
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalysis of a LiF-rich SEI by aromatic structure modified porous polyamine for stable all-solid-state lithium metal batteries†
Lijie
Dai
a,
Min
Cai
b,
Xuanyi
Zhou
a,
Weizhong
Liang
a,
Zishao
Zhao
a,
Zixiang
Xia
a,
Fenfen
Huang
a,
Jie
Jiang
 *ac,
Wenjuan
Jiang
*a,
Biao
Zhang
*ac and
Zengsheng
Ma
ac
*ac,
Wenjuan
Jiang
*a,
Biao
Zhang
*ac and
Zengsheng
Ma
ac
aSchool of Materials Science and Engineering, Xiangtan University, Xiangtan 411105, China. E-mail: jiangjie@xtu.edu.cn; wenjuanjiang@xtu.edu.cn; biaozhang@xtu.edu.cn
bChina Nuclear Power Engineering Co., Ltd., Beijing, 100048, China
cKey Laboratory of Low Dimensional Materials and Application Technology, Ministry of Education, Xiangtan University, Hunan 411105, China
First published on 8th January 2025
Abstract
Poly(ethylene oxide) (PEO)-based solid-state polymer electrolyte (SPE) is a promising candidate for the next generation of safer lithium-metal batteries. However, the serious side reaction between PEO and lithium metal and the uneven deposition of lithium ions lead to the growth of lithium dendrites and the rapid decline of battery cycle life. Building a LiF-rich solid electrolyte interface (SEI) layer is considered to be an effective means to solve the above problems. Here, porous organic polymers (POPs) with aromatic structures and non-aromatic structures were synthesized and introduced into the PEO-based SPE as fillers to explore the effect of aromatic structures on LiF-rich SEI formation. The results show that the POPs containing aromatic groups could catalyze the decomposition of LiTFSI to form a stable LiF-rich SEI layer and inhibit the growth of lithium dendrites. The discharge capacity of the LFP/Li battery is 103 mA h g−1 after 500 cycles at 1C (100 °C). It provides a promising way to improve the stability of the solid electrolyte matrix and SEI layer.
Introduction
All-solid-state lithium metal batteries (ASSLBs) are considered the next generation of high safety and high specific capacity energy storage devices due to the lowest potential of the lithium metal anode (−3.040 V vs. the standard hydrogen electrode) and high specific energy (3860 mA h g−1).1–3 Solid polymer electrolyte (SPE) is one of the important components widely used because of its high flexibility and easy preparation among all kinds of solid electrolytes.4–6 In particular, polyethylene oxide (PEO)-based SPEs have broad application prospects due to their low manufacturing cost, large permittivity, and good mechanical properties.7–9 However, the serious side reaction between PEO and lithium metal and the uneven deposition of lithium ions lead to the growth of lithium dendrites, resulting in significant capacity degradation and poor cycle performance.10,11 The practical application of PEO-based SPE in high-energy density solid-state lithium metal batteries (LMBs) is seriously hindered.Aiming at the tricky problem of the Li/PEO interface, a LiF-rich solid electrolyte interface (SEI) layer is constructed to solve the above problem.12 The low Li+ diffusion barrier and excellent electronic insulation of LiF are used to promote Li+ transfer and uniform deposition in LMBs.13 Zhao et al.14 enhanced battery performance by tuning the spatial site resistance of the electrolyte to produce LiF-rich SEIs. Zhang et al.15 formed a stable Li3N–LiF interface between PEO-based SPE and a lithium anode by adding a lithium nitrate (LiNO3) filler; LiNO3 promoted the uniform deposition of lithium. At the same time, the solid-state electrolyte itself cannot be ignored. Fang et al.16 improved battery performance through aluminum-oxo molecular clusters (AlOCs) used to reconstruct an efficient conductive network in the continuous amorphous structural domains of PEO. Lu et al.17 developed stimuli-responsive pre-grafted solid polymer electrolytes (PGSPEs) by utilizing the unique property of stimulus-induced phase transition in polymers to affect the interfacial structure. The polymer is capable of forming a void-free SSE electrode interface and an effective permeable ion transport network inside the electrode. These strategies have positive effects on the construction of PEO electrolytes and LiF-SEI layers. However, the regulating mechanism of filler chemical structures on the stability of SEI layers remains unclear, and few people discuss what kind of chemical structure is conducive to the construction of a LiF-rich SEI layer. The reason is that it is difficult to regulate the chemical structure of fillers. Therefore, a simple method is needed to adjust the chemical structure of fillers to improve the stability of the SEI layer.
Porous organic polymers (POPs) are widely used due to their large surface area, high stability, flexible synthesis strategy, and functional designability.18–21 POPs have porous structural controllability and functional designability and their surface structural properties can be regulated at the molecular level.22–25 Therefore, POP groups can be designed to interact with TFSI− to catalyze the generation of LiF and construct a LiF-rich SEI layer. Practice has proved that the introduction of POP fillers into PEO to prepare composite polymer electrolyte (CPE) is an effective way to improve the stability of the interface between PEO and the lithium anode.26,27
In this work, two kinds of POPs by polymerization of hydantoin and phenytoin with melamine were prepared as PEO-based CPE fillers (Fig. 1). The effect of aromatic structure of POPs on the stability of the SEI layer was studied. It was found that POPs containing aromatic groups could catalyze the decomposition of LiTFSI to form a stable LiF-rich SEI layer and inhibit the growth of lithium dendrites. Meanwhile, the rigid structure of the aromatic group in POPs can improve the thermal stability and structural stability of the electrolyte film and make the electrolyte film usable at high temperature. A Li/Li symmetric cell cycled for more than 2000 h at 0.1 mA cm−2 (60 °C). The initial discharge capacity of the assembled LiFePO4 (LFP)/Li battery is 159.7 mA h g−1 for 500 cycles at 1C at 100 °C. The CPE can still maintain its complete shape even at 120 °C. It provides a promising way to improve the stability and aspect of the solid electrolyte matrix and SEI layer.
