
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemodivergent dearomatization of benzene-linked O-oxime esters via EnT-induced radical cross-coupling†‡
Guohui
Zeng
 ,
Dongwen
Guo
,
Huanfeng
Jiang
and
Biaolin
Yin
*
,
Dongwen
Guo
,
Huanfeng
Jiang
and
Biaolin
Yin
*
Key Laboratory of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology (SCUT), Guangzhou 510640, China
First published on 9th January 2025
Abstract
Radical-mediated dearomatization strategies offer a blueprint for building value-added and synthetically valuable three-dimensional skeletons from readily available aromatic starting materials. Herein, we report a novel strategy by leveraging benzene-linked O-oxime esters as triply functionalized precursors to form two distinct persistent radicals under a chemodivergent pathway. These radicals then couple with a cyclohexadienyl radical for either carboamination or carbo-aminoalkylation. Remarkably, a series of 4-(2-aminoethyl)anilines derivatives featuring all-carbon quaternary centers, along with the formation of four different types of chemical bonds, are efficiently constructed through a unique rearomatization cascade in the carboamination. Importantly, employing DMPU as the hydrogen atom transfer (HAT) donor strategically diverts the reaction pathway from the C–N bond formation towards the C–C bond formation. Our mechanistic explorations support a sequential HAT/energy transfer (EnT)/HAT cascade as the key stage for carbo-aminoalkylation involving the N-center iminyl radical. Significantly, this work demonstrates the elegant expansion of divergent C–N and C–C bond formation using the imine moiety within O-oxime esters as the bifunctional reagent, and it broadens the chemical space of both benzenes and O-oxime esters in radical-mediated transformations.
Introduction
Phenyl rings, as readily available building blocks, undergo dearomative functionalization reactions to yield a variety of valuable skeletons that have garnered significant attention in organic synthesis.1–3 The chemodivergent dual-functionalization dearomatization strategy not only introduces two additional functional groups into the cyclic system but also substantially enhances the structural diversity of three-dimensional molecules.4 By leveraging an aromaticity disassembly-reconstruction process,5 peripheral editing of the benzene ring can be further achieved.6Compared to the traditional two-electron pathway for dearomatization of phenyl rings,7–9 radical-mediated dearomatizations are of interest as they tend to use mild conditions that overcome the unfavorable thermodynamics, especially for non-activated phenyl rings.10–12 The transience of the cyclohexadienyl radical (CHDR) intermediate during the dearomatization event, along with its high reactivity, are key factors for subsequent transformations. The radical-polar crossover (RPC)13–15 mechanism is well established for intercepting these radical species, leading to the irreversible generation of the corresponding ionic intermediates (Scheme 1A, upper left). Despite significant advances and their great potential, RPC strategies still have limited applications for diverse post-functionalization. For example, reductive RPC produces carbanion species, which primarily undergo protonation accompanied by few functionalizations,16–24 while oxidative RPC usually results in dienones via cation intermediates.25–31 Recently, a combination of CHDR and palladium catalyst to form the cyclohexadienyl Pd(II) species32 may be a potential direction for expanding CHDR chemistry (Scheme 1A, lower left). Nevertheless, it is still highly desirable to continue to seek efficient transformations using CHDRs.
 | ||
| Scheme 1 (A) Representative reaction modes of the cyclohexadienyl radical; (B) designed strategy and this work: divergent difunctionalization through dearomative radical-cross coupling of phenyl rings based on persistent radical effect; (C) selected examples including target skeletons in value-added molecules. | ||
The radical cross-coupling (RCC) strategy has emerged as a powerful tool for difunctionalization of alkenes and other unsaturated systems.33,34 We envisioned that this methodology can be implemented during the dearomative process of a non-activated phenyl ring (masked cyclic triene), namely, the CHDR is terminated by another radical, to greatly expand the dearomatization chemical library (Scheme 1A, right). It is intriguing to mention that some intramolecular radical cycloaddition strategies have already been established.35–39 However, several challenges should be highlighted, especially in intermolecular processes, such as chemo-/regioselectivity, reactivity and compatibility of the radical species, as well as undesirable side reactions.40–42 Encouragingly, this pathway might be facilitated by leveraging the persistent radical effect (PRE), which relies on a kinetic mechanism to govern the RCC process. In this process two different radicals with different lifetimes are simultaneously generated.43–45 Therefore, potential radical partners should possess persistence, stability, and a low rate of homocoupling.
With the above concept in mind, the persistent diphenyliminyl radical was considered an excellent choice as it not only efficiently undergoes heterocoupling, but it is also easily obtained from the homolytic cleavage of oxime esters via visible-light mediated triplet–triplet EnT.46–58 On the other hand, it was hypothesized that another persistent C-centered α-amino radical could be generated in situ from the diphenyliminyl radical through HAT/EnT/second HAT cascade under tunable conditions within the dearomative event.59 Herein, as part of our ongoing research into the dearomative difunctionalization of arenes,32,38,60 we report a chemodivergent RCC strategy that utilizes the PRE to recombine the cyclohexadienyl radical with the diphenyliminyl N-radical which ultimately leads to the dearomative carboamination, or to recombine the cyclohexadienyl radical with the α-amino C-radical generated from the same substrate, which ultimately leads to carbo-aminoalkylation of non-activated phenyl rings (Scheme 1B). All these core skeletons are versatile scaffolds commonly employed in pharmaceutical molecules (Scheme 1C).
