Fig. S15†), suggesting the presence of Fe–O–C bonds in the composites.51 XPS spectra of CNC-Fe-36 showed the characteristic peaks from C, O and Fe elements (Fig. S16a†). High-resolution Fe 2p XPS spectra (Fig. S16b†) showed Fe 2p1/2 and 2p3/2 peaks and two satellite peaks at 725.5, 711.9, 732.6 and 720.2 eV, respectively, corresponding to Fe3+ in β-FeOOH.52–54 In the XPS spectra of C 1s (Fig. S16c†), an obvious shift of the peak related to C–O bonds toward low binding energies in CNC-Fe-36 compared to CNCs could be attributed to the lower electronegativity of Fe atoms. The O 1s spectra of CNC-Fe-36 (Fig. S16d†) included a new Fe–O–C peak at 531.7 eV,55 confirming the existence of Fe–O–C bonds. The peak related to Fe–O–H presented an obvious shift toward high binding energies, a result also observed for β-FeOOH/graphene oxide composites.55 A typical lattice oxygen (Fe–O) peak52,56 at 530.3 eV was observed for CNC-Fe-36 compared to 531.3 eV for β-FeOOH.
The simulated absorption spectra of Fe3O6-3Glu differed from those of Glu (Fig. 3d and S13c†), extending into the longer wavelength range, closely correlated with the CD spectra (Fig. 3d). Fe3O6-3Glu generated chiroptical signals at 253, 349, and 439 nm due to LMCT, and the peaks at 600 nm and 800 nm are the result of d–d transitions according to hole–electron distributions57,58 (Fig. S17 and S18†). The HOMOs of Fe3O6-3Glu are situated not only at O atoms of Glu but also at lattice O atoms. LUMOs are mainly located at the d and p orbitals of the Fe3O6 centre, with a small portion distributed over O atoms in the chiral ligands (Fig. S19†). No CD signals could be observed for Fe3O6 due to its symmetrical structure (Fig. S20†). Despite the use of the simplified model, these results confirmed that glucose monomers could impart chirality to β-FeOOH through Fe–O–C bonds (Fig. 3d). Although there are noticeable similarities in the shapes of the experimental and calculated CD spectra, deviations are still apparent, likely stemming from the use of small molecular structures.5
According to the Rosenfeld equation, Roa = Im(|μ|·|m|![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ), optical activity (Roa) depends on the transition electric dipole moment (μ), the transition magnetic dipole moment (m), and the angle between them (θ). When θ exceeds 90°, Roa becomes negative, while it remains positive when θ is less than 90°. TD-DFT calculations indicated that interfacing CNCs with β-FeOOH led to the formation of new excited states (Fig. S21, Tables S2 and S3†), which are responsible for the shift from the positive peak observed in CNCs to the negative peak seen in β-FeOOH NWs upon chirality transfer (Fig. 3b).
cosθ), optical activity (Roa) depends on the transition electric dipole moment (μ), the transition magnetic dipole moment (m), and the angle between them (θ). When θ exceeds 90°, Roa becomes negative, while it remains positive when θ is less than 90°. TD-DFT calculations indicated that interfacing CNCs with β-FeOOH led to the formation of new excited states (Fig. S21, Tables S2 and S3†), which are responsible for the shift from the positive peak observed in CNCs to the negative peak seen in β-FeOOH NWs upon chirality transfer (Fig. 3b).
We did not observe any evidence of helical packing of β-FeOOH NWs with long-range order on the surface of CNCs. This can be explained by the fact that the direct growth of β-FeOOH NWs on the CNC surface occurs through random nucleation sites, which may disrupt the helical order. This contrasts with the self-assembly of NPs on a supramolecular template, where a more organized structure is typically achieved.3,30 Additionally, the presence of small NPs alongside the NWs further complicates the establishment of long-range order. FFT analysis of the top, middle, and bottom regions of a β-FeOOH NW revealed that the b-axis direction remained consistent at different positions (Fig. S22†), suggesting that no twisting occurred.