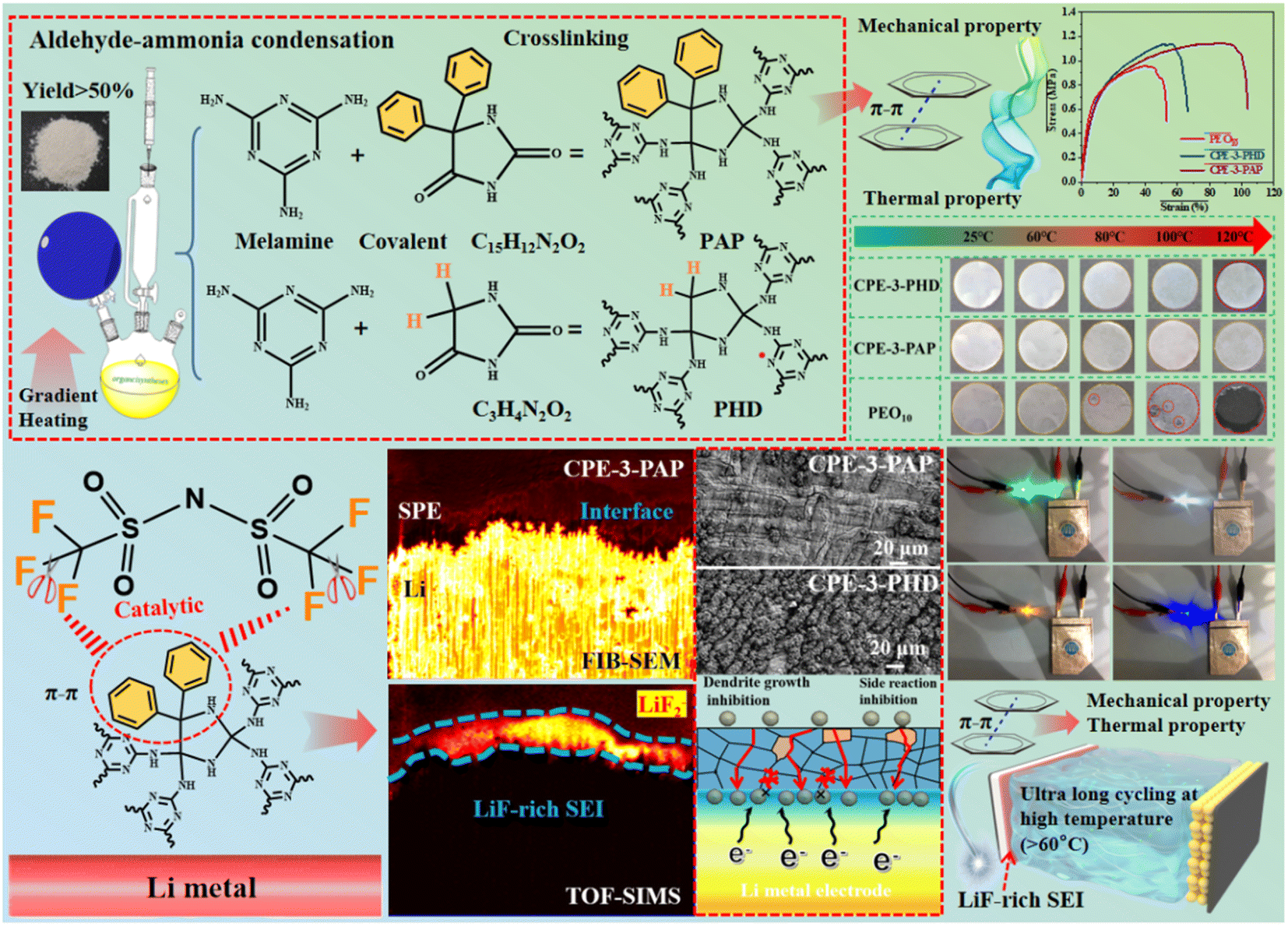 | ||
| Fig. 1 Design strategy of the PAP/PHD and CPE membrane preparation process and schematic illustration of the interface stabilization mechanism. | ||
Results and discussion
PAP/PHD structural characterization
The microstructure of PAP and PHD was investigated by SEM. The SEM images (Fig. 2a and b) show that the PAP and PHD powders have porous and loose structures. Fig. S1† shows the surface morphology of PAP and PHD powders at higher magnification. Fourier transform infrared spectroscopy (FTIR) confirmed that the carbonyl peak of phenytoin and hydantoin disappeared at 1700 and 1695 cm−1, and the peaks of PAP and PHD at 1547 and 1538 cm−1 proved the existence of the triazine ring which is derived from melamine (Fig. 2c and d).28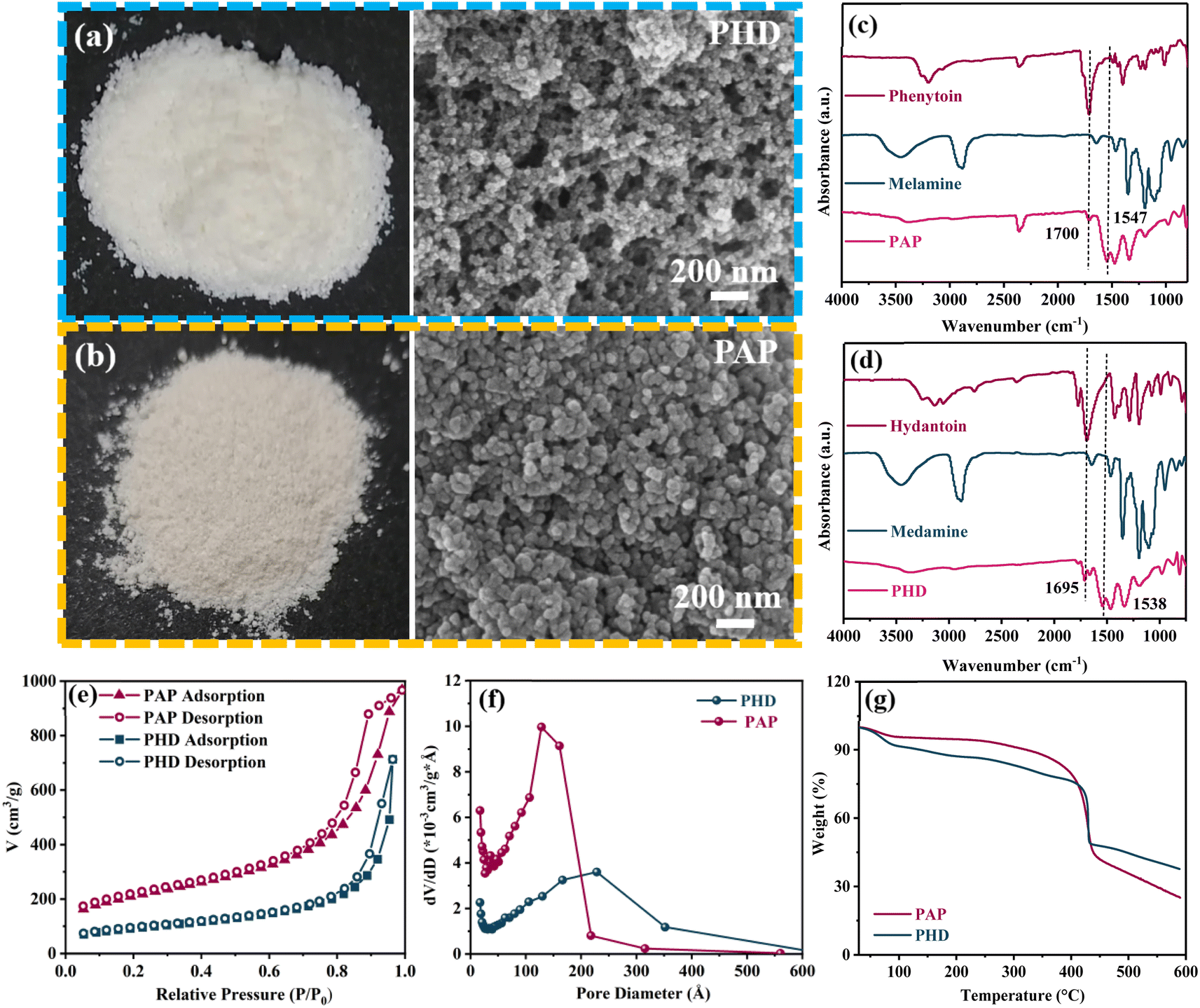 | ||
| Fig. 2 Photograph and SEM image of (a) PHD and (b) PAP. FTIR spectra in the wavenumber range of 4000–800 cm−1 of (c) PAP and (d) PHD. (e) Nitrogen isotherms at 298 K of PHD and PAP. (f) Pore size distribution of PHD and PAP. (g) TGA curves of PAP and PHD. | ||
Unlike PHD, the medium bimodal in Fig. 2c between 1400 and 1600 cm−1 refers to the characteristic infrared peak of the phenyl group, which is found for the phenytoin monomer and PAP. This confirms the successful synthesis of PAP equipped with a polar phenyl group. The introduction of the aromatic electron-withdrawing group, specifically