
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-site reduction of hexachlorophosphazene to low-valent PN heterocycles and extension to the reduction of poly-chlorophosphazene†
Etienne A.
LaPierre
 *a,
Roope A.
Suvinen
b,
Brian O.
Patrick
c,
Heikki M.
Tuononen
*b and
Ian
Manners‡
a
*a,
Roope A.
Suvinen
b,
Brian O.
Patrick
c,
Heikki M.
Tuononen
*b and
Ian
Manners‡
a
aDepartment of Chemistry, University of Victoria, 3800 Finnerty Rd, Victoria, British Columbia V8P 5C2, Canada. E-mail: elapierre@uvic.ca
bDepartment of Chemistry, NanoScience Centre, University of Jyväskylä, P. O. Box 35, FI-40014 Jyväskylä, Finland. E-mail: heikki.m.tuononen@jyu.fi
cDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada
First published on 24th April 2025
Abstract
Facile one and two site reduction of hexachlorophosphazene using cyclic (alkyl)(amino)carbene substituents are shown to yield P-CAACMe-cyclo-(PNP(Cl)2NP(Cl)2N) 1 and P,P′-bis-CAACMe-cyclo-(PNPNP(Cl)2N) 2 (CAACMe = 1-[2,6-bis(isopropyl)phenyl]-3,3,5,5-tetramethyl-2-pyrrolidinylidene), respectively. Compound 1 is characterized by its predominantly phosphorus-centered HOMO, which results in typical phosphine-type nucleophilic and reductive reactivity; however, the resultant compounds of such reactions feature properties distinct from their classical phosphine analogues due to the CAAC-centered LUMO, which acts as an acceptor for both intramolecular interactions and photophysical excitations. In contrast, compound 2 exhibits π-conjugation spanning the endocyclic PNP moiety and the two CAACMe substituents, despite its non-planar structure. Treatment of 2 with [Cp*RuCl]4 results in the electrophilic displacement of one of the CAACMe moieties by two Cp*RuCl fragments to yield the spirocyclic compound 3. Preliminary results show that the methodology used to reduce hexachlorophosphazene to 1 can be directly transposed to the regiospecific reduction of poly-chlorophosphazene, to yield poly-1, a fundamentally new class of inorganic polymer that possesses a phosphorus center with chemically active lone pairs in the main chain.
Introduction
First identified by von Liebig in 1834 as a product of the condensation of PCl5 with ammonia,1 hexachlorophosphazene, cyclo-(NPCl2)3, has attracted significant interest, both due to its unusual bonding and chemical properties as well as its practical utility in synthesis. Hexachlorophosphazene is notable for its planar structure, uniform bond lengths and angles, and resistance to hydrolysis by ambient moisture typical of chlorophosphoranes. While these properties were originally attributed to aromaticity arising from π-bonding invoking phosphorus d-orbitals,2 it is now widely accepted that phosphazenes do not exhibit such π-bonding, and these features arise instead from the negative hyperconjugation of the nitrogen-centered lone pair into phosphorus-chlorine σ* orbitals.3–5 The early chemistry of hexachlorophosphazene was meticulously investigated by Stokes in the 1890s,6–8 who noted the formation of a hydrolytically sensitive “inorganic rubber”, poly-chlorophosphazene, upon thermolysis. This material remained a curiosity due to its sensitivity until reports by Allcock and coworkers in the 1960s demonstrated the facile derivatization of the polymer chain by nucleophilic substitution of the phosphorus–chloride bonds to yield air and moisture stable high polymers.9,10 This process has been expanded to a wide variety of nucleophiles resulting in poly-phosphazenes with a broad range of properties and potential applications.11–17 In addition to its use as a polymer precursor, hexachlorophosphazene also possesses rich substitution chemistry,18 which has been utilized for the synthesis of numerous derivatives, notably well-defined phosphazene dendrimers.19,20In contrast to the well-developed polymerization and substitution chemistry, development of the controlled reduction chemistry of hexachlorophosphazene consists of a single example by Rivard and coworkers,21 whereby a single phosphorus center can be reduced in the presence of a stabilizing N-heterocyclic carbene (NHC) to compound A (Chart 1). Likewise, the reduction chemistry of the fluorine congener, hexafluorophosphazene, which is known to give more predictable reactivity with organometallic reagents, is similarly scant, being limited to single site reduction by formal reductive metalation using transition metal carbonyl derivatives, giving compounds B and C (Chart 1). However, even formal reduction in these cases is questionable given the ambiguity in oxidation state of the metal centers.15,22–24 Recently, we have explored the controlled reduction of linear chlorophosphazenes to give formally low-valent PN compounds stabilized by the cyclic (alkyl)(amino)carbene25 1-[2,6-bis(isopropyl)phenyl]-3,3,5,5-tetramethyl-2-pyrrolidinylidene (CAACMe).26,27 Notably, such species were found to exhibit delocalized π-bonding and share an isoelectronic relationship with conjugated hydrocarbons, unlike their high-valent analogues, as well as a rich-redox chemistry. Given these results, the general superiority of CAAC ligands in stabilizing low-valent species compared to NHCs,28–30 and the paucity of low-valent PN compounds,31–37 despite wide-spread interest,38–48 the suitability of CAACs for stabilizing the reduction products of hexachlorophosphazene, particularly in order to access cyclic conjugated species, such as cyclo-(PN)3, warrants investigation.
 | ||
| Chart 1 Examples of reduced cyclic six-membered phosphazene rings. | ||
Results and discussion
Synthesis and structure of 1
Firstly, the direct reaction of CAACMe with hexachlorophosphazene was explored in the absence of external reducing agents.26,49 Reaction of these compounds in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio led to immediate salt formation and development of a bright orange solution. Subsequent analysis by 31P{1H} NMR spectrometry revealed the consumption of one-half of hexachlorophosphazene and formation of a new AX2 spin system with δP = 104.3 (A) and 4.6 ppm (X), and a coupling constant of 87 Hz, consistent with the reduction of a single phosphorus center by two equivalents of CAACMe and concomitant formation of [CAAC–Cl][Cl] as a by-product. Accordingly, addition of a further equivalent of CAACMe resulted in full consumption of hexachlorophosphazene and complete and quantitative conversion to the novel orange product 1 (Scheme 1). This is in contrast to reports by Rivard and coworkers,21 where the lack of a strong external reductant, such as sodium metal, gave mixtures of products and only trace conversion to the desired reduction product, a difference likely arising due to the superior reducing power of CAACMe relative to NHCs.26,29,49
:1 ratio led to immediate salt formation and development of a bright orange solution. Subsequent analysis by 31P{1H} NMR spectrometry revealed the consumption of one-half of hexachlorophosphazene and formation of a new AX2 spin system with δP = 104.3 (A) and 4.6 ppm (X), and a coupling constant of 87 Hz, consistent with the reduction of a single phosphorus center by two equivalents of CAACMe and concomitant formation of [CAAC–Cl][Cl] as a by-product. Accordingly, addition of a further equivalent of CAACMe resulted in full consumption of hexachlorophosphazene and complete and quantitative conversion to the novel orange product 1 (Scheme 1). This is in contrast to reports by Rivard and coworkers,21 where the lack of a strong external reductant, such as sodium metal, gave mixtures of products and only trace conversion to the desired reduction product, a difference likely arising due to the superior reducing power of CAACMe relative to NHCs.26,29,49
 | ||
| Scheme 1 Reduction and ligation of hexachlorophosphazene by CAACMe to yield 1. | ||
The room temperature 1H NMR spectrum of 1 in C6D6 shows apparent Cs symmetry, featuring a single set of resonances corresponding to the CAACMe substituent. Similarly, the solid-state structure of 1, determined by single crystal X-ray diffraction studies, is approximately Cs symmetric (Fig. 1) with only minor puckering of the CAACMe ring. Density functional theory (DFT) calculations at the PBE0-GD3BJ/def2-TZVP level50–52 showed that the Gibbs free energy of activation for the CAACMe ring flip is only 3.33 kcal mol−1. As the barriers associated with the rotation of the CAACMe moiety are also small (vide infra), rapid interconversion between different conformers takes place, leading to spectroscopic data consistent with pseudo-Cs symmetry.