Results and discussion
N-benzoyl propanoic O-oxime ester 1-1 was used as the model substrate to investigate this chemodivergent reaction. 1-1 can generate a persistent iminyl radical and a transient CHDR intermediate through an intramolecular dearomative event by the EnT catalysts cascade. The desired RCC products were successfully obtained under the screening conditions outlined in Table 1. Initially the reaction was carried out in presence of 9H-thioxanthen-9-one (TXT) with MeCN as the solvent at room temperature providing only a 34% yield of 2-1 (entry 1). Through careful analysis of the reaction products (see ESI‡ for details), we believed that additional supplementation of the iminyl radical precursor would improve the C–N bond formation process. Subsequently, dioxime oxalate A-1 was chosen as the additional iminyl radical source to further screen solvents. As expected, a significantly increased 2-1 yield was observed (60%) when MeCN was the solvent (entry 4). Additionally, the mixed solvent (MeCN/EA = 2/1, v/v) had a slightly positive effect (entry 5). THF or DMF61 as potential HAT precursors are commonly employed in the formation of chemical bonds.62 These solvents were initially evaluated in the presence of A-1 to generate the desired product 3-1. Unfortunately, these experiments failed to produce 3-1, with the reaction favoring the formation of product 2-1 in low yields (entries 7 and 8). Notably, when the reaction was conducted in the presence of γ-terpinene (A-2)63 in EA (entry 9), the desired product 3-1 was afforded in a 25% yield, along with approximately a 15% yield of the CHDR homodimer. Gratifyingly, DMPU64 exhibited a significant enhancing effect (entry 10), especially in combination with EA as the mixed solvent, affording product 3-1 in 55% yield (entry 11). Benzophenone imine (A-3), a simpler additive, increased the yield of product 3-1 to 60% (entry 12). Furthermore, increasing the loading of A-3 from 1.0 to 2.0 equiv. resulted in a significant reaction efficiency improvement (entry 13) giving the target product 3-1 in 69% yield. Control experiments confirmed that both photoirradiation and the TXT catalyst were necessary for all the desired reactions (entries 6 and 14). Nevertheless, we recognized that the low cis/trans-selectivity for both transformations might arise from lower steric control during the radical–radical recombination events. Additionally, the increased proportion of cis-selectivity in carbo-aminoalkylation may be ascribed to the presence of intramolecular hydrogen bonding.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditions | Yieldb (%) | ||||
| Solvent | Additives | Variation | 2-1 | cis/trans | 3-1 | |
| a Standard conditions: the reactions were carried out under argon using 1-1 (0.1 mmol), Additive (1.0 equiv.), PC (5 mol%), Solvents for 2-1 (0.033 M), Solvents for 3-1 (0.05 M) at room temperature for 10 h. b The yield of 2-1 and 3-1 were based on 1H NMR analysis of the crude product using CH2Br2 as the internal standard. c Additive (2.0 equiv.). EA = ethyl acetate, DMPU = 1,3-dimethyltetrahydropyrimidin-2(1H)-one. | ||||||
| 1 | MeCN | — | PC-I | 34 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.0 :3.0 |
0 |
| 2 | DCE | A-1 | PC-I | 35 | 1:2.6 |
0 |
| 3 | EA | A-1 | PC-I | 39 | 1:3.6 |
0 |
| 4 | MeCN | A-1 | PC-I | 60 | 1:2.5 |
0 |
| 5 | MeCN/EA (2/1, v/v) | A-1 | PC-I | 62 | 1:2.4 |
0 |
| 6 | MeCN/EA (2/1, v/v) | A-1 | Other PC/no hv or PC | 0 | 0 | |
| 7 | THF | A-1 | PC-I | 33 | 1:2.9 |
0 |
| 8 | DMF | A-1 | PC-I | 24 | 1:2.7 |
0 |
| 9 | EA | A-2 | PC-I | 0 | 2.0:1 |
25 |
| 10 | DMPU | A-1 | PC-I | 0 | 1.7:1 |
37 |
| 11 | DMPU/EA (1/1, v/v) | A-1 | PC-I | 0 | 1.8:1 |
55 |
| 12 | DMPU/EA (1/1, v/v) | A-3 | PC-I | 0 | 1.8:1 |
60 |
| 13c | DMPU/EA (1/1, v/v) | A-3 | PC-I | 0 | 1.9:1 |
69 |
| 14 | DMPU/EA (1/1, v/v) | A-3 | Other PC/no hv or PC | 0 | 0 | |
|
||||||

With the optimal conditions in hand, the scope of the carboamination for non-activated phenyl substrates was investigated. As summarized in Scheme 2, various O-oxime esters derived from benzamidopropanoic acids afford the corresponding spiro-cyclohexadienyl imines in moderate to good yields. X-ray crystallographic analysis revealed the major product to be 2-1 (trans), with the amide carbonyl and imine moieties positioned opposite each other.65 The minor product, 2-1 (cis) was also identified. Compared to the unsubstituted phenyl product 2-1 (61% yield), the heterocoupling process showed a slight improvement in yields when substituents were present at the ortho- or meta-positions of the starting materials. Products 2-2 and 2-3 were obtained in 69% and 67% yield. Notably, it is easy to obtain cyclohexadienyl amine from 2 in the presence of weak acid conditions (see ESI‡ for detail). On the other hand, the electron-deficient groups such as F, Cl, Br on the 2- or 3-position of benzene ring were also compatible, giving corresponding products in 53% to 65% yields (2-4 to 2-8). The N-benzyl-derived malonate O-oxime ester also provided the dearomative product 2-9, albeit with an apparent decrease in yield, possibly due to the C–N bond is susceptible to hydrolysis, whereas steric bulk on the cyclohexadienyl amine can stabilize the C–N bond. This transformation also proceeded well when the reaction was performed on a starting material containing a thiophene ring, affording product 2-10 in a 55% yield. When N-propyl was used, the reaction yielded product 2-11 in 54% yield. It should be noted that the Thorpe–Ingold effect enhanced the dearomative event while there was minimal steric hindrance from the N-protected group (2-11). Notably, dearomative carboamination is completely inhibited by a substituent at the para-position of the phenyl ring or the 5-position of the thiophene ring (see ESI‡ for details).