the phenyl group, facilitates the polarization effect of lithium salts, thereby catalyzing the cleavage of the C–F bond in lithium salts and promoting the formation of inorganic LiF, which enhances the stability of the SEI.
Wide-angle X-ray diffraction (XRD) shows that the two polyamine networks have wide diffraction peaks, which proves that the two polyamine networks are essentially amorphous structures (Fig. S2†).29
The specific area and porous characteristics of PAP and PHD were also characterized by the N2 adsorption–desorption technique (Fig. 2e, f). The Brunauer–Emmett–Teller (BET) specific surface areas of PAP and PHD were 730 and 326.4 m2 g−1, and the pore volumes of PAP and PHD were 1.3 m3 g−1 and 0.8 m3 g−1. The pore size of PAP (12.8 nm) is smaller than that of PHD (23 nm) (Fig. 2f). It can be seen that PAP has a larger specific surface area, higher pore volume and a smaller pore diameter. The reason may be that the rigid benzene ring in PAP expands the pore volume and makes pore channels more abundant.23–25
Thermogravimetric (TG) analysis was used to test the thermal stability of PAP and PHD, as shown in Fig. 2g. PHD and PAP become weightless due to the evaporation of absorbed water below 100 °C. PAP and PHD showed a weight loss of 23.2 wt% and 20.5 wt% before 400 °C, and it can be seen that PAP is more stable than PHD, and PAP containing a benzene ring has high thermal stability.