 | ||
| Fig. 1 Molecular structure of 1 shown from two perspectives. Ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Select bond lengths (Å) and angles (°) for 1 [calculated values in square brackets]: P1–C1 1.912(1) [1.904]; C1–N4 1.301(2) [1.291]; P1–N1 1.6793(9) [1.665]; N1–P2 1.546(1) [1.553]; P2–N2 1.593(1) [1.588]; C1–P1–N1 98.14(5) [100.1]; N1–P1–N3 108.50(6) [109.3]. | ||
The CAACMe ligated phosphorus atom in 1 is bent away from the plane of the phosphazene ring, exhibiting pyramidalized geometry (Σ = 303.91°) and a long P–C bond length of 1.912(1) Å, longer than that found in A (1.8791(13) Å).21 The latter feature may be explained by the orthogonal disposition of the formally vacant orbital of the carbene carbon to the lone pair at phosphorus that prevents back-donation, an assertion supported by natural bond orbital (NBO) analyses that showed no significant donor–acceptor interactions between these orbitals. Further, the P–N bond lengths between the reduced phosphorus center and the adjacent nitrogen atoms are significantly elongated in 1 with respect to those in hexachlorophosphazene (1.6793(9) Å vs. 1.577(3) Å),53 while the P–N bond lengths between the nitrogen centers adjacent to the reduced phosphorus center and the PCl2 fragments are contracted. The former may be rationalized by reduced electrostatic attraction and lack of negative hyperconjugation, while the latter is likely due to increased negative hyperconjugation from the more electron rich nitrogen centers to the P–Cl σ* orbitals.
As most phosphorus(III) compounds, such as A, are colorless or weakly colored, we sought to investigate the reason for the striking orange coloration of 1. Analysis of 1 by UV-Vis spectrometry in THF (Fig. S1†) revealed a strong transition with λmax = 438 nm (ε438 = 4.65 × 103 M−1 cm−1), while similar analysis in toluene (Fig. S2†) showed the absorption properties of 1 to be solvent dependent, with a slight red-shifting of the absorption maximum (λmax = 442 nm) and a dramatic increase in the extinction coefficient (ε442 = 8.11 × 103 M−1 cm−1). Further, time-dependent density functional theory (TD-DFT) calculations employing implicit solvation for THF or toluene (polarizable continuum model using the integral equation formalism variant, IEFPCM)54 indicated that 1 should lack meaningful transitions in the visible light region, with an exceptionally weak oscillator strength (f = 0.0001) calculated for the S0→S1 HOMO to LUMO transition at around 500 nm. This agrees with the orthogonal orientation of the predominantly phosphorus lone pair HOMO and the carbene-centered C–N π* LUMO (Fig. 2), which prevents effective overlap between donor and acceptor orbitals. With this in mind, alternative energetically accessible conformations of 1 that better agree with the observed photophysical behavior were explored computationally.
 | ||
| Fig. 2 Frontier Kohn–Sham orbitals of two rotamers of 1 with pseudo-Cs (left, 1Cs) and C1 (right, 1C1) symmetry (isovalue = ±0.06 a.u.). Dipp = 2,6-diisopropylphenyl. | ||
A relaxed potential energy scan of the rotation of the CAAC moiety about the P–C bond in 1 identified several additional minima. Of lowest energy is 1C1 with the CAACMe moiety rotated ca. 90° about the P–C bond axis relative to the pseudo-Cs symmetric structure, denoted hereafter 1Cs (Fig. 2 and S3†). Interestingly, the asymmetric rotamer 1C1 is 4.55 kcal mol−1 lower in Gibbs free energy than the experimentally observed structure 1Cs and has a significantly shorter P–C bond; 1.790 Å in 1C1 compared to 1.904 Å in 1Cs. Accordingly, the frontier Kohn–Sham orbitals of 1C1 are distinct from those of 1Cs (Fig. 2), with the HOMO and LUMO of 1C1 being the in-phase and out-of-phase combinations of the phosphorus lone pair and the C–N π* orbital, respectively. Consequently, the HOMO of 1C1 is lower in energy by ca. 0.1 eV compared to 1Cs, while the LUMO is similarly destabilized. NBO analyses showed a markedly increased Wiberg bond index55 for the P–C bond in 1C1vs.1Cs (1.15 vs. 0.78), likely arising from the significant donor–acceptor interaction (53.05 kcal mol−1) between the phosphorus lone-pair and the C–N π* orbital in 1C1.
TD-DFT calculations for 1C1 using implicit solvation for THF revealed a strong (f = 0.154) electronic transition at 410 nm arising from the S0 → S1 HOMO to LUMO excitation, in good agreement with the experiments. Performing the calculations using implicit toluene solvation in lieu of THF accurately reproduces the slight red-shifting of the absorption maximum observed (416 nm), though the difference in the computed oscillator strength (f = 0.171) does not explain the difference in the observed absorptivity in these solvents. Consequently, the energy difference between the two rotamers in different environments was computed, revealing a greater preference for 1C1 in the gas phase (−4.55 kcal mol−1) or toluene (−3.37 kcal mol−1) than in THF (−2.30 kcal mol−1), which follows the experimental solvent dependence in absorptivity. Presumably, the preference for 1 to crystallize as 1Cs over the lower energy rotamer 1C1 arises from solid-state packing effects combined with the ease of 1Cs → 1C1 interconversion that has a calculated Gibbs free energy of activation of only 4.15 kcal mol−1 in the gas phase; the calculated Gibbs free energy of activation for the reverse process 1C1 → 1Cs is 8.70 kcal mol−1.
Oxidation and metalation of 1
With the HOMO of 1 being predominantly phosphorus lone pair in nature (Fig. 2), the ability of 1 to engage in archetypical phosphine reactivity was explored. As A was previously demonstrated to react with elemental sulfur to yield a formal phosphine sulfide, we explored the reactivity of 1 with various chalcogen transfer reagents (Scheme 2). This showed that 1 reacts with oxygen, sulfur, or selenium transfer reagents to generate the corresponding phosphorus chalcogenides 1Ch (Ch = O, S, or Se) in high yield. Compound 1O is colorless, with room temperature 1H and 31P{H} NMR spectra in CDCl3 consistent with pseudo-Cs symmetry. Notably, the 31P NMR resonance corresponding to the oxidized phosphorus center is shifted dramatically upfield to δP = −16.0 ppm, in contrast to organophosphines that typically shift downfield. The 31P{1H} NMR spectra of 1S and 1Se likewise feature a smaller upfield shift of the resonance corresponding to the oxidized phosphorus center, which in 1Se also shows distinct satellites (J = 710 Hz) with an area (7:93) consistent with 77Se coupling;56 correspondingly, the 77Se NMR spectrum of 1Se consists of a doublet at δSe = 215.7 ppm. While the magnitude of the 1JP–Se coupling constant has been used to evaluate the bonding in phosphine selenides,57 the presence of confounding intramolecular interactions in 1Se (vide infra), which have been shown to influence the coupling constant and 77Se NMR chemical shift in phosphine58 and related carbene selenides,59 preclude such analysis. Saliently, the resonances for the PCl2 centers in the 31P{1H} NMR spectrum of both 1S and 1Se are broadened, indicative of inequivalent sites in dynamic exchange. The 1H NMR spectra of 1S and 1Se are similarly broadened, indicative of dynamic exchange and consistent with hindered rotation about the C–P bond. In further contrast with 1O, both 1S and 1Se are colored, the former yellow and the latter orange.