We employed the g-factor to assess the optical activity of CNC/β-FeOOH dispersions obtained at different reaction times to eliminate the influence of concentration. Our primary focus was on the negative peak occurring at approximately 350 nm, as it represents the most intense transition associated with chirality transfer. The g-factor exhibited an increase from 12 to 36 hours, followed by a negligible change at the 48-hour mark (Fig. 4a). This change in the g-factor closely paralleled the variations in the ARs of β-FeOOH (Fig. 4a). Notably, as ARs increased from 2.6 to 4.4, we observed a nearly 34-fold increase in the g-factor (from 1.7 × 10−4 to 5.7 × 10−3) (Fig. 4a). The absolute value of 5.7 × 10−3 is one order of magnitude higher than that of CdSe nanorods (5 × 10−4),59 representing a fairly high g-factor achieved for the dispersion of chiral iron oxides, which is typically in the range from 1 × 10−4 to 2 × 10−3. For example, chiral superparamagnetic Fe3O4 supraparticles with a g-factor of ∼2 × 10−3 were fabricated by Li et al.2 Polyacetylene macromolecular helicity endowed Fe3O4 nanoparticles with a dissymmetric factor of ∼1.3 × 10−3.60 Magnetic nanoparticles decorated with D-cysteine molecules were demonstrated to have a g-factor of 1.69 × 10−3 by Visheratina et al.61 In addition, regioselective magnetization endowed one-dimensional ZnxCd1−xS-Ag2S/Au@Fe3O4 hybrid nanorods with a g-factor of 0.15 × 10−3.62
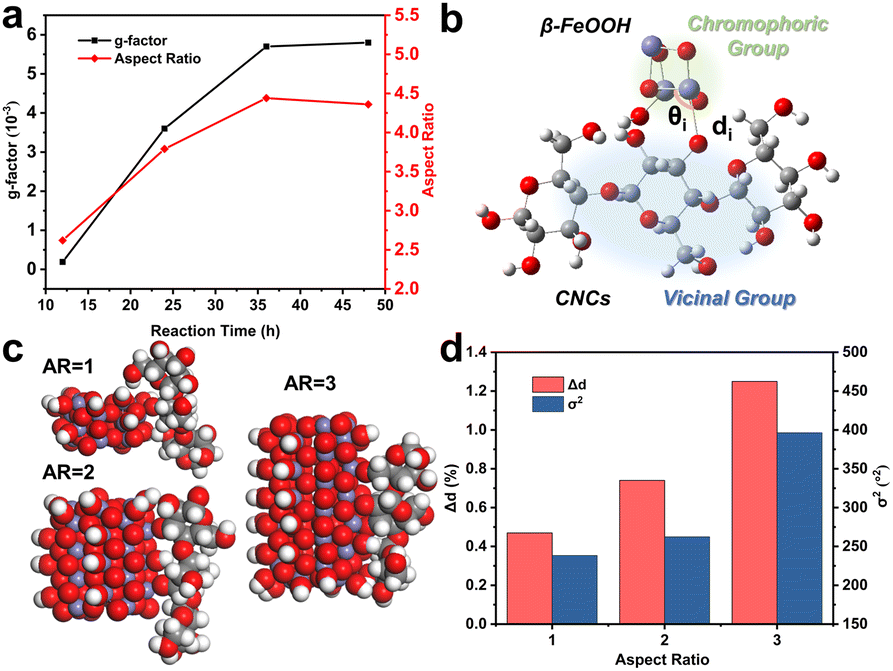 | ||
| Fig. 4 (a) The change of the g-factor and ARs during the reaction. (b) Schematic illustration of the division of two groups in one-electron theory. (c) Space-filling models of iron oxide clusters with different ARs connected with β-1,4-linked anhydro-D-glucose from three monomers through Fe–O–C3. (d) The effect of ARs on Δd and σ2. | ||
The enlargement of the g-factor of CdSe nanorods was explained by a non-degenerate coupled-oscillator model59 for surface-related CD and linear polarization63,64 for excitonic CD. Since anisotropic structures often possess large DMs for strong interactions with chiral molecules, one-electron theory65 based on the static coupling mechanism (ESI, Section S2†) provides a comprehensive explanation for the correlation between the g-factor and ARs. In this theory, the perturbation of electronic states of achiral chromophores by vicinal actions results in chiroptical activity. The rotatory power and g-factor, therefore, depend on vicinal actions, which are proportional to the amount of atomic displacement distorting the structural framework.