 | ||
| Scheme 2 Substrate scopes for carboamination. Reaction conditions: O-oxime ester 1 (0.2 mmol), A-1 (0.2 mmol, 1.0 equiv.), TXT (5 mol%) in 6 mL of MeCN/EA (0.033 M, v/v = 2/1) under Ar atmosphere at room temperature and irradiated with visible LEDs (2 × 15 W, 405 nm) for 10 h. Isolated yields are reported as the average of four experiments. | ||
To make the method more useful and develop the product for new and diverse applications, we envisioned that the amine-substituted benzene ring can be achieved through a rearomative strategy,66 yielding 4-(2-aminoethyl)aniline derivatives. These derivatives are crucial building blocks for pharmaceuticals and liquid crystal materials. Traditionally, the pathway for introducing an amino group on the phenyl ring involves challenging nitration and reduction steps. Upon treatment of 4-bromobenzoyl-linked O-oxime ester under the standard carboamination conditions, no formation of rearomative product was observed. Interestingly, when this process was switched to an intermolecular approach, namely using 4 and 5 as starting materials, the desired product 6 was successfully obtained under modified conditions (Scheme 3). To probe this transformation's functional group tolerance, this reaction was carried with 4-1 (X = F) as the substrate. The desired product 6-1 was obtained in 64% yield, and its configuration confirmed through X-ray crystallographic analysis.67 It was found that the steric hindrance from the β-position of allyl (4, R1 = Me) is compatible, giving all-carbon quaternary center68,69 products in moderate to good yields (6-2, 41% to 60% yields). The effect of different halogens (X = F, Cl, Br) was also investigated. Interestingly, a decrease in the yield was observed with increasing atomic radius of the halogen. Next, several substrates 4 with different substitutions on the phenyl ring were examined. They produced the corresponding all-carbon quaternary center products in good yields (6-3 to 6-7). Significantly, rearomative cyclization product 6-6 was achieved in 62% yield, this outcome strongly indicates that a Friedel–Crafts cyclization occurred between the electron-rich phenyl ring and the amide carbocation during the cascade process. Finally, a range of O-picolinoyl oxime substrates 5 bearing different substitutions on the pyridyl ring were investigated, and the corresponding all-carbon quaternary products (6-7 to 6-10) were obtained. Note that the yields of products decreased when there was steric hindrance at the C3 position of the pyridyl ring (30% yields for both 6-8 and 6-9).
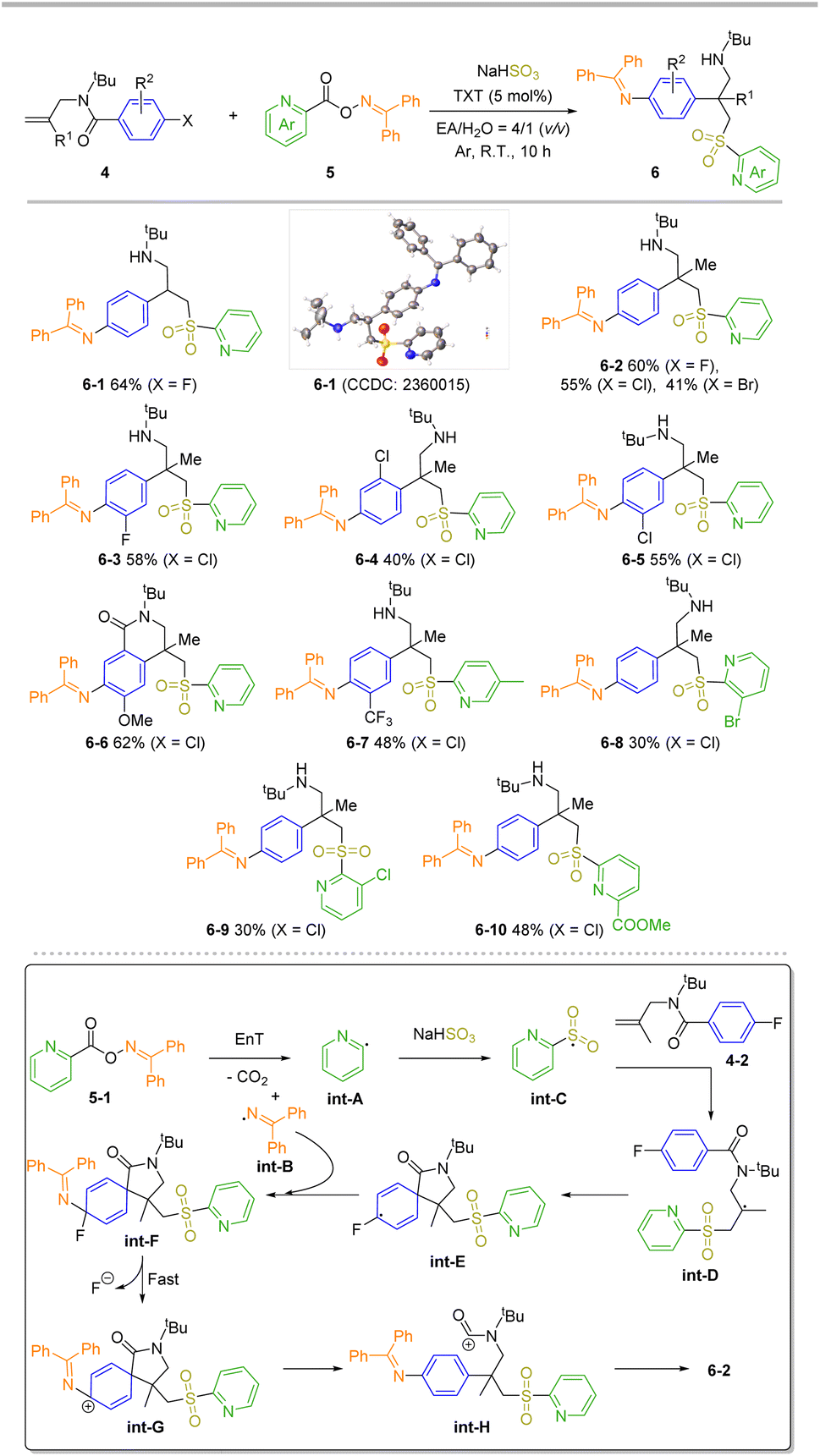 | ||
| Scheme 3 Substrate scopes for transient dearomatize-tion and plausible mechanism. Reaction conditions: N-allyl-N-(tert-butyl)benzamide 4 (0.2 mmol), O-oxime ester 5 (0.34 mmol, 1.7 equiv), NaHSO3 (0.6 mmol, 3.0 equiv.), TXT (5 mol%) in 5 mL of EA/H2O (v/v = 4/1) under Ar atmosphere at room temperature and irradiated with visible LEDs (2 × 15 W, 405 nm) for 10 h. Isolated yields are reported as the average of two experiments. | ||
Based on the experiment outcomes, and our recently reported results,66 a plausible reaction mechanism for this transient dearomatization process is proposed in Scheme 3 (bottom). N–O bond cleavage occurs in the triplet state of 5-1 (generated by EnT process with excited TXT), followed by the release of CO2 to yield int-A and int-B. int-A quickly reacts with SO2 to form a sulfonyl radical int-C, which then affords the int-D through radical addition with alkene 4-2.70,71 The key intermediate, int-F forms via a radical-induced dearomatization/RCC process cascade from int-D. Then, a rapid dehalogenation process takes place in situ to yield int-G, which provides the 4-(2-aminoethyl)-aniline derivatives 6-2 through the rearomative cation int-H.72 When a more electron-rich phenyl ring is involved, a Friedel–Crafts cyclization event will take place.