Physical and electrochemical performance characterization of the CPEs
Different qualities of PAP/PHD were introduced into the PEO polymer matrix to prepare CPEs. Fig. 3a, S3 and S4† show the surface morphologies of CPE-3-PAP, CPE-3-PHD, and PEO10. CPE has no obvious cavities or cracks proving that PAP and PHD have good dispersion in the polymer matrix. The corresponding EDS mapping shows that elements C and N are evenly distributed which indicated that PAP and PHD are uniformly dispersed in the polymer matrix without obvious agglomeration. | ||
| Fig. 3 SEM morphology of (a) surface and (b) cross section of the CPE-3-PAP membrane. (c) EDS mapping of the CPE-3-PAP membrane. (d) X-ray diffraction, (e) DSC curves, (f) ionic conductivity, and (g) FT-IR spectra at 1400–1150 cm−1 of CPE-3-PAP, CPE-3-PHD, and PEO10. (h) Chronoamperometry curve for CPE-3-PAP at 60 °C with a polarization voltage of 10 mV and EIS plots before and after the polarization (inset). (i) LSV curves of CPE-3-PAP, CPE-3-PHD and PEO10 at a scanning rate of 10 mV s−1 at 60 °C. (j) Photographs of CPE-3-PAP, CPE-3-PHD and PEO10 after heat treatment at various temperatures for 1 h. | ||
Conductivity is an important parameter of polymer electrolyte and can reflect the transport capacity of Li+. The conductivity of CPEs with different PHD and PAP contents is shown in Fig. S5.† The ionic conductivity of CPE-3-PAP and CPE-3-PHD is higher than that of CPE-x-PAP and CPE-x-PHD (x = 1, 5). The small amount of PAP and PHD packing is not enough to build a continuous Li+ transport channel when too little packing is added. And there are fewer PEO amorphous regions, resulting in an insignificant increase in ionic conductivity. In contrast, when the filler addition was too high, a large amount of PAP and PHD fillers would reduce the free volume for the movement of PEO segments and hinder the movement of PEO segments, which was unfavorable for Li+ migration.27,30 In general, the transport capacity of Li+ is not only related to the conductivity of CPEs, but also related to the crystallinity of the polymer. Ion migration in PEO-based electrolytes occurs primarily in the amorphous region. Conventional PEOs have high crystallinity and reduced amorphous regions, which hinders Li+ transport and results in poor ionic conductivity. Fillers help reduce the crystallinity of PEO and increase the amorphous region of PEO, which helps ionic conductivity and improves Li+ transport.30 XRD was used to characterize the motility of PEO chains. XRD patterns in Fig. 3d reflect that the strong characteristic crystallization peaks of PEO are located at 19.3° and 23.2°, indicating that PEO10 has high crystallinity. No obvious crystallization peaks were observed in the spectra of CPE-3-PHD and CPE-3-PAP, due to the decrease of PEO crystallinity. This phenomenon was further supported by DSC measurements. As shown in Fig. 3e, the glass transition temperature of CPE-3-PAP was −42.28 °C which was below that of CPE-3-PHD (−41.5 °C) and PEO10 (−41.2 °C). It was confirmed that the addition of PAP disrupts the crystallization of the –EO chain segment and promotes the movement of the PEO chain, resulting in a decrease in crystallinity and the “Tg” value. On the other hand, the Tm of CPE-3-PAP was 49.82 °C, while the Tm of CPE-3-PHD and PEO10 was 59.04 and 59.98 °C, respectively. Table S1† shows the ionic conductivity of CPE-3-PAP, CPE-3-PHD, and PEO10 at different temperatures. Compared with PEO10 (1.2 × 10−6 S cm−1 at 25 °C), the ionic conductivities of CPE-3-PHD and CPE-3-PAP were 9.86 × 10−6 S cm−1 and 2.49 × 10−5 S cm−1 (Fig. 3b). The ionic conductivity of CPE-3-PHD and CPE-3-PAP increased to 2.07 × 10−4 S cm−1 and 3.32 × 10−4 S cm−1, respectively, while the ionic conductivity of PEO10 was only 1.11 × 10−4 S cm−1 at 60 °C.
In addition to the motility of the PEO chain segment affecting the conductivity of lithium ions, the number of free lithium ions is also one of the factors. The interaction between fillers and LiTFSI was analyzed using FTIR spectra and Gauss–Lorentz fitting lines. As shown in Fig. 3g, PEO10 had the characteristic peak of –SO2 stretching at 1331.4 cm−1, while in the FTIR spectra of CPE-3-PAP and CPE-3-PHD, the characteristic peak of –SO2 stretching moved to 1334.9 cm−1 and 1334.4 cm−1. At the same time, –CF3 symmetrical stretch shifts from 1243.1 cm−1 (PEO10) to 1249 (CPE-3-PHD) and 1249.9 (CPE-3-PAP) cm−1. In addition, –CF3 asymmetrically stretched from 1200.1 cm−1 (PEO10) to 1198.1 cm−1 (CPE-3-PHD) and 1197.1 cm−1 (CPE-3-PAP).27,30 PAP causes a more obvious peak position shift of –SO2 and –CF3, indicating that the aromatic ring in PAP has strong interaction with LiTFSI, which results in an increase of free Li+ and improves ionic conductivity.