Applying this theory to our case, we regarded β-FeOOH as the chromophoric group and CNCs as the vicinal groups (Fig. 4b). To estimate the effect of vicinal actions, the distortion of coordination geometry was characterized by changes in the bond length (Δd) and bond angle variance (σ2).66 An increase in Δd and σ2 as calculated by the PM6 method was observed when the ARs of β-FeOOH increased (Fig. 4c and d) or when increasing the number of glucose monomers while maintaining the same binding site with Fe3O6 (Fig. S23†). Because the increase of ARs of iron oxide clusters and the number of glucose monomers accompanied the increase of DMs (Fig. S9 and S24†), strengthened dipolar interactions (Fig. 2g) play a significant role in enhancing vicinal actions. This effect provides a strong short-range field perturbation for inducing structural distortions, ultimately contributing to a larger g-factor.67,68 It is important to mention that oriented attachment may also result in an augmented density of crystal defects69,70 (Fig. S25†), further distorting the coordination geometry.
The chiral β-FeOOH could co-assemble with CNCs to produce iridescent photonic films. A consistent directional arrangement was observed on the film surface of CNCs (Fig. S26a†), which diminished for CNC-Fe-6 (Fig. S26b†), probably due to the disturbance of interfibrillar interactions by Fe3+. The arrangement gradually improved from CNC-Fe-12 to CNC-Fe-36, but the trend reversed for CNC-Fe-48 (Fig. S26c–f†). A longer reaction likely transformed Fe3+ into thin β-FeOOH NWs, which could conform to the assembly of CNCs. However, when β-FeOOH NWs became too large, the conformality decreased, affecting the assembly again (Fig. S26f†). The observed optimal loading of inorganic NPs is common in CNC-based chiral photonic films.15,16 Polarized optical microscopy (POM) images (Fig. S26,† insets) showed obvious birefringence, consistent with the anisotropic structure. The colour of the images gradually changed from green to yellow and then to orange, indicating a change in the packing of CNCs.71 The cross-sectional SEM images revealed a lamellar architecture (Fig. S27†), a characteristic feature of the cholesteric arrangement.72
All these films exhibited positive CD peaks due to the preferential reflection of left-handed circularly polarized light (CPL) by a left-handed helical structure73 (Fig. S28†). Slight changes in CD spectra during sample rotation and “front-back” measurements confirmed that linear dichroism (LD) and linear birefringence (LB) as potential artifacts could be ruled out (Fig. S29†).74,75 As more β-FeOOH was incorporated, CD peaks redshifted about 150 nm from 6 to 48 hours (Fig. S28†) due to the higher average refractive indices76 and the larger helical pitch.23 The redshift was similarly observed when increasing the film thickness (Fig. S30†), which, however, can be attributed to the varied assembly kinetics.77,78 In line with the optimal helical organization (Fig. S26†), the highest CD intensity was observed for CNC-Fe-36 (Fig. S28†). In diffusion reflectance circular dichroism (DRCD) spectra, in addition to the peaks associated with the photonic bandgap, the characteristic peaks from chiral β-FeOOH NWs were also evident due to the selective absorption of diffused CPL (Fig. S31†), which is consistent with the CD spectra from the solution (Fig. 3b).
To control the optical properties of CNC-based materials, an external magnetic field represents an effective way. CNCs with anisotropic diamagnetism can be regulated directly,79–81 while the addition of magnetic NPs allows more facile structural control.23,73,82 These efforts focused on the interference with the dynamic self-assembly of CNCs by the external magnetic field. Little attention was paid to the modulation of the optical properties of CNC-based photonic films after EISA, which holds great promise for the real-time control of light-matter interactions.83–86 We studied the optical modulation of photonic films through a magnetic field. Different from previous work on the magnetic effect on chiral NPs6,87 or molecules,88,89 here, a helical medium dominated the response. The real-time modulation of CD spectra with a weak magnetic field (50 mT) aligned parallel or anti-parallel to the incident light and the helical axis was performed (Fig. 5a). We observed that intensity increases for CNC films (Fig. 5b) and decreases for CNC/β-FeOOH films (Fig. 5c and S32†). Cellulose exhibits negative diamagnetic anisotropy,81 meaning ø| – ø⊥ < 0, where ø| and ø⊥ represent the diamagnetic susceptibilities in the directions parallel and perpendicular to the fiber axis, respectively (Fig. 5b, inset). The magnetic field enhances selective reflection by aligning the magnetic dipole at different domains with the helical axis, consistent with the emergence of well-defined absorption bands for CNC films (Fig. S33a†). Conversely, the aligned paramagnetic β-FeOOH NWs (Fig. S34†) may alter magnetic anisotropy90 (Fig. 5c, inset). The magnetic field then tends to align the magnetic dipole perpendicular to the incident light, reducing the absorption of CNC-Fe-x films due to misalignment with the helical axis (Fig. S33b–f†).