Next, an investigation into the substrate's compatibility for the carbo-aminoalkylation reaction was conducted and the results summarized in Scheme 4. The standard product 3-1 was obtained in 68% yield (cis/trans = 1.7/1). X-ray crystallographic analysis confirmed a cis configuration for the major diastereomer which exhibits low polarity.73 A large variety of substituents on the phenyl ring gave the corresponding spiro carbo-aminoalkylation products (3-2 to 3-9) in moderate to good yields. Notably, different electronic properties such as methoxy (3-2), 2,3-dimethyl (3-3), trifluoromethoxy (3-4), isobutenyl (3-5) and halides (3-6 to 3-9) were well tolerated. In addition, a malonic acid derived O-oxime reagent afforded product 3-10 in 60% yield, showing the formal transposition of the carbonyl group relative to the standard product. Gratifyingly, as mentioned earlier, when employing a thiophene ring substrate, product 3-11 was obtained in excellent yield (up to 89%). ortho-Aryl biphenyl O-oxime ester substrates bearing a broad range of substituents on the aryl position, including methyl (3-13 and 3-14), trifluoromethoxy (3-15), and halides (3-16 and 3-17), underwent the reaction to provide the desired products in moderate yields (48% to 56%). Additionally, this reaction tolerated other aryl substituents at the ortho-position, such as 3-thiophenyl (3-18) and 2-naphthalenyl (3-19). However, the yield was significantly affected by the steric hindrance of the aryl moiety, with bulkier substituents leading to lower yields. Notably, the reaction exhibited a remarkable preference for the trans configuration due to the steric hindrance imposed by the ortho-aryl group. Furthermore, the electronic effect of the symmetrical benzene rings on the diphenyl imine radical was studied. Increasing product yield was observed as the substituents on the diphenyl ring evolved from electron-donating methoxy (3-20, 38% yield) and methyl (3-21, 41% yield) groups to electron-withdrawing fluoro (3-22, 55% yield), chloro (3-23, 69% yield), and bromo (3-24, 71% yield) groups. This observation underscores the profound impact of electronic effects on the outcomes of the reaction. Finally, the reaction successfully incorporated a non-symmetric diphenyl imine radical, affording product 3-25 in a moderate 41% yield.
 | ||
| Scheme 4 Substrate scope for carbo-aminoalkylation. Reaction conditions: a O-oxime ester 1 (0.2 mmol), A-3 (0.4 mmol, 2.0 equiv.), TXT (5 mol%) in 4 mL of DMPU/EA (0.05 M, v/v = 1/1) under Ar at room temperature and irradiated with visible LEDs (2 × 15 W, 405 nm) for 10 h. Isolated yields are reported as the average of four experiments. bMajor products were visualized. cThe corresponding imine additive was used instead of A-3. | ||
Inspired by the well-studied tetraphenylethylene (TPE) AIE materials, we realized that the conjugated triphenylcyclo-hexadiene product 7 (Scheme 5), likely formed via NH3 elimination from 3, is expected to exhibit a similar AIE effect. To investigate this hypothesis further, a new molecule, 7-1, was synthesized. It's fluorescence image (λirr = 365 nm) at different compositions is shown in Scheme 5 (bottom) and supports the AIE property.