As an important parameter of battery electrochemistry, the Li+ transference number (tLi+) also plays a critical role in improving the electrochemical performance. Fig. 3h shows that the tLi+ of CPE-3-PAP is 0.36, while the tLi+ of PEO10 and 3-PHD is only 0.19 and 0.2 (Fig. S6†). The electrons on TFSI− can conjugate with the benzene ring, which can weaken the interaction between Li+ and TFSI− and promote the migration of Li+.31–33
The electrochemical stability of CPEs is studied through the linear sweep voltammetry (LSV) test. As shown in Fig. 3i, compared to CPE-3-PHD (4.7V) and PEO10 (4.1V), CPE-3-PAP can be stably charged up to above 4.8 V. Lewis acid–base interaction may occur between PAP and PEO, which lowered the highest occupied molecular orbital (HOMO) of PEO and increased the antioxidant stability of CPE.34
The extensibility and elasticity of CPE will affect the battery stability. An ideal CPE must have sufficient mechanical properties to inhibit the puncture of Li dendrites. Fig. S7† shows the strain stress curve of CPE. The maximum strain of CPE-3-PAP, CPE-3-PHD, and PEO10 is 95%, 66.5%, and 40.5%. CPE-3-PAP has higher tensile strength and better ductility compared with CPE-3-PHD and PEO10, because PAP contains conjugated electron-rich benzene rings that can interact with PEO, leading to potential physical crosslinking and formation of a strong network between PEO and PAP.35,36
Besides good mechanical strength, thermal stability is another important indicator of CPE for practical application in ASSLBs. The thermogravimetric analysis (TGA) was used to evaluate the thermal stability of the electrolytes at 50–600 °C. As shown in Fig. S8,† PEO10 and CPE-3-PHD decompose after 366 °C and 397 °C, while CPE-3-PAP decomposition occurred after 402 °C. The rigid framework of the benzene ring in PAP delays the decomposition of PEO and improves the thermal stability of CPE. Furthermore, Fig. 3j shows the photos of CPEs at 25, 60, 80, 100, and 120 °C. PEO10 partially melts at 80 °C. In addition, CPE-3-PHD also melts and becomes transparent at 100 °C, and its shape shrinks rapidly and melts at 120 °C. In contrast, CPE-3-PAP retains its original form and does not melt, indicating that the introduction of aromatic groups in CPE significantly improved the thermal stability of CPE.
Li plating/stripping stability and dendrite suppression capability of CPEs
The interfacial compatibility between CPEs and lithium metal was investigated using the Li/CPEs/Li cell. Fig. 4a exhibits the plating/stripping curves of the Li/CPEs/Li coin cell at a current density of 0.1 mA cm−2 and an aerial capacity of 0.1 mA h cm−2. The initial polarization voltages of CPE-3-PAP, CPE-3-PHD, and PEO10 were 30 mV, 70 mV, and 80 mV, respectively. CPE-3-PAP exhibits excellent long-term cycling stability for over 2000 h, while CPE-3-PHD and PEO10 had short circuit after 860 and 340 hours. The results show that the aromatic groups in PAP have a better optimization effect on the anode interface of CPE than the groups in PHD while PHD also shows a better performance than PEO10. | ||
| Fig. 4 (a) Constant current polarization curve fragment of CPE-3-PAP, CPE-3-PHD, and PEO10 measured in a Li/CPE/Li coin cell at 0.1 mA cm−2 at 60 °C. (b–d) Corresponding partial enlarged images at different times. (e) Constant current polarization curve fragment of CPE-3-PAP, CPE-3-PHD, and PEO10 measured at 0.1 mA cm−2 at 100 °C. (f) Rate performance of symmetric lithium cells with CPE-3-PAP, CPE-3-PHD and PEO10. (g–i) Surface morphologies of the Li electrode obtained from Li/CPE-3-PAP/Li, Li/CPE-3-PHD/Li, and Li/PEO10/Li symmetric cells cycled for about 400 h. (j) Voltage profiles of the Li/Cu deposition process. (k) Tafel plot for Li plating/stripping of the Li/Li symmetric cells. | ||
We believe that CPE-3-PHD shows improved electrochemical performance and thermal stability compared to PEO, even though CPE-3-PHD has lower conductivity and lithium ion mobility than CPE-3-PAP. It should be that the SEI component of PHD is also improved; just compared with PAP it is worse. However, compared with PEO, there is still a significant improvement. Therefore, the CPE-3-PHD samples still have good long cycle stability at 100 °C. The thermal stability of the CPE-3-PHD samples is also improved. The amplified voltage curve (Fig. 4b–d) shows that the CPE-3-PAP batteries exhibit a flat voltage platform throughout the plating/stripping process, with an overpotential of approximately 40 mV, lower than that of CPE-3-PHD and PEO10, demonstrating the highly reversible and stable lilting/stripping process. Besides, CPE-3-PAP exhibits better interfacial stability than CPE-3-PHD at 100 °C (Fig. 4e). CPE-3-PAP exhibits excellent long-term cycling stability and small overpotential for over 1000 h while PEO10 was short-circuited at 288 h. Moreover, the time–voltage curves of lithium symmetric cells cycled at different current densities (0.05, 0.1, 0.2, and 0.3 mA cm−2) are shown in Fig. 4f. With the increase of current density, all voltage platforms in CPE-3-PAP are stable without significant fluctuation while CPE-3-PHD and PEO10 have large polarization and fluctuation.
In order to explore the reasons for the difference in the cycle times of Li/CPEs/Li symmetric batteries cells, the surface morphology of lithium metal after 400 h of cycling was observed by SEM. As shown in Fig. 4g–i. The lithium surface of CPE-3-PHD has a large number of lithium dendrites growing like weeds. The lithium surface of PEO10 has obvious lithium dendrites, resulting from uneven and loose deposition of lithium ions. This means that lithium dendrites grow uncontrollably, which eventually lead to a short circuit in the battery. In contrast, CPE-3-PAP has no obvious lithium dendrites. It was confirmed that PAP makes it possible to form a more stable SEI layer on the CPE/Li surface which could promote the uniform deposition of lithium.