 | ||
| Fig. 5 (a) Schematic of the experimental set-up of CD spectral measurement under a magnetic field parallel or antiparallel to the direction of incident light and the helical axis of chiral photonic films. The effect of the magnetic field on the CD spectra of (b) CNC and (c) CNC-Fe-36 films. Insets show the magnetic anisotropy of CNCs and β-FeOOH modified CNCs, respectively. | ||
Conclusions
In summary, oriented attachment of β-FeOOH NPs on CNCs led to the formation of highly aligned chiral NWs. Their g-factor increased substantially along the elongation process, and the observed correlation between the g-factor and ARs could be attributed to enhanced dipolar interactions, responsible for the induced structural distortions. The static coupling mechanism based on one-electron theory can be readily extended to comprehend chirality transfer in various hybrid systems. The observed anisotropy-dependent chiromagnetic properties hold great promise for technological advances in field-controlled optical devices.Experimental section
Materials
Iron trichloride hexahydrate and potassium hydroxide were purchased from Sinopharm Chemical Reagent Co., Ltd. 6 wt% CNC solution was purchased from CelluForce. Deionized (DI) water (Millipore, Milli-Q grade) was used in all experiments. All chemical solvents and reagents were used without further purification.Synthesis of CNC-Fe-x dispersions and films
We prepared CNC-Fe-x dispersions (where x = 6, 12, 24, 36, and 48, representing the reaction time) by adding 1 mL of a 2 wt% CNC solution and 250 μL of a 10 mg per mL KOH solution to 50 mL of a 20 mM FeCl3 solution. The mixture was continuously stirred for various durations (6, 12, 24, 36, and 48 hours) at room temperature. The solution was centrifuged three times at 8000 rpm for 10 minutes. After centrifugation, the resulting material was redispersed in 2.5 mL of DI water and placed in Petri dishes with a diameter of 30 mm. Different films were formed through evaporation-induced self-assembly (EISA) at room temperature. The synthesis of β-FeOOH NWs followed a similar process to CNC-Fe-x, without the addition of CNCs.Theoretical calculation
| ΔG = Gab − Ga − Gb |
| G = E + C |
In this equation, Edd represents the pair of permanent dipole–dipole interactions. The variable “r” denotes the distance between the centers of two dipoles, while θ and θ′ represent the angles formed by μ and μ′ with the central line. The variable “ζ” indicates the angle between the planes formed by the axes of dipoles μ and μ′ with the line of centers. In this specific scenario, ζ = 0 since all the lines are in the same plane. The positions of central axes and the values of r, cos(θ), cos(θ′), sin(θ), and sin(θ′) can be determined by assuming that the dipoles are situated in a 3D coordinate system.


and
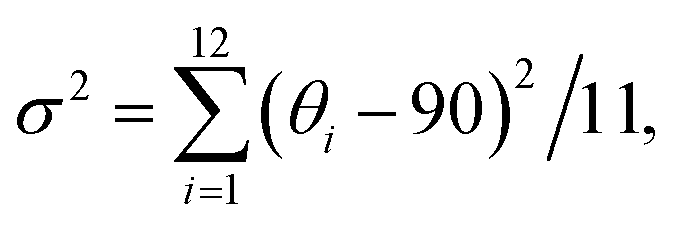
and
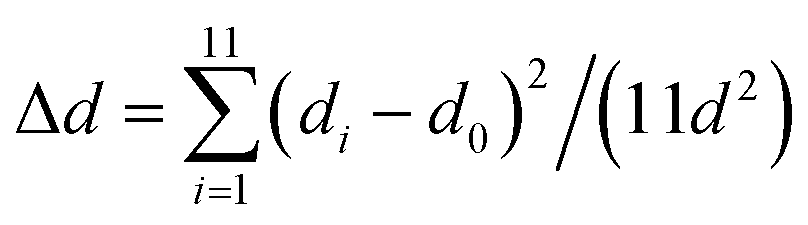
where di is the specific Fe–O bond length, d0 is the average Fe–O bond length and θi is the O–Fe–O bond angle in Fe–O tetrahedrons. 11 is the number of bonds and 14 is the number of angles minus 1 in Fe3O6.