 | ||
| Scheme 2 Chalcogenation of 1 by PhIO or elemental chalcogens. | ||
The solid-state structures of all compounds in the series 1Ch were examined by X-ray crystallography. Unfortunately, single crystals of 1O of suitable quality for full analysis could not be obtained; however, connectivity could be established (Fig. S4†), which indicates that 1O crystallizes with approximate Cs symmetry, similarly to 1. In contrast, the structures of the heavier congeners 1S and 1Se show the CAACMe moiety to be rotated by ca. 90° about the P–C bond (Fig. 3), similarly to the calculated structure 1C1. The P–Ch distances are comparable to those in respective triphenylphosphine analogues (1.9487(6) vs. 1.950(3) Å (ref. 60) and 2.096(2) vs. 2.106(1) Å (ref. 61) for S and Se, respectively). In both 1S and 1Se, the C–P–Ch bond angle is narrowed considerably from idealized tetrahedral geometry (∠C–P–S = 99.13(6)°, ∠C–P–Se = 98.2(2)°), in contrast to sulfurized A (Chart 1), which features a more typical C–P–S angle of 105.02(6).21 Given the orientation of the CAACMe fragment, these relatively acute angles suggest an interaction between the heavy chalcogen and the formally vacant orbital on carbon. The C⋯Ch distances are 2.898(2) and 2.990(5) Å for S and Se, respectively, both significantly shorter than sum of van der Waals radii for the corresponding elements.
 | ||
| Fig. 3 Molecular structures of 1S (left) and 1Se (right). Ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Select bond lengths (Å) and angles (°) for 1S: P1–C1 1.858(2) [1.833]; P1–S1 1.82965 [1.947]; C1–N4 1.305(2) [1.303]; P1–N1 1.625(1) [1.613]; N1–P2 1.558(2) [1.561]; P2–N2 1.591(2) [1.585]; C1–P1–S1 99.13(6) [94.4]; C1–P1–N1 108.60(8) [108.4]; N1–P1–N3 111.42(8) [112.7]. Select bond lengths (Å) and angles (°) for 1Se: P1–C1 1.850(5) [1.830]; P1–Se1 2.098(2) [2.103]; C1–N4 1.306(7) [1.307]; P1–N1 1.613(4) [1.612]; N1–P2 1.556(5) [1.563]; P2–N2 1.580(5) [1.584]; C1–P1–Se1 98.2(2) [94.4]; C1–P1–N1 109.2(2) [108.4]; N1–P1–N3 111.7(2) [113.1]. | ||
Analysis of the calculated Kohn–Sham frontier orbitals for both 1S and 1Se showed that the HOMO−1 and the LUMO correspond to the in-phase and out-of-phase combinations of a chalcogen lone pair and the C–N π* orbital, respectively, while the HOMO is another lone pair on the chalcogen element (Fig. 4). NBO analyses revealed sizeable through-space C⋯Ch donor–acceptor interactions in both 1S and 1Se (14.08 and 13.95 kcal mol−1, respectively), which, combined with the steric profile imposed by the large chalcogen atom, provide a rationale for restricted rotation about the P–C bond observed by NMR spectrometry (vide supra). Further, the presence of this interaction explains the unexpected coloration of both compounds. The UV-Vis spectra of 1S and 1Se in THF feature strong absorptions at 340 nm (ε340 = 4.52 × 103 M−1 cm−1) and 387 nm (ε387 = 5.34 × 103 M−1 cm−1), respectively, that TD-DFT calculations with implicit THF solvation suggest to arise from S0 → S2 transitions of primarily HOMO−1 to LUMO character.
 | ||
| Fig. 4 Top: Frontier Kohn–Sham molecular orbitals of 1Se (isovalue = ±0.04 a.u.). Bottom: Natural transition orbitals (NTOs; isovalue = ±0.04 a.u.) and the electron density difference map (isovalue = ±0.006 a.u.; red = depletion, teal = accumulation) for the S0 → S2 transition of 1Se. The orbitals of 1S are qualitatively similar. | ||
The solution dynamics of 1S and 1Se were further explored by variable temperature NMR spectrometry in CDCl3. At 213 K, both 1S and 1Se exhibit 31P{1H} and 1H NMR spectra that are consistent with C1 symmetry and show no evident line broadening, indicative of the existence of a single conformer at this temperature (Fig. S5–S8†); in contrast, 1O shows spectra consistent with a pseudo-Cs symmetric structure at the same temperature (Fig. S9 and S10†). For both 1S and 1Se, 1H NMR spectra were acquired at temperatures between 273 and 313 K and modelled to extract rate constants via line shape analysis. Eyring analysis (Fig. S11 and S12†) reveals positive enthalpies of activation for bond rotation, ΔH‡ = 9.69 ± 0.17 and 9.50 ± 0.36 kcal mol−1 for 1S and 1Se, respectively, with significant negative entropies of activation, ΔS‡ = −9.56 ± 0.60 and −12.9 ± 1.21 cal K−1 mol−1 for 1S and 1Se, respectively, indicative of an ordered transition state in both cases. DFT analysis of the rotation of the CAAC moiety about the P–C bond in 1Ch with implicit solvation model for CDCl3 gave ΔH‡ (at 298 K and 1 atm) of 7.48, 12.42, and 13.78 kcal mol−1 for the C1 → Cs interconversion of 1O, 1S, and 1Se, respectively; calculated enthalpies of activation for the reverse rotation Cs → C1 are 6.39, 7.61, and 7.93 kcal mol−1, respectively. The DFT data show the expected trend and suggest largely unhindered rotation about the P–C bond for 1O at 213 K, while the same rotation in 1S and 1Se is clearly restricted with almost twice the barrier height. Interestingly, the calculated entropies of activation are negative but negligible for all compounds 1Ch, suggesting that explicit interactions with solvent molecules play a large role in modulating the entropies.
The properties of 1 as a ligand in transition metal complexes was explored by the synthesis of analogues of classical mono-phosphine complexes (Scheme 3). The gold chloride adduct of 1, 1Au, is readily accessed by the reaction of 1 with one equivalent of ClAu·SMe2. The room temperature NMR spectra of 1Au in CDCl3 are broad, similarly to those of 1Sand 1Se, and indicate restricted rotation about the P–C bond. DFT analysis with implicit solvation model for CDCl3 showed that the rotation of the CAAC moiety about the P–C bond has a barrier of 13.83 kcal mol−1, in agreement with experimental observations. The solid-state structure of 1Au (Fig. 5, top left) resembles more 1Cs than 1C1 and shows close association of the gold center with the phenyl ring of the diisopropylphenyl moiety (distance from Au to ring centroid is 3.333 Å), consistent with a gold⋯arene interaction that is a hallmark of many effective gold catalysts.62,63 The steric profile of 1 was evaluated by calculation of the topographic steric map and percent buried volume using the SambVca 2.1 tool developed by Cavallo and coworkers (Fig. 5, bottom).64 Compound 1 provides considerable steric bulk (%Vbur = 47.7%), comparable to Buchwald-type biaryl phosphine ligands (cf. %Vbur of 46.7% and 50.9% for CyJohnPhos and JohnPhos),65 with a highly asymmetric steric profile due to the presence of the aforementioned gold⋯arene interaction.