In order to explore the influence of SEI layers formed at different Li/CPE interfaces on Li+ nucleation and Li+ conduction, the interfacial transfer kinetics of Li+ in the SEI layers were systematically studied (Fig. 4j). The initial overpotential of CPE-3-PAP, CPE-3-PHD, and PEO10 is 13, 30, and 59 mV, respectively. The CPE-3-PAP SEI layer shows the lowest initial overpotential, indicating that the SEI layer formed at the Li/CPE-3-PAP interface can reduce the energy barrier of Li+ interface travel, which is conducive to nucleation in the initial Li plating stage and greatly enhances the interface stability of subsequent Li growth.37,38
The exchange current density (i0) is calculated by fitting with the Tafel plot of the three electrolytes to further investigate Li+ transfer kinetics at the SEI layer (Fig. 4k). The exchange current density of the CPE-3-PAP SEI layer (I0 = 0.34 mA cm−2) was much higher than that of the CPE-3-PHD SEI layer (I0 = 0.19 mA cm−2) and PEO10 SEI layer (I0 = 0.14 mA cm−2), indicating faster interfacial transport of Li+. This further confirmed that the CPE-3-PAP SEI can promote rapid Li+ transfer kinetics and inhibited the growth of lithium dendrites.14,39
To further investigate the stability of the SEI, the interfacial resistance of the Li/Li symmetric battery is summarized in Fig. S9.† After the 1, 5, 10, 20, and 50 cycles, CPE-3-PAP maintains a relatively stable interfacial resistance and shows a small value (90 Ω) compared to CPE-3-PHD (265 Ω). As the cycle time increased, the impedance of CPE-3-PAP was more stable than CPE-3-PHD, indicating that the SEI of CPE-3-PAP prevented further decomposition of electrolyte and promoted uniform lithium deposition, thereby reducing interfacial impedance.
Characterization of the components of the SEI
To explore the reasons for SEI stability, the chemical composition of the SEI layer on the lithium anode was detected by X-ray photoelectron spectroscopy (XPS) (Fig. 5a). The F 1s spectrum has two peaks at 684.5 eV and 688.7 eV, corresponding to LiF and C–F bonds.40,41Fig. 5b shows that the LiF ratio of CPE-3-PAP, CPE-3-PHD, and PEO10 SEI layer is 66%, 57%, and 14%, respectively. It can be seen that the different structures of PAP and PHD fillers have different effects on LiF content. The results indicate that the introduction of a benzene ring in POPs may stimulate the decomposition of LiTFSI and generate LiF in the SEI layer. | ||
| Fig. 5 (a) XPS characterization of the Li anode after 200 cycles of Li/CPE-3-PAP/Li, Li/CPE-3-PHD/Li, and Li/PEO10/Li. (b) The content comparison of different chemical species calculated from the fitted XPS spectra. (c–e) Corresponding TOF-SIMS LiF2− species 2D surface distribution. (f–h) Corresponding TOF-SIMS LiF2− species cross-section distribution spectra. (i) Corresponding TOF-SIMS depth profiles of LiF2− species intensity distribution (inset: 3D rendering). Multilayer microstructure SEM images of the cycled cell from (j) FIB-prepared cross-section. (k) TOF-SIMS signals of LiF2−. (l) The schematic diagram of the SEI layer containing LiF generated by adding PAP to promote the dissociation of LiTFSI. | ||
Time of Flight Secondary Ion Mass Spectrometry (TOF-SIMS) analysis was employed to further detect the interface chemical species. Relevant ion signals are fed back through continuous face scanning of the ion beam at the cycled Li metal interface. Fig. 5c–h display the surface (Fig. 5c–e), cross-section (Fig. 5f–h) and 3D rendering (Fig. 5i) distribution spectrum of chosen species, LiF2−, which are representatives of LiF. The higher intensity of LiF2− fragments in the TOF-SIMS spectra of Li/CPE-3-PAP than that in Li/CPE-3-PHD and Li/PEO10 confirms it as the leading component on the Li metal surface of the SEI. According to the 3D reconstruction of the sputtered volume for LiF2− species, the SEI layer of Li/CPE-3-PAP possesses distinct advantages of density and uniformity over the other two Li/CPEs. To directly detect the distribution of the composition on the Li/CPE-3-PAP interface, we tested a fresh and flat cross-section interface by the focused ion beam (FIB) technique (Fig. 5j). Fig. 5i shows the high lateral resolution mapping of the Li/CPE-3-PAP interface cross section, and the TOF-SIMS analysis provided a clear localization of LiF2− at the interface, indicating that the addition of the PHD promoted the formation of an artificial SEI layer due to the catalysis interaction of aromatic groups. Fig. 5l shows the SEI layer with LiF generated by adding PAP to promote the dissociation of LiTFSI.
To further validate, first principles calculations were conducted based on density functional theory (DFT) to elucidate the interaction between aromatic groups and TFSI anions. We have listed the bond lengths of C–F in TFSI− in the table in Fig. 6. Compared with PHD without aromatic groups (C9N9H8F6O4S2) and original molecular C2F6O4S2N, the addition of PAP with aromatic groups (C21N9H16F6O4S2) results in longer C–F bonds for TFSI−. The longest C–F bond in PHD is 1.37 Å, while the PAP bond is 1.38 Å in length. To better illustrate the changes in the C–F bond, we calculated the electron localization function of the structure before and after the group approaches the C–F bond as shown in Fig. 6d–f. When the aromatic group approaches the C–F bond, the white area becomes smaller or even disappears, indicating a weakened interaction between C and F. Consistent with our experimental results, the addition of aromatic groups is more conducive to the cleavage of C–F bonds, thereby promoting the formation of more LiF.