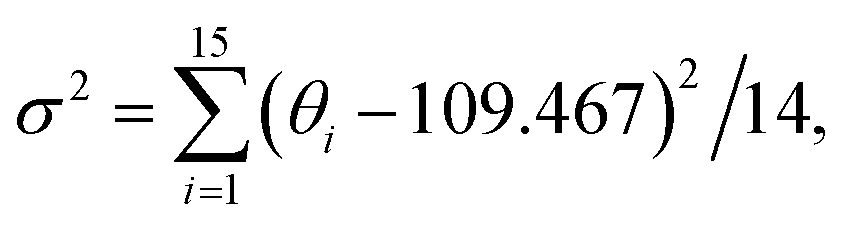
Data availability
The data that support the findings of this study are available from the corresponding author, M. Y., upon reasonable request.Author contributions
J. Tang: data curation, formal analysis, investigation, methodology, and writing – original draft; S. Feng: supervision; M. Yang: conceptualization, funding acquisition, project administration, supervision, validation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Y. thanks the National Natural Science Foundation of China (grant nos. 22071075 and 22090044) and the Research Foundation of the Education Bureau of Jilin Province (grant no. JJKH20231134CY) for financial support.References
- W. Ma, L. Xu, A. F. De Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- C. Li, J. Zhao, X. Gao, C. Hao, S. Hu, A. Qu, M. Sun, H. Kuang, C. Xu and L. Xu, Adv. Mater., 2023, 35, 2308198 CrossRef CAS PubMed.
- J. Lu, Y. Xue, K. Bernardino, N. N. Zhang, W. R. Gomes, N. S. Ramesar, S. Liu, Z. Hu, T. Sun, A. F. de Moura, N. A. Kotov and K. Liu, Science, 2021, 371, 1368–1374 CrossRef CAS PubMed.
- H. E. Lee, H. Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–365 CrossRef CAS PubMed.
- Y. Li, J. Cheng, J. Li, X. Zhu, T. He, R. Chen and Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 10236–10240 CrossRef CAS PubMed.
- J. Yeom, U. S. Santos, M. Chekini, M. Cha, A. F. De Moura and N. A. Kotov, Science, 2018, 359, 309–314 CrossRef CAS PubMed.
- Z. Li, Q. Fan, Z. Ye, C. Wu, Z. Wang and Y. Yin, Science, 2023, 380, 1384–1390 CrossRef CAS PubMed.
- T. Bai, J. Ai, J. Ma, Y. Duan, L. Han, J. Jiang and S. Che, Angew. Chem., Int. Ed., 2021, 60, 20036–20041 CrossRef CAS PubMed.
- J. Cai, J. Zhao, X. Gao, W. Ma, D. Meng, H. Zhang, C. Hao, M. Sun, H. Kuang, C. Xu and L. Xu, ACS Nano, 2022, 16, 11066–11075 CrossRef CAS PubMed.
- T. Bai, J. Ai, Y. Duan, L. Han and S. Che, Small, 2022, 18, 2104509 CrossRef CAS PubMed.
- J. L. Greenfield, J. Wade, J. R. Brandt, X. Shi, T. J. Penfold and M. J. Fuchter, Chem. Sci., 2021, 12, 8589–8602 RSC.
- S. F. C. S. Chan, G. De Stasio, S. A. Welch, M. Girasole, B. H. Frazer, M. V. Nesterova and J. F. Banfield, Science, 2004, 1656, 1656–1658 CrossRef PubMed.
- E. D. Sone, S. Weiner and L. Addadi, J. Struct. Biol., 2007, 158, 428–444 CrossRef CAS PubMed.
- W. Huang, D. Montroni, T. Wang, S. Murata, A. Arakaki, M. Nemoto and D. Kisailus, Acc. Chem. Res., 2022, 55, 1360–1371 CrossRef CAS PubMed.
- A. Querejeta-Fernández, G. Chauve, M. Methot, J. Bouchard and E. Kumacheva, J. Am. Chem. Soc., 2014, 136, 4788–4793 CrossRef PubMed.
- A. Querejeta-Fernández, B. Kopera, K. S. Prado, A. Klinkova, M. Methot, G. Chauve, J. Bouchard, A. S. Helmy and E. Kumacheva, ACS Nano, 2015, 9, 10377–10385 CrossRef PubMed.
- X. Gao, B. Han, X. Yang and Z. Tang, J. Am. Chem. Soc., 2019, 141, 13700–13707 CrossRef CAS PubMed.
- W. J. Kauzmann, J. E. Walter and H. Eyring, Chem. Rev., 1940, 26, 339–407 CrossRef.