 | ||
| Scheme 3 Metalation reactions of 1. | ||
 | ||
| Fig. 5 Molecular structures of 1Au (top, left) and 1Ru (top, right), and topographic steric map for calculation of %Vbur of 1Au using the shown orientation (bottom). Ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Select bond lengths (Å) and angles (°) for 1Au [calculated values in square brackets]: Au1–P1 2.2184(6) [2.223]; Au1–Cl1 2.2874(8) [2.284]; P1–C1 1.888(2) [1.878]; C1–N4 1.287(3) [1.291]; P1–N1 1.633(2) [1.628]; N1–P2 1.554(2) [1.561]; P2–N2 1.586(2) [1.589]; C1–P1–Au1 117.62(7) [114.7]; C1–P1–N1 102.73(9) [103.7]; N1–P1–N3 111.8(1) [111.7]. Select bond lengths (Å) and angles (°) for 1Ru [calculated values in square brackets]: Ru1–P1 2.3283(5) [2.315]; Ru1–Cl1 2.3805(6) [2.359]; P1–C1 1.866(2) [1.850] C1–N4 1.312(2) [1.307]; P1–N1 1.633(2) [1.651]; N1–P2 1.552(1) [1.557]; P2–N2 1.588(2) [1.587]; C1–P1–Ru1 104.45(5) [104.1]; C1–P1–N1 101.96(7) [102.4]; N1–P1–N3 110.08(7) [111.0]; P1–Ru1–Cl1 106.51(2) [106.9]; Ru1–P1–N1 116.42(5) [115.7]. | ||
Having established the steric properties of 1, we sought to evaluate the donor ability by synthesis of an appropriate metal carbonyl complex.66 Unfortunately, attempts to coordinate 1 to rhodium(I) precursor [(CO)2RhCl]2 led to degradation of 1 and no apparent formation of any rhodium phosphine complexes, as indicated by the lack of AX2 or AXY spin systems or readily apparent coupling to rhodium in the 31P{1H} NMR spectra of the reaction mixtures. Owing to toxicity concerns with Ni(CO)4, the donor ability of 1 was evaluated in silico using DFT by calculation of the IR spectra of the putative nickel tricarbonyl complex with 1, denoted 1Ni, and comparing the result to data for known tricarbonyl complexes investigated at the same level of theory (Fig. S13†).67 This methodology indicates that 1 has a computed Tolman electronic parameter (cTEP) of 2070 cm−1, which is comparable to triaryl phosphines lacking donor substituents, suggestive of modest donor ability. Exploiting the flexibility afforded by computational protocols, the effect of the phosphazene chloride substituents on donor ability was also investigated by replacing them with modestly donating methyl substituents (Fig. S14†). This modification had significant influence on the cTEP value, with a red-shift of 21 cm−1 to 2049 cm−1, suggesting that the framework of 1 may have utility as an electronically modular ligand platform.
Lastly, as phosphines are known to effectively bind the Cp*RuCl fragment68,69 to give synthetically useful compounds with metal to ligand stoichiometry depending on the bulk of the ligand, the reactivity of 1 with [Cp*RuCl]4 (ref. 70) was explored. Compound 1 was found to react with 0.25 eq. [Cp*RuCl]4 in THF with immediate formation of a dark purple solution (Scheme 3).The room temperature 31P{1H} NMR spectrum of 1 in C6D6 indicated the presence of a single AMX spin system, consistent with the formation of an asymmetric 1:1 adduct of 1 with Cp*RuCl, 1Ru. The composition of 1Ru was further supported by determination of its solid-state structure by single crystal X-ray diffraction studies, indicating that the chloride ligand at ruthenium orients itself above the CAACMe moiety with a C⋯Cl distance of 3.554(2) Å, that is, only slightly longer than the sum of van der Waals radii for the respective elements. Given the acceptor nature of the CAACMe fragment in 1S and 1Se, the possibility of a weak halogen⋯π interaction was raised. However, computational analyses gave no support for such phenomenon, suggesting that the preferred ligand orientation in 1Ru more likely results from a combination of electrostatic H⋯Cl interactions as well as steric factors.
The UV-Vis spectrum of 1Ru in THF (Fig. S15†) features a broad asymmetric absorption band at 492 nm (ε492 = 4.67 × 103 M−1 cm−1), with a visible shoulder at ca. 570 nm. This is atypical for analogous complexes featuring classical phosphines that often show relatively weak d → d transitions at wavelengths greater than 550 nm.69 Accordingly, TD-DFT calculations with implicit THF solvation were performed for 1Ru, illustrating that the strongest absorption in the visible region arises from the S0 → S4 transition at 470 nm, in good agreement with the experiment. Calculation of the natural transition orbitals (NTOs)71 for this excitation are suggestive of a metal-to-ligand charge-transfer (MLCT), with the electron NTO having clear ruthenium d-orbital character, while the hole NTO is predominantly the C–N π*-orbital at the CAAC moiety (Fig. 6); similar features are seen in the associated electron density difference plot. Further evidence for the assignment is provided by calculation of its DCT index that quantifies the distance of charge-transfer as the distance between barycenters of electron depletion and augmentation of a given transition.72,73 For the S0 → S4 transition, a DCT index of 2.545 Å was obtained, consistent with charge-transfer, serving to highlight the photophysical non-innocence of 1 as a ligand, which is unusual for monodentate phosphines.
 | ||
| Fig. 6 Natural transition orbitals (NTOs, isovalue = ±0.08 a.u.) and the electron density difference map (isovalue = ±0.008 a.u.; red = depletion, teal = accumulation) for the S0 → S4 transition of 1Ru. | ||
Reduction of 1
Having explored the oxidation and metalation chemistry of 1, the ability of CAACMe to mediate multi-site reduction of hexachlorophosphazene was explored. Treating 1 with comparatively mild metal-based reductants zinc and manganese in the presence of one or two additional equivalents of CAACMe gave an intractable mixture of products, while the use of a stronger reductant magnesium led to complete loss of all resonances in the 31P{1H} NMR spectrum. In contrast, treatment of 1 with two equivalents of stoichiometric strong reductants, such as KC8 or alkali metal naphthalenides, in the presence of excess CAACMe led to partial consumption of 1, as assayed by 31P{1H} NMR spectrometry that showed the formation of a new A2X spin system with δP = 105.8 (A) and −4.3 (X) ppm, that is, only slightly shifted from those of 1 (δP = 104.3 and 4.6 ppm), suggestive of partial reduction of 1 to give a new species 2. Treatment of the reaction mixture with additional reductant and CAACMe did not lead to full conversion of 1 to 2 but to complete loss of signals in the 31P{1H} NMR spectrum, which, combined with the incomplete conversion when using appropriate stoichiometry, may be indicative of kinetically competitive over-reduction to an unidentified species.As silylated pyrazines have previously been demonstrated to effect the controlled reduction of phosphazenes,26 1,4-bis(trimethylsilyl)-dihydropyrazine (TMS-DHP, also called Mashima's reagent)74,75 was explored as a potential reductant for the selective generation of hemi-reduced species 2 (Scheme 4). Treatment of 1 with one equivalent of TMS-DHP and two equivalents of CAACMe in THF gave smooth conversion to 2, which could be isolated as a navy blue microcrystalline solid in good yield. Conveniently, 2 could also be prepared directly from hexachlorophosphazene without isolation of 1 in comparable yields (Scheme 4). The room temperature 1H NMR spectrum of 2 in C6D6 possesses only a single set of resonances for the CAACMe substituents, which suggests symmetric orientation of pyrrolidine rings or a fast dynamic process by which the faces of the rings interconvert (vide infra). Attempts to extend the synthetic methodology by either reducing 1 in the presence of an NHC, 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene,76 or by reducing A with CAACMe and external reductants were met with failure.
 | ||
| Scheme 4 Reduction of 1 to 2 using 1,4-bis(trimethylsilyl)-dihydropyrazine (TMS-DHP) in the presence of excess CAACMe and the direct synthesis of 2 from hexachlorophosphazene with TMS-DHP and CAACMe. | ||
The solid-state structure of 2, as determined by single crystal X-ray diffraction experiments, revealed an asymmetric structure with the two CAACMe moieties trans-oriented with respect to the phosphazene ring and with different relative orientation of their diisopropylphenyl rings (Fig. 7). Unlike in 1, the phosphazene ring is effectively planar and possesses notably short bonds between the reduced phosphorus centers and the nitrogen atom between them, P1–N1 = 1.610(2) Å and P2–N1 = 1.624(2) Å, which are shorter than those in 1 and suggest partial multiple bond character.77,78 Saliently, the reduced phosphorus centers exhibit highly inequivalent P–C bond lengths of 1.752(2) Å and 1.825(2) Å. While this feature is reproduced by DFT optimization of the structure, the difference in calculated bond lengths is much smaller, only 0.04 Å, suggesting that the experimentally observed disparity is not only due to the steric strain caused by the orientation of the diisopropylphenyl rings but also because of crystallographic packing effects.