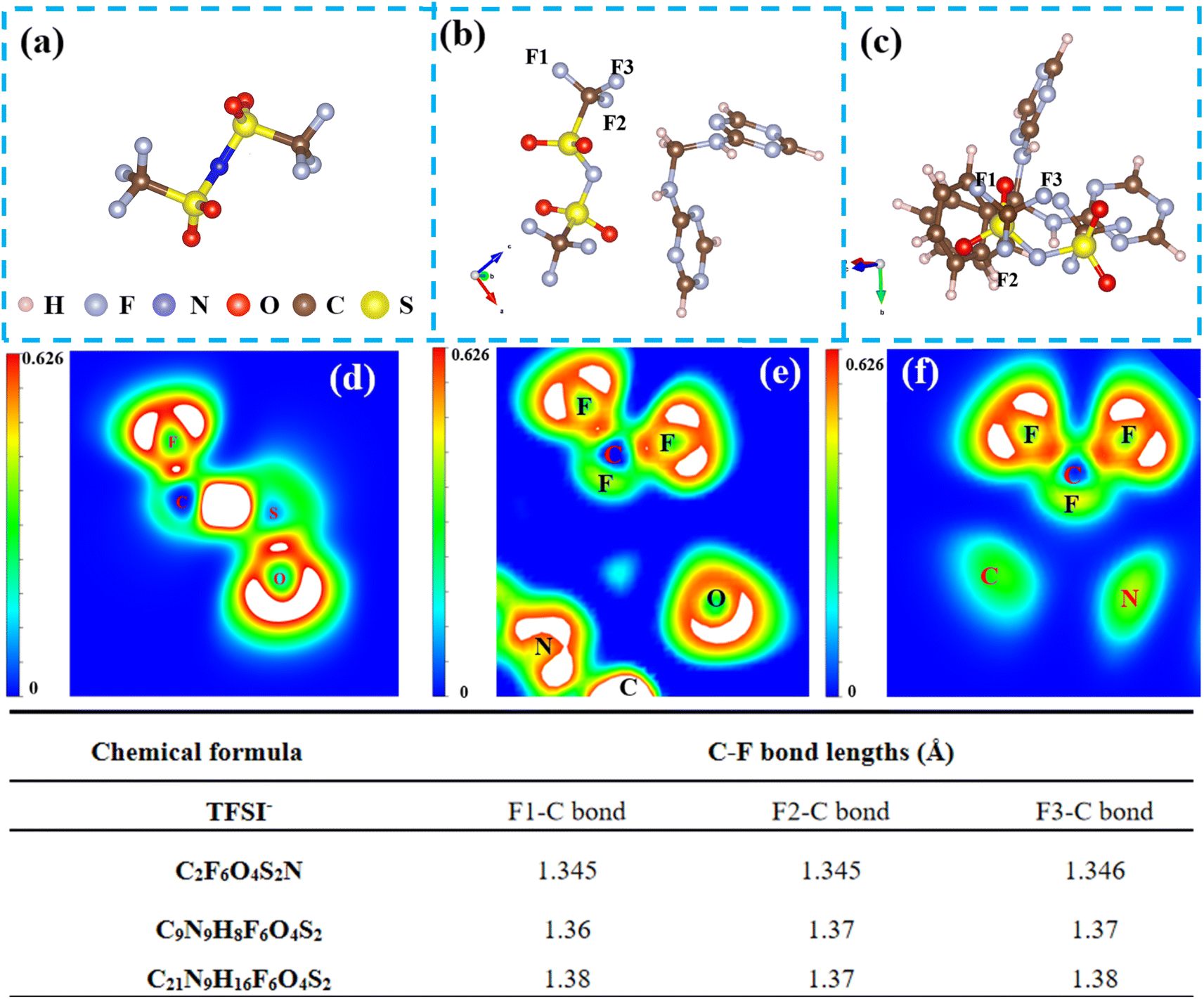 | ||
| Fig. 6 Molecular structures of (a) C2F6O4S2N, (b) C9N9H8F6O4S2, and (c) C21N9H16F6O4S2. The electron localization function (ELF) of (d) C2F6O4S2N, (e) C9N9H8F6O4S2, and (f) C9N9H8F6O4S2. Inset: The C–F bond length after being fully optimized. | ||
Electrochemical performance of the LMBs
The LFP/Li batteries were installed to evaluate the cycling stability of CPEs. The discharge capacities of CPE-3-PAP and CPE-3-PHD are 106.9 and 14.2 mA h g−1 after 800 cycles at 0.5C, while PEO10 is short-circuited after 96 cycles (Fig. S10†). At 1C (Fig. 7a), the initial discharge capacity of LFP/CPE-3-PAP/Li is 139.8 mA h g−1, and the discharge capacity is 108 mA h g−1 after 600 cycles with about 100% coulombic efficiency. In contrast, LFP/CPE-3-PHD/Li and LFP/PEO10/Li failed after 600 and 76 cycles with a remaining specific capacity of 42.2 and 86.4 mA h g−1. Fig. 7b shows LFP/CPE-3-PAP/Li and LFP/CPE-3-PHD/Li voltage platforms at the 1st and 600th cycles at 1C (60 °C). The charge–discharge characteristic platform of LFP/CPE-3-PAP/Li is more stable compared with LFP/CPE-3-PHD/Li, and the polarization did not increase with the increase of the cycle, which further realizes the stable charge–discharge process. Moreover, the LFP/CPE-3-PAP/Li cells also offer good rates of cycle stability