- G. González-Rubio, J. Mosquera, V. Kumar, A. Pedrazo-Tardajos, P. Llombart, D. M. Solís, I. Lobato, E. G. Noya, A. Guerrero-Martínez, J. M. Taboada, F. Obelleiro, L. G. MacDowell, S. Bals and L. M. Liz-Marzán, Science, 2020, 368, 1472–1477 CrossRef PubMed.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J. L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9, 57–65 CrossRef CAS PubMed.
- M. Giese, L. K. Blusch, M. K. Khan and M. J. MacLachlan, Angew. Chem., Int. Ed., 2015, 54, 2888–2910 CrossRef CAS PubMed.
- Z. Cheng, Y. Ma, L. Yang, F. Cheng, Z. Huang, A. Natan, H. Li, Y. Chen, D. Cao, Z. Huang, Y. H. Wang, Y. Liu, R. Yang and H. Zhu, Adv. Opt. Mater., 2019, 7, 1801816 CrossRef.
- T. Chen, Q. Zhao, X. Meng, Y. Li, H. Peng, A. K. Whittaker and S. Zhu, ACS Nano, 2020, 14, 9440–9448 CrossRef CAS PubMed.
- S. Kang, G. M. Biesold, H. Lee, D. Bukharina, Z. Lin and V. V. Tsukruk, Adv. Funct. Mater., 2021, 31, 2104596 CrossRef CAS.
- S. Zhao, H. Zhu, J. Lu, M. Li, L. Zhao, L. Zhou and L. Gao, Adv. Funct. Mater., 2022, 32, 2201927 CrossRef CAS.
- C. Wang, S. R. Mouchet, O. Deparis, J. Li, E. Paineau, D. Dragoe, H. Remita and M. N. Ghazzal, Small, 2024, 20, 2402211 CrossRef CAS PubMed.
- W. Zhang, X. Cheng, S. H. Chen and M. Anthamatten, Langmuir, 2023, 39, 9180–9185 CrossRef CAS PubMed.
- C. Wang, J. Li, E. Paineau, A. Laachachi, C. Colbeau-Justin, H. Remita and M. N. Ghazzal, J. Mater. Chem. A, 2020, 8, 10779–10786 RSC.
- T. D. Nguyen, E. Lizundia, M. Niederberger, W. Y. Hamad and M. J. Maclachlan, Chem. Mater., 2019, 31, 2174–2181 CrossRef CAS.
- J. Majoinen, J. Hassinen, J. S. Haataja, H. T. Rekola, E. Kontturi, M. A. Kostiainen, R. H. A. Ras, P. Törmä and O. Ikkala, Adv. Mater., 2016, 28, 5262–5267 CrossRef CAS PubMed.
- A. Chakraborty, Nonappa, B. Mondal, K. Chaudhari, H. Rekola, V. Hynninen, M. A. Kostiainen, R. H. A. Ras and T. Pradeep, J. Phys. Chem. C, 2021, 125, 3256–3267 CrossRef CAS.
- H. Yu, H. Huang, J. Liang and J. Deng, Nanoscale, 2018, 10, 12163–12168 RSC.
- K. Ståhl, K. Nielsen, J. Jiang, B. Lebech, J. C. Hanson, P. Norby and J. van Lanschot, Corros. Sci., 2003, 45, 2563–2575 CrossRef.
- C. Luna, M. Ilyn, V. Vega, V. M. Prida, J. González and R. Mendoza-Reséndez, J. Phys. Chem. C, 2014, 118, 21128–21139 CrossRef CAS.
- L. B. Mao, H. L. Gao, H. Bin Yao, L. Liu, H. Cölfen, G. Liu, S. M. Chen, S. K. Li, Y. X. Yan, Y. Y. Liu and S. H. Yu, Science, 2016, 354, 107–110 CrossRef CAS PubMed.
- S. Llàcer Navarro, K. Nakayama, A. Idström, L. Evenäs, A. Ström and T. Nypelö, Cellulose, 2021, 28, 9633–9644 CrossRef.
- S. Mann, C. C. Perry and J. Webb, Proc. R. Soc. London, Ser. B, 1986, 227, 179–190 CAS.
- R. M. H. Rumney, S. C. Robson, A. P. Kao, E. Barbu, L. Bozycki, J. R. Smith, S. M. Cragg, F. Couceiro, R. Parwani, G. Tozzi, M. Stuer, A. H. Barber, A. T. Ford and D. C. Górecki, Nat. Commun., 2022, 13, 3753 CrossRef CAS PubMed.