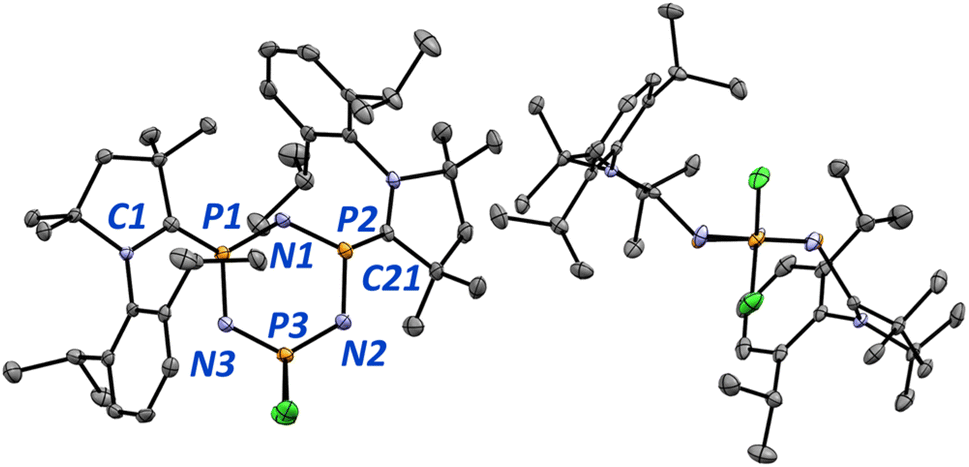 | ||
| Fig. 7 Molecular structure of 2 shown from two perspectives. Ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Select bond lengths (Å) and angles (°) for 2 [calculated values in square brackets]: P1–C1 1.752(2) [1.736]; P2–C21 1.825(2) [1.772]; C1–N4 1.356(3) [1.346]; C21–N5 1.328(3) [1.336]; P1–N1 1.610(2) [1.617]; N1–P2 1.624(2) [1.615]; P2–N2 1.658(2) [1.644]; N2–P3 1.560(2) [1.568]; C1–P1–N1 112.3(1) [114.1]; C21–P2–N1 112.2(1) [115.5]; P1–N1–P2 124.8(1) [122.1]; N1–P1–N3 116.4(1) [116.4]; C1–P1–N3 110.9(1) [112.0]. | ||
Given the planar arrangement of the phosphazene ring in 2, its unusual coloration, and its NMR spectra, which are incongruent with its solid-state structure, further computational analyses were performed. Despite the CAACMe fragments not being co-planar with the phosphazene ring, the Kohn–Sham HOMO and LUMO of 2 represent those of a seven-membered π-system comprising the carbon and nitrogen atoms of both CAACMe substituents, the reduced phosphorus centers, and the nitrogen atom connecting them (Fig. 8). This electronic structure rationalizes the intense coloration of 2, whose UV-Vis absorption spectrum in toluene (Fig. S16†) is characterized by the presence of a strong absorption centered at 561 nm (ε561 = 1.29 × 104 M−1 cm−1), as is typical for a π → π* transition. This assertion is further supported by TD-DFT calculations with implicit solvation for toluene, which show a high-intensity band at 536 nm with HOMO → LUMO character.
 | ||
| Fig. 8 Frontier Kohn–Sham orbitals of 2 (isovalue ±0.04 a.u.). | ||
Further experimental work was performed to rationalize the spectroscopic data for 2. First, low-temperature NMR studies were performed at 198 K in toluene-d8, revealing an exceptionally broad 1H NMR spectrum, while the 31P{1H} NMR spectrum shows decoalescence of the resonances attributed to the reduced phosphorus centers (Fig. S17 and S18†), with the resonance for the PCl2 moiety remaining sharp. These results indicate that the faces of the pyrrolidine rings interconvert at higher temperatures, the chemically most reasonable process for it being inversion about phosphorus. The plausibility of such process was probed with DFT calculations employing implicit solvation for toluene that identified a second isomer for 2 in which the two CAACMe moieties are cis-oriented with respect to the phosphazene ring (Fig. S19†). Because the two diisopropylphenyl rings have nearly identical relative orientation, this isomer has overall pseudo-Cs symmetry, 2Cs, and Gibbs free energy that is only 3.25 kcal mol−1 higher than the asymmetric global minimum, denoted hereafter 2C1. While phosphorus does not normally invert, the presence of strongly π-accepting substituents, such as CAAC, can significantly lower the energy of planar intermediates or transition states.79 In good agreement with the above, the Gibbs free energy of activation for inversion about phosphorus was calculated to be only 5.04 kcal mol−1 for 2Cs.
The reactivity of 2 was examined first by attempting to effect reduction of the final PCl2 moiety. Unfortunately, all attempts using a wide variety of reducing agents (Li, Na, K, Na/K, potassium naphthalenide, KC8, Mg, Mn, KFp, LiHBEt3, or Na2Fe(CO)4) under various conditions, in the presence or absence of additional equivalent(s) of CAACMe, led to degradation of 2, consistent with observations made during its attempted synthesis. Therefore, other potential reactivity was explored. As the lone pairs on phosphorus in 2 are engaged in conjugation, their chemical availability is questionable; thus, attempts to illicit oxidative chemistry analogous to that of 1 was explored (Scheme 5). Treatment of 2 with an excess of selenium resulted in rapid color change from blue to orange. Room temperature 31P{1H} NMR spectrometry in CDCl3 indicated complete consumption of 2 and formation of a new A2X spin system with δP = 16.3 (A) and 4.2 (X) ppm, the former resonance possessing distinct 77Se satellites that appear as multiplets. This may be due to second order coupling owing to magnetic inequivalence in the 77SeXXSe isotopologues, resulting in an ABMX spin system. The 77Se NMR spectrum of the product consists of a doublet of broad resonances centered at δSe = 230.5 ppm with a 1JP–Se coupling constant of 648 Hz. Subsequent structural analysis by single crystal X-ray diffraction confirmed the product to be the doubly oxidized 2Se (Fig. 9). Both the 1H NMR data and the solid-state structure of 2Se are consistent with C2 molecular symmetry, indicating that the CAACMe moieties are locked in the same rotational orientation. The C⋯Se distance in 2Se is slightly shorter than in 1Se, 2.911(2) Å, indicative of a sizable through-space interaction. The UV-Vis spectrum of 2Se in THF is analogous to that of 1Se, with an identical λmax = 387 nm and an extinction coefficient approximately double that of 1Se (ε387 = 1.15 × 104 M−1 cm−1), congruous with the presence of two chromophores per molecule of 2Se.
 | ||
| Scheme 5 Reactivity of 2 towards selenium and [Cp*RuCl]4. | ||
 | ||
| Fig. 9 Molecular structure of 2Se. Ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Select bond lengths (Å) and angles (°) for 2Se: P1–Se1 2.1212(8) [2.120]; P1–C1 1.845(2) [1.833]; C1–N4 1.305(3) [1.307]; P1–N1 1.601 [1.596]; P1–N2 1.620(2) [1.614]; P2–N2 1.565 [1.567]; C1–P1–Se1 94.18(7) [93.1]; C1–P1–N1 106.3 [106.7]; P1–N1–P1′ 124.0 [124.7]; N1–P1–N2 114.6 [114.6]. | ||
With the above reaction demonstrating the availability of the phosphorus lone pairs of 2 to participate in chemical reactions, the ability of 2 to serve as a ligand in transition metal chemistry was explored. Treatment with two equivalents of ClAu·SMe2 results in the rapid formation of a gold mirror and degradation of 2 to a brown intractable material; accordingly, 2 is also degraded by mild one electron oxidants such as trityl cation and ferrocenium, consistent with its extreme electron richness. In contrast, treatment of 2 with a half an equivalent of [Cp*RuCl]4 results in conversion to a new product 3, characterized by an AMX spin system in the 31P{1H} NMR spectrum of the reaction mixture. While an AMX spin system could be indicative of formation of a ruthenium phosphine complex akin to 1Ru, the dramatic downfield chemical shift of one of the resonances (δP = 254.1, 108.2, and 1.5 ppm) is inconsistent with this formulation (cf. δP = 99.1 ppm for 1Ru). Further, the 1H NMR spectrum of 3 is indicative of at least two Cp* containing compounds and three CAAC containing species, two major and one minor. One of the major products could be identified as free CAACMe, while the minor product was identified as [(CAACMe)(Cp*)RuCl], by comparison of known NMR spectra and independent synthesis (Fig. S20†), respectively.