 | ||
| Scheme 5 The synthesis of a novel AIEgens structure and fluorescence experiment. Reaction conditions: 3-13 (0.3 mmol), in 3 mL of HOAc/Ac2O (v/v = 1/2) under Ar atmosphere at 110 °C for 20 h. Isolated yield is reported. | ||
To gain insights into the reaction mechanism, a series of control experiments were conducted under specific conditions, and the corresponding results are provided in Scheme 6. Careful analysis of the initial reaction mixture by HRMS and NMR shows the desired product 2-1 was present along with dimer byproducts Dimer-1 and Dimer-2 (Scheme 6A). This observation suggests the involvement of the cyclohexadienyl and the iminyl radical as reaction intermediates. Switching from benzamide O-oxime ester to picolinamide O-oxime ester did not yield the desired product, however, the reaction surprisingly afforded a key intermediate DMPU-1 in 15% yield (Scheme 6B). This result strongly implies a HAT process under the standard conditions, potentially with DMPU acting as the hydrogen atom donor or a mediator in the process. To validate our hypothesis, a cross-validation experiment, reacting 1-20 with A-3 under standard conditions, was performed. NMR analysis of the inseparable reaction products revealed 3-1 as the major component and 3-20 as a minor component (Scheme 6C). This finding illustrates that the α-amino carbon-centered radical arises from the transformation of A-3via a triplet EnT/HAT cascade process. UV-vis characterization of 1-1 and PC-1 revealed that 1-1 cannot be directly excited by visible-light. Furthermore, the Stern–Volmer experiments elucidated that an EnT process between 1-1 and excited-state PC-1 (Scheme 6D).
 | ||
| Scheme 6 Control experiments. | ||
Based on the above experimental results, a proposed photocatalytic pathway for the chemodivergent radical cross-coupling is outlined in Scheme 7. Initially, O-oxime ester 1-1 undergoes excitation via an EnT process, followed by N–O bond homolysis of triplet state 1-1*. This event, coupled with releasing CO2 and an intramolecular dearomatization process, leads to the formation of the N-centered radical int-1 and the CHDR int-2. These two radicals readily couple together, leading to the formation of product 2-1 (route A). However, the presence of a suitable HAT donor, such as DMPU, disrupts this established coupling process, leading to the formation of imine int-3via an alternative pathway. Subsequently, imine int-3 can be directly excited by excited TXT to generate its triplet state (int-4),74 followed by a second HAT process with DMPU, yielding int-5. Ultimately, the radical–radical cross-coupling between int-2 and int-5 affords product 3-1 (route B), forming a new C(sp3)–C(sp3) bond instead of the above C(sp3)–N(sp2) bond.
 | ||
| Scheme 7 Plausible mechanism. | ||
Conclusions
In summary, an energy transfer catalysis strategy was developed to enable chemodivergent dearomatization of benzene rings. This strategy harnesses the PRE to accomplish both carboamination and carbo-aminoalkylation through RCC. Employing this protocol, four structurally distinct molecular frameworks were synthesized via divergent pathways. We believe this discovery represents a groundbreaking advancement in C–C bond formation by utilizing O-oxime esters. It marks a significant departure from the traditional paradigm of C–N bond formation and holds immense potential for expanding the chemical library.Data availability
Supporting data or code have been included in the article's ESI‡.Author contributions
This work was conceptualized by G. Z. and prof. B. Y. experimentation was performed by G. Z. and D. G. The first draft of the manuscript was prepared by G. Z. and the final version was edited and revised by prof. B. Y. and prof. H. J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Guangdong Province (no. 2023A1515010771). We also thank Prof. Honggen Wang (Sun Yat-Sen University) and Prof. Pengju Feng (Jinan University) for helpful discussions.Notes and references
- S. P. Roche and J. A. Porco, Dearomatization Strategies in the Synthesis of Complex Natural Products, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed.
- W. C. Wertjes, E. H. Southgate and D. Sarlah, Recent advances in chemical dearomatization of nonactivated arenes, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC.
- C. J. Huck and D. Sarlah, Shaping Molecular Landscapes: Recent Advances, Opportunities, and Challenges in Dearomatization, Chem, 2020, 6, 1589–1603 CAS.
- I. P. Beletskaya, C. Nájera and M. Yus, Chemodivergent reactions, Chem. Soc. Rev., 2020, 49, 7101–7166 RSC.
- L. Li, M. Yang, Q. He and R. Fan, Conversion of anilines to chiral benzylic amines via formal one-carbon insertion into aromatic C–N bonds, Nat. Commun., 2020, 11, 4805–4813 CrossRef CAS PubMed.
- Z. Fan, X. Chen, K. Tanaka, H. S. Park, N. Y. S. Lam, J. J. Wong, K. N. Houk and J.-Q. Yu, Molecular editing of aza-arene C–H bonds by distance, geometry and chirality, Nature, 2022, 610, 87–93 CrossRef CAS PubMed.
- C. Zheng and S.-L. You, Advances in Catalytic Asymmetric Dearomatization, ACS Cent. Sci., 2021, 7, 432–444 CrossRef CAS PubMed.
- L. M. Comparini and M. Pineschi, Recent Progresses in the Catalytic Stereoselective Dearomatization of Pyridines, Molecules, 2023, 28, 6186–6215 CrossRef CAS PubMed.
- M. Escolano, D. Gaviña, G. Alzuet-Piña, S. Díaz-Oltra, M. Sánchez-Roselló and C. d. Pozo, Recent Strategies in the Nucleophilic Dearomatization of Pyridines, Quinolines, and Isoquinolines, Chem. Rev., 2024, 124, 1122–1246 CrossRef CAS PubMed.
- M. Okumura and D. Sarlah, Visible-Light-Induced Dearomatizations, Eur. J. Org Chem., 2019, 2020, 1259–1273 CrossRef PubMed.
- Y.-Z. Cheng, Z. Feng, X. Zhang and S.-L. You, Visible-light induced dearomatization reactions, Chem. Soc. Rev., 2022, 51, 2145–2170 Search PubMed.
- D. H. Liu and J. Ma, Recent Advances in Dearomative Partial Reduction of Benzenoid Arenes, Angew. Chem., Int. Ed., 2024, 63, e202402819 CrossRef CAS PubMed.
- R. J. Wiles and G. A. Molander, Photoredox-Mediated Net-Neutral Radical/Polar Crossover Reactions, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS PubMed.
- S. Sharma, J. Singh and A. Sharma, Visible Light Assisted Radical-Polar/Polar-Radical Crossover Reactions in Organic Synthesis, Adv. Synth. Catal., 2021, 363, 3146–3169 CrossRef CAS.