- S. Li, C. Huang, J. Hao and C. Wang, J. Comput. Chem., 2014, 35, 415–426 CrossRef CAS PubMed.
- G. Zhu, M. L. Sushko, J. S. Loring, B. A. Legg, M. Song, J. A. Soltis, X. Huang, K. M. Rosso and J. J. De Yoreo, Nature, 2021, 590, 416–422 CrossRef CAS PubMed.
- J. F. Banfield, S. A. Welch, H. Zhang, T. T. Ebert and R. L. Penn, Science, 2000, 289, 751–754 CrossRef CAS PubMed.
- B. C. Park, M. J. Ko, Y. K. Kim, G. W. Kim, M. S. Kim, T. M. Koo, H. E. Fu and Y. K. Kim, Nat. Commun., 2022, 13, 1144 CrossRef CAS PubMed.
- S. Dong and M. Roman, J. Am. Chem. Soc., 2007, 129, 13810–13811 CrossRef CAS PubMed.
- U. A. Agú, M. I. Oliva, S. G. Marchetti, A. C. Heredia, S. G. Casuscelli and M. E. Crivello, J. Magn. Magn. Mater., 2014, 369, 249–259 CrossRef.
- D. M. Sherman and T. D. Waite, Am. Mineral., 1985, 70, 1262–1269 CAS.
- H. Yao and Y. Ishikawa, J. Phys. Chem. C, 2015, 119, 13224–13230 CrossRef CAS.
- B. Zhu, F. Wang, C. Wang, Y. Cao, L. Guo, J. Zhang and Y. Gu, J. Phys.:Condens. Matter, 2016, 28, 285801 CrossRef PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- D. Wang, S. Feng and M. Yang, ACS Nano, 2024, 18, 29636–29647 CrossRef CAS PubMed.
- A. Kumar and S. K. Gupta, J. Nanopart. Res., 2013, 15, 1466 CrossRef.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- J. Hu, S. Li, J. Chu, S. Niu, J. Wang, Y. Du, Z. Li, X. Han and P. Xu, ACS Catal., 2019, 9, 10705–10711 CrossRef CAS.
- J. Zhao, B. Wu, X. Huang, Y. Sun, Z. Zhao and M. Ye, Adv. Sci., 2022, 9, 2201678 CrossRef CAS PubMed.
- X. Chen, Y. Zeng, Z. Chen, S. Wang and C. Xin, Front. Chem., 2020, 8, 328 CrossRef CAS PubMed.
- H. Qi, L. Cao, J. Li, J. Huang, Z. Xu, Y. Cheng, X. Kong and K. Yanagisawa, ACS Appl. Mater. Interfaces, 2016, 8, 35253–35263 CrossRef CAS PubMed.
- J. Liu, M. Zheng, X. Shi, H. Zeng and H. Xia, Adv. Funct. Mater., 2016, 26, 919–930 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- X. Gao, X. Zhang, K. Deng, B. Han, L. Zhao, M. Wu, L. Shi, J. Lv and Z. Tang, J. Am. Chem. Soc., 2017, 139, 8734–8739 CrossRef CAS PubMed.
- H. Chen, J. Zhou and J. Deng, Polym. Chem., 2016, 7, 125–134 RSC.
- A. K. Visheratina, F. Purcell-Milton, R. Serrano-García, V. A. Kuznetsova, A. O. Orlova, A. V. Fedorov, A. V. Baranov and Y. K. Gun’ko, J. Mater. Chem. C, 2017, 5, 1692–1698 RSC.
- T. T. Zhuang, Y. Li, X. Gao, M. Wei, F. P. García de Arquer, P. Todorović, J. Tian, G. Li, C. Zhang, X. Li, L. Dong, Y. Song, Y. Lu, X. Yang, L. Zhang, F. Fan, S. O. Kelley, S. H. Yu, Z. Tang and E. H. Sargent, Nat. Nanotechnol., 2020, 15, 192–197 CrossRef CAS PubMed.
- J. S. Kamal, R. Gomes, Z. Hens, M. Karvar, K. Neyts, S. Compernolle and F. Vanhaecke, Phys. Rev. B:Condens. Matter Mater. Phys., 2012, 85, 035126 CrossRef.
- J. Hu, L. S. Li, W. Yang, L. Manna, L. W. Wang and A. P. Alivisatos, Science, 2001, 292, 2060–2063 CrossRef CAS PubMed.
- E. U. Condon, W. Altar and H. Eyring, J. Chem. Phys., 1937, 5, 339–407 CrossRef.