The formulation and structure of 3 was determined by fractional crystallization from the reaction mixture and subsequent X-ray diffraction studies. As implied by the spectroscopic data, 3 arises from the formal loss of CAACMe from 2, followed by geminal dimetallation of the phosphorus center by a [Cp*RuCl]2 fragment to yield a spirocyclic bimetallic complex bridged by two chlorides and the endocyclic phosphorus center (Fig. 10). This unusual reaction is, to our knowledge, the first formal electrophilic displacement of a carbene. While the mechanism of the reaction was not probed with experiments, we note that 2 is stable for at least two weeks in C6D6 solution at room temperature and overnight at 353 K, and is unlikely to spontaneously release CAACMe. In agreement with this result, DFT calculations probing the dissociation of CAACMe from 2 resulted in a highly endergonic Gibbs free energy change of 21.57 kcal mol−1, indicating that the associated transition state, if any, would be even higher in energy. Therefore, one may postulate that the loss of CAACMe is induced by the introduction of [Cp*RuCl]4. A reasonable mechanistic proposal involves the dissociation of [Cp*RuCl]4 in solution to two units of [Cp*RuCl]2, as this process has a calculated Gibbs free energy change of only 13.88 kcal mol−1 in the gas phase. Subsequent association of the ruthenium centers in [Cp*RuCl]2 with the lone pairs on phosphorus and nitrogen in 2 would weaken the π-component of the C–P bond, leading to release of CAACMe and subsequent formation of 3 upon rearrangement of the bound [Cp*RuCl]2 fragment. Overall, the calculated Gibbs free energy change for the process 2 + 0.5 [Cp*RuCl]4 → 3 + CAACMe is slightly exergonic, −3.61 kcal mol−1. A full computational study of the mechanism of this process is beyond the scope of this work.
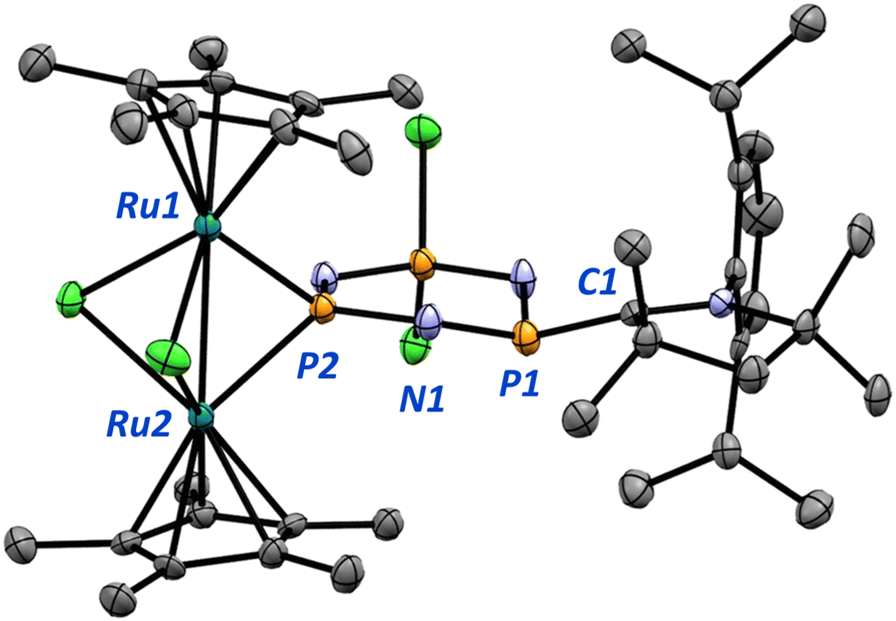 | ||
| Fig. 10 Molecular structure of 3. Ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity. Select bond lengths (Å) and angles (°) [calculated values in square brackets]: P1–C1 1.761(4) [1.722]; C1–N4 1.345(6) [1.344]; P1–N1 1.634(4) [1.609]; N1–P2 1.609(4) [1.603]; P2–Ru1 2.328(1) [2.312]; Ru1–Ru2 2.7339(6) [2.724]; Ru1–Cl3 2.4560(9) [2.498]; Ru1–P1–Ru2 71.89(3) [72.0]; Ru1–Cl3–Ru2 66.87(3) [65.9]. | ||
Thermal properties of 1 and 2
Having explored the small molecule chemistry of 1 and 2, and given the known utility of phosphazenes containing endocyclic heteroatoms as precursors to main-chain substituted poly-phosphazenes accessible by thermal ring opening polymerization (ROP),80–82 the thermal properties of 1 and 2 were explored by differential scanning calorimetry (DSC) to determine their viability as polymer precursors. The differential thermogram of 1 reveals a single exothermic event at 161 °C (onset, peak = 168 °C), which is not observed in subsequent heating cycles (Fig. S21†), indicative of either decomposition or thermal ROP without melting; in contrast, the differential thermogram of 2 features an irregular exothermic peak at 137 °C (onset, peak = 151 °C), followed by several others at higher temperatures, strongly suggestive of decomposition (Fig. S22†). Accordingly, heating 1 or 2 to near the exotherm onset temperature (165 °C and 140 °C, respectively) in a flame sealed Pyrex tube under vacuum resulted in the formation of a glassy red-orange solid and a black tar, respectively. While the latter is consistent with decomposition and was not further examined, the former may be indicative of the desired reactivity and attempts to analyze the material were made. Unfortunately, the material accessed by thermal treatment of 1 was completely insoluble in all common solvents and highly sensitive, fuming in air, which precluded further investigation. Hypothesizing that the intractability of this material may be due to its uncontrolled synthesis, an alternative route to the desired polymer was sought.Synthesis and reactivity of poly-1
As the reductive protocol to access 1 is exceptionally mild, the ability of CAACMe to effect the direct reduction of poly-chlorophosphazene was examined. End-capped linear poly-chlorophosphazene was synthesized by the living cationic polymerization of P,P,P-trichloro-N-trimethylsilyl-phosphoranimine83,84 by modification of literature procedures85,86 and immediately reacted with 2.2 eq. of CAACMe upon synthesis (Scheme 6). This resulted in immediate formation of a precipitate and a color change to bright orange, akin to the synthesis of 1. Following filtration to remove the precipitate, a bright yellow solid could be isolated from the reaction mixture by dropwise addition to n-hexane. Analysis of the material by 31P{1H} NMR spectrometry revealed absence of poly-chlorophosphazene and the presence of two broad resonances in a 1:2 ratio at δP = 77.1 and −31.6 ppm, that are shifted upfield to that of 1, which is consistently observed when comparing the chemical shift of phosphazene polymers to their (formal) cyclic monomers.80–82 This spectral data is suggestive of the regiospecific, direct reduction of poly-chlorophosphazene to the desired product poly-1. The regiospecificity and stoichiometry of the reduction can be readily explained on the basis of reduction potentials, whereby the reduction and ligation of one phosphorus center renders the two proximal PCl2 units along the main chain too electron rich for CAAC to effect their reduction; accordingly, addition of further CAACMe was not observed to bring about additional reduction events. Likewise, the 1H NMR spectrum of poly-1 features broad resonances attributable to embedding a CAAC fragment into a polymer backbone; however, CAAC derived small molecule impurities are also noted, which may arise due to free CAACMe and/or [CAACMe–Cl][Cl] becoming entrained in the polymer, and could unfortunately not be removed by reprecipitation of the polymer.
 | ||
| Scheme 6 Reduction of poly-chlorophosphazene to poly-1 and its subsequent reactivity with excess S8 and ClAu·SMe2. Dipp = 2,6-diisopropylphenyl. | ||
The UV-Vis spectrum of poly-1 is in good agreement with that observed for 1, with a λmax = 437 nm (cf. 438 nm for 1), suggesting that the chromophore of 1, that is, the P-CAACMe fragment, is present in poly-1; however, the extinction coefficient is reduced by an order of magnitude (ε437 = 3.5 × 102 M−1 cm−1), which can be explained by the different rotational orientations of the CAACMe moieties adopted by the flexible open chain poly-1, compared to 1. Examination of the DSC thermogram of poly-1 reveals a nearly identical exothermic event to 1 (onset = 161 °C) that is lost in subsequent heating cycles (Fig. S23†). Heating poly-1 to this temperature leads to formation of a red-brown glassy insoluble solid, as occurs when heating 1. While assigning the exact chemical events that occur are fraught, the crosslinking of poly-phosphazenes at or near the ROP temperature is a common issue11,80,81 and would be consistent with the observed macroscopic outcomes, which in turn strongly suggest that ROP of 1 is not a viable route to poly-1.