- L. Pitzer, J. L. Schwarz and F. Glorius, Reductive radical-polar crossover: traditional electrophiles in modern radical reactions, Chem. Sci., 2019, 10, 8285–8291 Search PubMed.
- A. R. Flynn, K. A. McDaniel, M. E. Hughes, D. B. Vogt and N. T. Jui, Hydroarylation of Arenes via Reductive Radical-Polar Crossover, J. Am. Chem. Soc., 2020, 142, 9163–9168 Search PubMed.
- Y. Masuda, H. Tsuda and M. Murakami, Photoinduced Dearomatizing Three-Component Coupling of Arylphosphines, Alkenes, and Water, Angew. Chem., Int. Ed., 2020, 60, 3551–3555 Search PubMed.
- R. C. McAtee, E. A. Noten and C. R. J. Stephenson, Arene dearomatization through a catalytic N-centered radical cascade reaction, Nat. Commun., 2020, 11, 2528–2535 CrossRef CAS PubMed.
- H. Takeuchi, S. Inuki, K. Nakagawa, T. Kawabe, A. Ichimura, S. Oishi and H. Ohno, Total Synthesis of Zephycarinatines via Photocatalytic Reductive Radical ipso-Cyclization, Angew. Chem., Int. Ed., 2020, 59, 21210–21215 CrossRef CAS PubMed.
- Y. Gao, H. Wang, Z. Chi, L. Yang, C. Zhou and G. Li, Spirocyclizative Remote Arylcarboxylation of Nonactivated Arenes with CO2 via Visible-Light-Induced Reductive Dearomatization, CCS Chem., 2022, 4, 1565–1576 Search PubMed.
- Y. Wang, W.-Y. Zhang, Z.-L. Yu, C. Zheng and S.-L. You, SmI2-mediated enantioselective reductive dearomatization of non-activated arenes, Nat. Synth., 2022, 1, 401–406 CrossRef CAS.
- Q.-Q. Zhao, J. Rehbein and O. Reiser, Thermoneutral synthesis of spiro-1,4-cyclohexadienes by visible-light-driven dearomatization of benzylmalonates, Green Chem., 2022, 24, 2772–2776 RSC.
- C. Zhou, A. Shatskiy, A. Z. Temerdashev, M. D. Kärkäs and P. Dinér, Highly congested spiro-compounds via photoredox-mediated dearomative annulation cascade, Commun. Chem., 2022, 5, 92–99 CrossRef CAS PubMed.
- J. Li, A. Kumar and J. C. Lewis, Non-native Intramolecular Radical Cyclization Catalyzed by a B12-Dependent Enzyme**, Angew. Chem., Int. Ed., 2023, 62, e202312893 CrossRef CAS PubMed.
- S. Manna, P. K. Someswara Ashwathappa and K. R. Prabhu, Visible light-mediated ipso-annulation of activated alkynes: access to 3-alkylated spiro[4,5]-trienones, thiaspiro[4,5]-trienones and azaspiro[4,5]-trienones, Chem. Commun., 2020, 56, 13165–13168 RSC.
- P. Chen, J. Xie, Z. Chen, B. Q. Xiong, Y. Liu, C. A. Yang and K. W. Tang, Visible-Light-Mediated Nitrogen-Centered Radical Strategy: Preparation of 3-Acylated Spiro[4,5]trienones, Adv. Synth. Catal., 2021, 363, 4440–4446 CrossRef CAS.
- W. Dong, Y. Yuan, C. Liang, F. Wu, S. Zhang, X. Xie and Z. Zhang, Photocatalytic Radical Ortho-Dearomative Cyclization: Access to Spiro[4.5]deca-1,7,9-trien-6-ones, J. Org. Chem., 2021, 86, 3697–3705 CrossRef CAS PubMed.
- C. R. Azpilcueta-Nicolas, D. Meng, S. Edelmann and J. P. Lumb, Dearomatization of Biaryls through Polarity Mismatched Radical Spirocyclization, Angew. Chem., Int. Ed., 2022, 62, e202215422 CrossRef PubMed.
- L. Li, Z.-W. Hou, P. Li and L. Wang, Electrochemical Dearomatizing Spirocyclization of Alkynes with Dimethyl 2-Benzylmalonates to Spiro[4.5]deca-trienones, J. Org. Chem., 2022, 87, 8697–8708 CrossRef CAS PubMed.
- M. Yang, J. Hua, H. Wang, T. Ma, C. Liu, W. He, N. Zhu, Y. Hu, Z. Fang and K. Guo, Photomediated Spirocyclization of N-Benzyl Propiolamide with N-Iodosuccinimide for Access to Azaspiro[4.5]deca-6,9-diene-3,8-dione, J. Org. Chem., 2022, 87, 8445–8457 CrossRef CAS PubMed.
- S. Samanta and D. Sarkar, Photoredox-Catalyzed Thiocyanative Cyclization of Biaryl Ynones to Thiocyanated Spiro[5.5]trienones: An External-Oxidant- and Transition-Metal-Free Approach, ChemPhotoChem, 2023, 7, e202200335 CrossRef CAS.
- Q. Fan, K. Jiang, B. Liu, H. Jiang, X. Cao and B. Yin, Radical-Dearomative Generation of Cyclohexadienyl Pd(II) toward the 3D Transformation of Nonactivated Phenyl Rings, Adv. Sci., 2023, 11, 2307074–2307082 CrossRef PubMed.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer, Chem. Rev., 2021, 122, 1626–1653 CrossRef PubMed.
- L. Li, Q. Pang, B. Chen, Y. Liu, Y. Zhao, J. Wu, K. Ge, J. Shen and P. Zhang, A General Approach for the Synthesis of Cyanoisopropyl Bicyclo[1.1.1]pentane (BCP) Motifs by Energy Transfer Process, Org. Lett., 2024, 26, 7060–7065 CrossRef CAS PubMed.