- S. Ma, Y. K. Jung, J. Ahn, J. Kyhm, J. Tan, H. Lee, G. Jang, C. U. Lee, A. Walsh and J. Moon, Nat. Commun., 2022, 13, 3259 CrossRef CAS PubMed.
- H. Lu, C. Xiao, R. Song, T. Li, A. E. Maughan, A. Levin, R. Brunecky, J. J. Berry, D. B. Mitzi, V. Blum and M. C. Beard, J. Am. Chem. Soc., 2020, 142, 13030–13040 CrossRef CAS PubMed.
- M. Kepenekian, R. Robles, C. Katan, D. Sapori, L. Pedesseau and J. Even, ACS Nano, 2015, 9, 11557–11567 CrossRef CAS PubMed.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969–971 CrossRef CAS PubMed.
- M. H. Tsai, S. Y. Chen and P. Shen, Nano Lett., 2004, 4, 1197–1201 CrossRef CAS.
- D. Qu, H. Zheng, H. Jiang, Y. Xu and Z. Tang, Adv. Opt. Mater., 2019, 7, 1801395 CrossRef.
- S. N. Fernandes, P. L. Almeida, N. Monge, L. E. Aguirre, D. Reis, C. L. P. de Oliveira, A. M. F. Neto, P. Pieranski and M. H. Godinho, Adv. Mater., 2017, 29, 1603560 CrossRef PubMed.
- P. Li, L. Li, K. J. Jeong, X. Yu, X. Yu and Y. Xu, Adv. Opt. Mater., 2022, 10, 2102616 CrossRef CAS.
- X. Wang, Q. Wang, X. Zhang, J. Miao, J. Cheng, T. He, Y. Li, Z. Tang and R. Chen, Adv. Opt. Mater., 2022, 10, 2200761 CrossRef CAS.
- A. Salij, R. H. Goldsmith and R. Tempelaar, J. Am. Chem. Soc., 2021, 143, 21519–21531 CrossRef CAS PubMed.
- H. Maeda and Y. Maeda, Langmuir, 2011, 27, 2895–2903 CrossRef CAS PubMed.
- D. Lu, X. Yu, S. Zhu and Y. Xu, Cellulose, 2023, 30, 8325–8338 CrossRef CAS.
- S. Beck, J. Bouchard, G. Chauve and R. Berry, Cellulose, 2013, 20, 1401–1411 CrossRef CAS.
- B. Frka-Petesic, G. Guidetti, G. Kamita and S. Vignolini, Adv. Mater., 2017, 29, 1701469 CrossRef PubMed.
- T. Nypelö, J. Mater. Chem. C, 2022, 10, 805–818 RSC.
- F. Kimura, T. Kimura, M. Tamura, A. Hirai, M. Ikuno and F. Horii, Langmuir, 2005, 21, 2034–2037 CrossRef CAS PubMed.
- X. Zhang, S. Kang, K. Adstedt, M. Kim, R. Xiong, J. Yu, X. Chen, X. Zhao, C. Ye and V. V. Tsukruk, Nat. Commun., 2022, 13, 5804 CrossRef CAS PubMed.
- X. Liu, Y. Du, S. Mourdikoudis, G. Zheng and K. Y. Wong, Adv. Opt. Mater., 2023, 11, 2202859 CrossRef CAS.
- K. J. Jeong, D. K. Lee, V. T. Tran, C. Wang, J. Lv, J. Park, Z. Tang and J. Lee, ACS Nano, 2020, 14, 7152–7160 CrossRef CAS PubMed.
- B. Han, X. Gao, J. Lv and Z. Tang, Adv. Mater., 2020, 32, 1801491 CrossRef CAS PubMed.
- T. Ji, S. Che and Y. Duan, Acc. Mater. Res., 2023, 4, 1057–1067 CrossRef CAS.
- X. Wu, C. Hao, L. Xu, H. Kuang and C. Xu, Small, 2020, 16, 1905734 CrossRef CAS PubMed.
- Y. Kitagawa, H. Segawa and K. Ishii, Angew. Chem., Int. Ed., 2011, 50, 9133–9136 CrossRef CAS PubMed.
- G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390, 493–494 CrossRef CAS.
- B. J. Lemaire, P. Davidson, J. Ferré, J. P. Jamet, P. Panine, I. Dozov and J. P. Jolivet, Phys. Rev. Lett., 2002, 88, 125507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07747d |
| This journal is © The Royal Society of Chemistry 2025 |