With poly-1 in hand, the viability of transposing the reactivity of 1 to the polymeric system to enable further derivatization was explored. Firstly, attempts to further reduce poly-1 were unsuccessful, as treatment with TMS-DHP in the presence of CAACMe led to an intractable mixture of products. In contrast, the oxidation of poly-1 with an excess of sulfur proceeds smoothly and rapidly, with quantitative conversion achieved within 5 minutes to yield poly-1S. The 31P{1H} NMR spectrum of poly-1S is consistent with oxidation at the phosphorus(III), as evidenced by the large upfield shift of the resonance to δP = 10.4 ppm, as was observed in the oxidation of 1 to 1S. The UV-Vis spectrum of poly-1S matches that of 1S, with a λmax = 341 nm, though with a reduced extinction coefficient (ε341 = 1.49 × 102 M−1 cm−1). The magnitude of the reduction, in comparison to the difference between the extinction coefficients of 1 and poly-1, is smaller, which may be caused by a more rigid structure facilitated by intramolecular through-space C⋯S interactions. The lone pairs in poly-1 are also chemically available for metalation reactions, as evidenced by reaction with ClAu·SMe2 that quantitatively converts poly-1 to a new species poly-1Au, as determined by 31P{1H} NMR spectrometry. Unfortunately, poly-1Au is highly light sensitive, readily depositing a gold mirror, which hinders its isolation and complete characterization. Despite this, the formation of poly-1Au provides evidence that poly-1 may be utilized as platform for the synthesis of metallopolymers that could be used as heterogenous catalysts or ceramic precursors.
Conclusions
In summary, this work presents the cyclic (alkyl)(amino)carbene mediated single-site and unprecedented multi-site reduction of hexachlorophosphazene, as well as a thorough experimental and computational study of the reactivity, structure, and bonding of the reduced species. The single-site reduced 1 exhibits oxidation and metalation chemistry typical of phosphines, however, the CAAC-centered LUMO bestows the resultant compounds with unusual properties, capable of engaging in acceptor-type (Z-type) secondary coordination sphere interactions, as well as serving as an acceptor orbital in photophysical transitions. Further, 1 may engage in more classical metal⋯arene secondary coordination donor-type interactions, possesses an encumbering steric profile, and should be electronically tunable by distal modification of the phosphazene ring, all promising traits for the use of 1 and its derivatives in transition metal catalysis. Further reduction of 1 in the presence of CAACMe results in the formation of 2, which features a non-planar π-system spanning the two CAACMe moieties and the reduced PNP fragment, that underscores the ability of reduced phosphazenes to engage in conjugation. Treatment of 2 with [Cp*RuCl]4 results in the formation of a spirocyclic bimetallic complex 3 arising from the unusual electrophilic displacement of a stable carbene. Importantly, the reductive methodology utilized in the synthesis of 1 can be readily applied to the regiospecific controlled reduction of poly-chlorophosphazene to poly-1, which represents a new class of inorganic polymers. The lone pair of the reduced phosphorus center in poly-1 is chemically available, as demonstrated by successful chemical oxidation and metalation in direct analogy to the chemistry of 1, which underscores the utility of this ring system to serve as an effective model for the chemistry of poly-1. Optimization of the synthesis of poly-1, as well as further studies of its properties and chemistry are currently underway.Data availability
The data supporting this article have been included as part of the ESI.† Experimental and characterization details for all new compounds, including spectroscopic data, ESI figures,† and full computational details (PDF). Coordinates for all optimized structures are available as a separate file (XYZ). Crystallographic data for complexes 1, 1S, 1Se, 1Au, 1Ru, 2, 2Se, 3, and [(CAAC)(Cp*)RuCl] are provided as a CIF file, or can be obtained free of charge from https://www.ccdc.cam.ac.uk/structures/ CCDC: 2383407–2383415 (CIF).Author contributions
E. L. designed and carried out the synthetic work. B. P. acquired and processed the SC-XRD data, E. L. carried out the structure refinement. DFT studies were performed by R. S., E. L. and H. T. Supervision was shared between E. L., I. M. and H. T. The manuscript was drafted by E. L. and revised by E. L. and H. T., and approved by all authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
I. M. thanks the Canadian Government for a Canada 150 Research Chair, NSERC for a Discovery Grant, and the University of Victoria for start-up funds. E. L. thanks NSERC for a Postdoctoral Fellowship. H. M. T. and R. S. thank the University of Jyväskylä for funding. Digital Research Alliance of Canada and the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533) are acknowledged for access to computing resources.References
- J. von Liebig, Ann. Pharm., 1834, 11–12 Search PubMed.
- D. P. Craig and N. L. Paddock, Nature, 1958, 181, 1052–1053 Search PubMed.
- A. E. Reed and P. V. Schleyer, J. Am. Chem. Soc., 1990, 112, 1434–1445 Search PubMed.
- A. B. Chaplin, J. A. Harrison and P. J. Dyson, Inorg. Chem., 2005, 44, 8407–8417 Search PubMed.
- H. Sun, J. Am. Chem. Soc., 1997, 119, 3611–3618 Search PubMed.
- H. Stokes, Am. Chem. J., 1895, 17, 275–291 Search PubMed.
- H. Stokes, Am. Chem. J., 1896, 18, 629–663 Search PubMed.
- H. Stokes, Am. Chem. J., 1897, 19, 782–796 Search PubMed.
- H. R. Allcock, R. L. Kugel and K. J. Valan, Inorg. Chem., 2002, 5, 1709–1715 Search PubMed.
- H. R. Allcock and R. L. Kugel, J. Am. Chem. Soc., 2002, 87, 4216–4217 Search PubMed.
- S. Rothemund and I. Teasdale, Chem. Soc. Rev., 2016, 45, 5200–5215 Search PubMed.
- K. S. Ogueri, K. S. Ogueri, H. R. Allcock and C. T. Laurencin, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.:Mater., Process., Meas., Phenom., 2020, 38, 030801 Search PubMed.
- X. Zhou, S. L. Qiu, X. W. Mu, M. T. Zhou, W. Cai, L. Song, W. Y. Xing and Y. Hu, Composites, Part B, 2020, 202, 108397 Search PubMed.
- H. R. Allcock and N. L. Morozowich, Polym. Chem., 2012, 3, 578–590 Search PubMed.
- H. R. Allcock, Polymer, 2022, 249, 124761 Search PubMed.
- H. R. Allcock, Chem. Rev., 1972, 72, 315 Search PubMed.
- H. R. Allcock and C. Chen, J. Org. Chem., 2020, 85, 14286–14297 Search PubMed.
- C. W. Allen, Chem. Rev., 2002, 91, 119–135 Search PubMed.
- A. M. Caminade, Chem. Soc. Rev., 2016, 45, 5174–5186 Search PubMed.
- H. Henke, O. Bruggemann and I. Teasdale, Macromol. Rapid Commun., 2017, 38, 1600644 Search PubMed.
- S. M. Al-Rafia, M. J. Ferguson and E. Rivard, Inorg. Chem., 2011, 50, 10543–10545 Search PubMed.
- H. R. Allcock, L. J. Wagner and M. L. Levin, J. Am. Chem. Soc., 2002, 105, 1321–1327 Search PubMed.
- P. P. Greigger and H. R. Allcock, J. Am. Chem. Soc., 2002, 101, 2492–2493 Search PubMed.
- P. R. Suszko, R. R. Whittle and H. R. Allcock, J. Chem. Soc., Chem. Commun., 1982, 649–650 Search PubMed.