- J. J. Ma, F. Strieth-Kalthoff, T. Dalton, M. Freitag, J. L. Schwarz, K. Bergander, C. Daniliuc and F. Glorius, Direct Dearomatization of Pyridines via an Energy-Transfer-Catalyzed Intramolecular [4+2] Cycloaddition, Chem, 2019, 5, 2854–2864 CAS.
- J. Proessdorf, C. Jandl, T. Pickl and T. Bach, Arene Activation through Iminium Ions: Product Diversity from Intramolecular Photocycloaddition Reactions, Angew. Chem., Int. Ed., 2022, 61, e202208329 CrossRef CAS PubMed.
- M. Chiminelli, A. Serafino, D. Ruggeri, L. Marchiò, F. Bigi, R. Maggi, M. Malacria and G. Maestri, Visible-Light Promoted Intramolecular para-Cycloadditions on Simple Aromatics, Angew. Chem., Int. Ed., 2023, 62, e202216817 CrossRef CAS PubMed.
- G. Zhen, G. Zeng, F. Wang, X. Cao and B. Yin, Direct Construction of Fused/Bridged 3D Rings via Visible-Light Induced Dearomative Cycloaddition of Arenes, Adv. Synth. Catal., 2023, 365, 43–52 Search PubMed.
- M. Zhu, Y. J. Gao, X. L. Huang, M. Z. Li, C. Zheng and S. L. You, Photo-induced intramolecular dearomative [5+4] cycloaddition of arenes for the construction of highly strained medium-sized-rings, Nat. Commun., 2024, 15, 2462–2469 CrossRef CAS PubMed.
- B. Altundas, E. Alwedi, Z. H. Song, A. R. Gogoi, R. Dykstra, O. Gutierrez and F. F. Fleming, Dearomatization of aromatic asmic isocyanides to complex cyclohexadienes, Nat. Commun., 2022, 13, 6444–6451 Search PubMed.
- Y. Zhang, D. Yang, D. Lu and Y. Gong, Photoredox-Enabled Dearomatization of Protected Anilines: Access to Cyclohexadienone Imines with Contiguous Quaternary Centers, Org. Lett., 2023, 25, 1320–1325 CrossRef CAS PubMed.
- P. F. Yuan, X. T. Huang, L. Long, T. Huang, C. L. Sun, W. Yu, L. Z. Wu, H. Chen and Q. Liu, Regioselective Dearomative Amidoximation of Nonactivated Arenes Enabled by Photohomolytic Cleavage of N-nitrosamides, Angew. Chem., Int. Ed., 2024, 63, e202317968 CrossRef CAS PubMed.
- D. Leifert and A. Studer, The Persistent Radical Effect in Organic Synthesis, Angew. Chem., Int. Ed., 2019, 59, 74–108 CrossRef PubMed.
- K. Yang, J. Liu, D. Fu, L. Niu, S.-J. Li and Y. Lan, A theoretical study of selective radical relay and coupling reactions for alkene difunctionalization, Org. Chem. Front., 2023, 10, 4336–4341 RSC.
- H. Fischer, The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- V. K. Soni, S. Lee, J. Kang, Y. K. Moon, H. S. Hwang, Y. You and E. J. Cho, Reactivity Tuning for Radical–Radical Cross-Coupling via Selective Photocatalytic Energy Transfer: Access to Amine Building Blocks, ACS Catal., 2019, 9, 10454–10463 CrossRef CAS.
- T. Patra, P. Bellotti, F. Strieth-Kalthoff and F. Glorius, Photosensitized Intermolecular Carboimination of Alkenes through the Persistent Radical Effect, Angew. Chem., Int. Ed., 2020, 59, 3172–3177 Search PubMed.
- R. I. Rodríguez, M. Sicignano, M. J. García, R. G. Enríquez, S. Cabrera and J. Alemán, Taming photocatalysis in flow: easy and speedy preparation of α-aminoamide derivatives, Green Chem., 2022, 24, 6613–6618 RSC.
- G. Tan, M. Das, H. Keum, P. Bellotti, C. Daniliuc and F. Glorius, Photochemical single-step synthesis of β-amino acid derivatives from alkenes and (hetero)arenes, Nat. Chem., 2022, 14, 1174–1184 CrossRef CAS PubMed.
- Y. Zheng, Z. J. Wang, Z. P. Ye, K. Tang, Z. Z. Xie, J. A. Xiao, H. Y. Xiang, K. Chen, X. Q. Chen and H. Yang, Regioselective Access to Vicinal Diamines by Metal-Free Photosensitized Amidylimination of Alkenes with Oxime Esters, Angew. Chem., Int. Ed., 2022, 61, e202212292 CrossRef CAS PubMed.
- L. Geniller, M. Taillefer, F. Jaroschik and A. Prieto, Photocatalyzed Amination of Alkyl Halides to Access Primary Amines, J. Org. Chem., 2023, 89, 656–664 Search PubMed.
- S.-S. Li, Y.-S. Jiang, X.-L. Luo, X. Ran, Y. Li, D. Wu, C.-X. Pan and P.-J. Xia, Photocatalytic vinyl radical-mediated multicomponent 1,4-/1,8-carboimination across alkynes and olefins/(hetero)arenes, Sci. China:Chem., 2023, 67, 558–567 CrossRef.
- F. Paulus, C. Stein, C. Heusel, T. J. Stoffels, C. G. Daniliuc and F. Glorius, Three-Component Photochemical 1,2,5-Trifunctionalizations of Alkenes toward Densely Functionalized Lynchpins, J. Am. Chem. Soc., 2023, 145, 23814–23823 Search PubMed.