- V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 Search PubMed.
- E. A. LaPierre, L. K. Watanabe, B. O. Patrick, J. M. Rawson, H. M. Tuononen and I. Manners, J. Am. Chem. Soc., 2023, 145, 9223–9232 Search PubMed.
- E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2024, 146, 6326–6335 Search PubMed.
- Y. Wang and G. H. Robinson, J. Am. Chem. Soc., 2023, 145, 5592–5612 Search PubMed.
- D. Munz, Organometallics, 2018, 37, 275–289 Search PubMed.
- S. Kumar Kushvaha, A. Mishra, H. W. Roesky and K. Chandra Mondal, Chem.–Asian J., 2022, 17, e202101301 Search PubMed.
- A. K. Eckhardt, M. Y. Riu, M. Ye, P. Muller, G. Bistoni and C. C. Cummins, Nat. Chem., 2022, 14, 928–934 Search PubMed.
- C. Hering, A. Schulz and A. Villinger, Chem. Sci., 2014, 5, 1064–1073 Search PubMed.
- R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 Search PubMed.
- E. Niecke, M. Nieger and F. Reichert, Angew. Chem., Int. Ed., 2003, 27, 1715–1716 Search PubMed.
- J. L. Martinez, S. A. Lutz, D. M. Beagan, X. Gao, M. Pink, C. H. Chen, V. Carta, P. Moenne-Loccoz and J. M. Smith, ACS Cent. Sci., 2020, 6, 1572–1577 Search PubMed.
- A. K. Eckhardt, M. Y. Riu, P. Muller and C. C. Cummins, Inorg. Chem., 2022, 61, 1270–1274 Search PubMed.
- A. Velian and C. C. Cummins, Science, 2015, 348, 1001–1004 Search PubMed.
- R. Ahlrichs, M. Bär, H. S. Plitt and H. Schnöckel, Chem. Phys. Lett., 1989, 161, 179–184 Search PubMed.
- I. K. Ahmad and P. A. Hamilton, J. Mol. Spectrosc., 1995, 169, 286–291 Search PubMed.
- J. Curry, L. Herzberg and G. Herzberg, J. Chem. Phys., 1933, 1, 749 Search PubMed.
- J. Curry, L. Herzberg and G. Herzberg, Z. Phys., 1933, 86, 348–366 Search PubMed.
- A. A. Starikova, N. M. Boldyreva, R. M. Minyaev, A. I. Boldyrev and V. I. Minkin, ACS Omega, 2018, 3, 286–291 Search PubMed.
- F. C. Wyse, W. Gordy and E. L. Manson, J. Chem. Phys., 1972, 57, 1106 Search PubMed.
- R. M. Atkins and P. L. Timms, Inorg. Nucl. Chem. Lett., 1978, 14, 113–115 Search PubMed.
- C. Zhu, A. K. Eckhardt, A. Bergantini, S. K. Singh, P. R. Schreiner and R. I. Kaiser, Sci. Adv., 2020, 6, eaba6934 Search PubMed.
- C. Zhu, A. K. Eckhardt, S. Chandra, A. M. Turner, P. R. Schreiner and R. I. Kaiser, Nat. Commun., 2021, 12, 5467 Search PubMed.
- D. Tofan and A. Velian, ACS Cent. Sci., 2020, 6, 1485–1487 Search PubMed.
- B. E. Turner and J. Bally, Astrophys. J., 1987, 321, L75 Search PubMed.
- S. Roy, K. C. Mondal, S. Kundu, B. Li, C. J. Schurmann, S. Dutta, D. Koley, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Chem.–Eur. J., 2017, 23, 12153–12157 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 Search PubMed.
- S. W. Bartlett, S. J. Coles, D. B. Davies, M. B. Hursthouse, H. Ibisoglu, A. Kilic, R. A. Shaw and I. Un, Acta Crystallogr., Sect. B, 2006, 62, 321–329 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 Search PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 Search PubMed.
- J. Meija, T. B. Coplen, M. Berglund, W. A. Brand, P. De Bièvre, M. Gröning, N. E. Holden, J. Irrgeher, R. D. Loss, T. Walczyk and T. Prohaska, Pure Appl. Chem., 2016, 88, 293–306 Search PubMed.
- D. W. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51–54 Search PubMed.
- U. Beckmann, D. Süslüyan and P. C. Kunz, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2061–2070 Search PubMed.
- G. P. Junor, J. Lorkowski, C. M. Weinstein, R. Jazzar, C. Pietraszuk and G. Bertrand, Angew. Chem., Int. Ed., 2020, 59, 22028–22033 Search PubMed.
- P. W. Codding and K. A. Kerr, Acta Crystallogr., Sect. B, 1978, 34, 3785–3787 Search PubMed.
- P. W. Codding and K. A. Kerr, Acta Crystallogr., Sect. B, 1979, 35, 1261–1263 Search PubMed.
- P. Perez-Galan, N. Delpont, E. Herrero-Gomez, F. Maseras and A. M. Echavarren, Chem.–Eur. J., 2010, 16, 5324–5332 Search PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 Search PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 Search PubMed.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 Search PubMed.
- A. R. Chianese, X. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663–1667 Search PubMed.
- L. Perrin, E. Clot, O. Eisenstein, J. Loch and R. H. Crabtree, Inorg. Chem., 2001, 40, 5806–5811 Search PubMed.
- T. D. Tilley, R. H. Grubbs and J. E. Bercaw, Organometallics, 1984, 3, 274–278 Search PubMed.
- B. K. Campion, R. H. Heyn and T. D. Tilley, J. Chem. Soc., Chem. Commun., 1988, 278–280 Search PubMed.
- P. J. Fagan, M. D. Ward and J. C. Calabrese, J. Am. Chem. Soc., 1989, 111, 1698–1719 Search PubMed.
- J. M. Herbert, Phys. Chem. Chem. Phys., 2024, 26, 3755–3794 Search PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 Search PubMed.
- C. Adamo, T. Le Bahers, M. Savarese, L. Wilbraham, G. García, R. Fukuda, M. Ehara, N. Rega and I. Ciofini, Coord. Chem. Rev., 2015, 304, 166–178 Search PubMed.
- H. Tsurugi and K. Mashima, Acc. Chem. Res., 2019, 52, 769–779 Search PubMed.
- W. Kaim, J. Am. Chem. Soc., 1983, 105, 707–713 Search PubMed.
- L. Jafarpour, E. D. Stevens and S. P. Nolan, J. Organomet. Chem., 2000, 606, 49–54 Search PubMed.
- P. Pyykko, J. Phys. Chem. A, 2015, 119, 2326–2337 Search PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 Search PubMed.
- C. C. Levin, J. Am. Chem. Soc., 1975, 97, 5649–5655 Search PubMed.
- I. Manners, H. R. Allcock, G. Renner and O. Nuyken, J. Am. Chem. Soc., 1989, 111, 5478–5480 Search PubMed.
- J. A. Dodge, I. Manners, H. R. Allcock, G. Renner and O. Nuyken, J. Am. Chem. Soc., 1990, 112, 1268–1269 Search PubMed.
- M. Liang and I. Manners, J. Am. Chem. Soc., 1991, 113, 4044–4045 Search PubMed.
- E. Niecke and W. Bitter, Inorg. Nucl. Chem. Lett., 1973, 9, 127–129 Search PubMed.
- B. Wang, E. Rivard and I. Manners, Inorg. Chem., 2002, 41, 1690–1691 Search PubMed.
- C. H. Honeyman, I. Manners, C. T. Morrissey and H. R. Allcock, J. Am. Chem. Soc., 1995, 117, 7035–7036 Search PubMed.
- H. R. Allcock, C. A. Crane, C. T. Morrissey, J. M. Nelson, S. D. Reeves, C. H. Honeyman and I. Manners, Macromolecules, 1996, 29, 7740–7747 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2383407–2383415. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07559e |
| ‡ Ian Manners: deceased December 3rd, 2023. |
| This journal is © The Royal Society of Chemistry 2025 |