- G. Tan, F. Paulus, A. Petti, M.-A. Wiethoff, A. Lauer, C. Daniliuc and F. Glorius, Metal-free photosensitized radical relay 1,4-carboimination across two distinct olefins, Chem. Sci., 2023, 14, 2447–2454 RSC.
- R. Laskar, S. Dutta, J. C. Spies, P. Mukherjee, Á. Rentería-Gómez, R. E. Thielemann, C. G. Daniliuc, O. Gutierrez and F. Glorius, γ-Amino Alcohols via Energy Transfer Enabled Brook Rearrangement, J. Am. Chem. Soc., 2024, 146, 10899–10907 CrossRef CAS PubMed.
- M.-J. Zheng, X.-K. Qi, C. Yang, L. Guo, Y. Zhao and W. Xia, Photoinduced amination of iodoalkanes enabled by bifunctional O-benzoyl oxime, Org. Chem. Front., 2024, 11, 1949–1954 Search PubMed.
- Y. Zhong, Z. Zhuang, X. Zhang, B. Xu and C. Yang, Difunctionalization of gem-difluoroalkenes for amination and heteroarylation via metal-free photocatalysis, Chem. Commun., 2024, 60, 4830–4833 Search PubMed.
- D. S. Lee, V. K. Soni and E. J. Cho, N–O Bond Activation by Energy Transfer Photocatalysis, Acc. Chem. Res., 2022, 55, 2526–2541 CrossRef CAS PubMed.
- M. C. Nicastri, D. Lehnherr, Y.-h. Lam, D. A. DiRocco and T. Rovis, Synthesis of Sterically Hindered Primary Amines by Concurrent Tandem Photoredox Catalysis, J. Am. Chem. Soc., 2020, 142, 987–998 CrossRef CAS PubMed.
- G. Zeng, H. Luo, K. Jiang, J. Cai and B. Yin, Fully atom-economic access to spiro-cyclic skeletons through photoredox-induced hydrogen transfer/Giese addition/dearomative cyclization/protonation cascade, Org. Chem. Front., 2024, 11, 3165–3172 RSC.
- L. Chen, K. Jiang, G. Zeng and B. Yin, Photoinduced Pd-Catalyzed Csp2–H/Csp3–H Dehydrocoupling Reaction Employing Polyhaloaromatics as the Dehydrogenating Agent, Org. Lett., 2022, 24, 9071–9075 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration, Chem. Rev., 2021, 122, 1875–1924 CrossRef PubMed.
- D. H. Liu, K. Nagashima, H. Liang, X. L. Yue, Y. P. Chu, S. Chen and J. Ma, Chemoselective Quinoline and Isoquinoline Reduction by Energy Transfer Catalysis Enabled Hydrogen Atom Transfer, Angew. Chem., Int. Ed., 2023, 62, e202312203 CrossRef CAS PubMed.
- S. H. Cai, D. X. Wang, L. Ye, Z. Y. Liu, C. Feng and T. P. Loh, Pyrroline Synthesis via Visible-Light-Promoted Hydroimination of Unactivated Alkenes with N,N′-Dimethylpropylene Urea as H-Donor, Adv. Synth. Catal., 2018, 360, 1262–1266 CrossRef CAS.
- Deposition Number CCDC 2343819 cis-2-1 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- J. Cai, G. Zeng, K. Jiang, H. Luo and B. Yin, Intramolecular Cobalt/Visible Light Cocatalyzed Reductive Coupling of Unactivated Arenes with Unactivated Alkenes, Org. Lett., 2024, 26, 327–331 CrossRef CAS PubMed.
- Deposition Number CCDC 2360015 6-1 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- C. Hervieu, M. S. Kirillova, T. Suárez, M. Müller, E. Merino and C. Nevado, Asymmetric, visible light-mediated radical sulfinyl-Smiles rearrangement to access all-carbon quaternary stereocentres, Nat. Chem., 2021, 13, 327–334 CrossRef CAS PubMed.
- X.-c. Gan, B. Zhang, N. Dao, C. Bi, M. Pokle, L. Kan, M. R. Collins, C. C. Tyrol, P. N. Bolduc, M. Nicastri, Y. Kawamata, P. S. Baran and R. Shenvi, Carbon quaternization of redox active esters and olefins by decarboxylative coupling, Science, 2024, 384, 113–118 CrossRef CAS PubMed.
- M. Chen, W. Sun, J. Yang, L. Yuan, J.-Q. Chen and J. Wu, Metal-free photosensitized aminosulfonylation of alkenes: a practical approach to β-amido sulfones, Green Chem., 2023, 25, 3857–3863 RSC.
- X.-K. Qi, M.-J. Zheng, C. Yang, Y. Zhao, L. Guo and W. Xia, Metal-Free Amino(hetero)arylation and Aminosulfonylation of Alkenes Enabled by Photoinduced Energy Transfer, J. Am. Chem. Soc., 2023, 145, 16630–16641 CrossRef CAS PubMed.
- C. Faderl, S. Budde, G. Kachkovskyi, D. Rackl and O. Reiser, Visible Light-Mediated Decarboxylation Rearrangement Cascade of ω-Aryl-N-(acyloxy)phthalimides, J. Org. Chem., 2018, 83, 12192–12206 CrossRef CAS PubMed.
- Deposition Number CCDC 2343822 cis-3-1 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- S. K. Ghosh, L. He, Z. Tang and R. J. Comito, Selective and Functional-Group-Tolerant Photoalkylation of Imines by Energy-Transfer Photocatalysis, J. Org. Chem., 2023, 88, 15209–15217 CrossRef CAS PubMed.
Footnotes |
| † This article is dedicated to professor Yulin Wu to express our deep memory. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds, and copies of 1H, 13C and 19F NMR spectra. See DOI: https://doi.org/10.1039/d4sc07681h |
| This journal is © The Royal Society of Chemistry 2025 |