at 60 °C (Fig. 7c). The discharge specific capacities of the LFP/Li cell are 167.6, 157.9, 145.9, 130.4 and 110.9 mA h g−1 at 0.1C, 0.2C, 0.5C, 1C, and 2C, respectively. In contrast, the LFP/CPE-3-PHD/Li and LFP/PEO10/Li rate cycles have lower discharge specific capacities. To verify the cycling performance of CPEs at high load, the LFP active material load is increased to 6 mg cm−2 and the LFP/CPEs/Li battery is subjected to a cycle test at 60 °C. As shown in Fig. 7d, the initial discharge capacity of LFP/CPE-3-PAP/Li is 137.1 mA h g−1 at 0.2C, and the discharge capacity after 170 cycles is 123.4 mA h g−1. The capacity retention rate is 90%. In contrast, LFP/CPE-3-PHD/Li and LFP/PEO10/Li short-circuited after 23 and 15 cycles. Fig. 7e shows the LFP/CPE-3-PAP/Li voltage platform at the 1st and 180th cycles at 0.2C at a high mass loading of 6 mg cm−2(60 °C).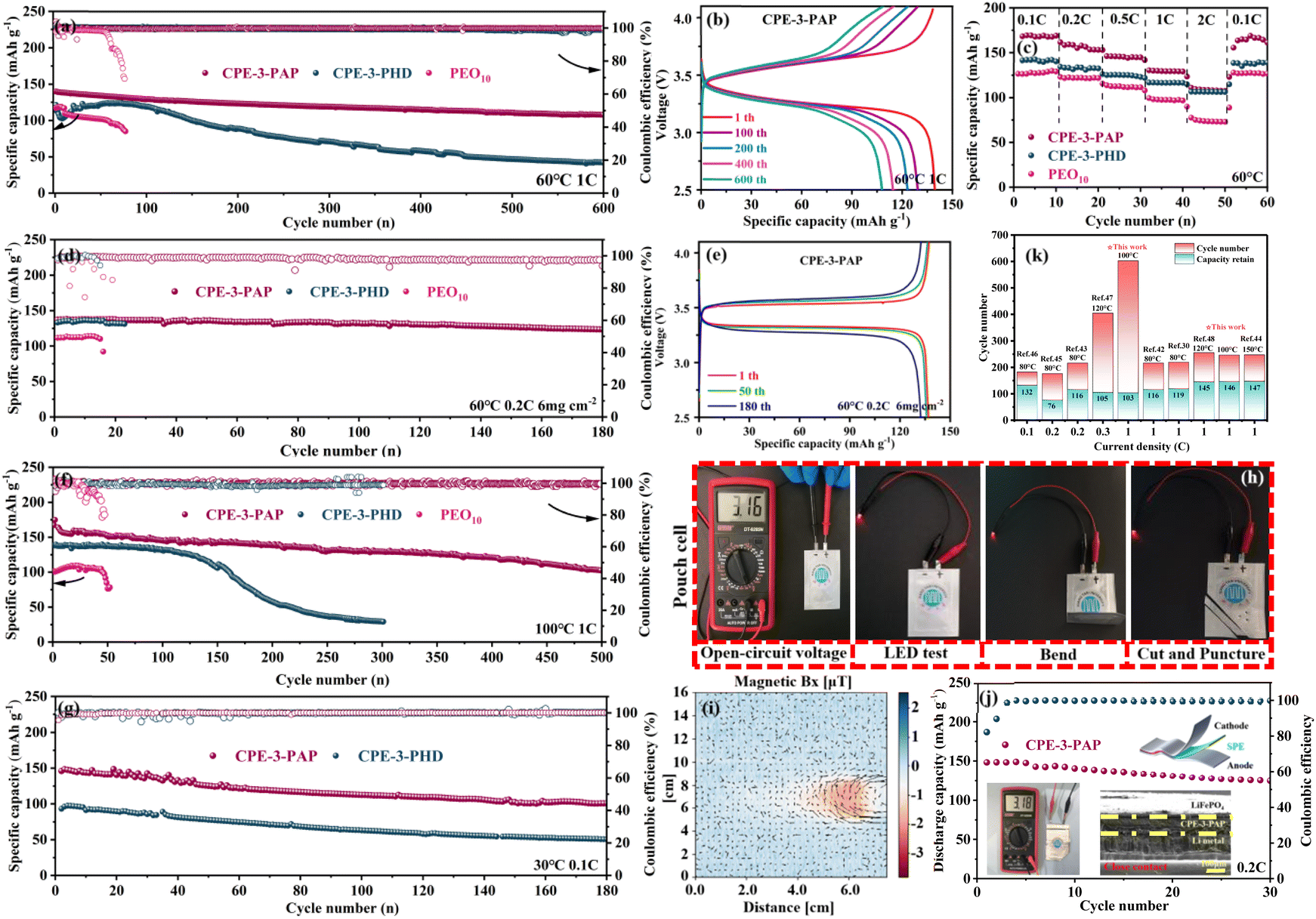 | ||
| Fig. 7 (a) Cycling performance of the LFP/Li battery at 60 °C and 1C. (b) Charge/discharge curves during the 1st and 600th cycles of LFP/CPE-3-PAP/Li at 1C. (c) Rate performance of the LFP/Li battery at 60 °C. (d) Cycling performance of the LFP/Li battery at 60 °C and 6 mg cm−2 at 0.02C. (e) Charge/discharge curves during the 1st and 180th cycles of LFP/CPE-3-PAP/Li at 60 °C and 6 mg cm−2 at 0.02C. (f) Cycling performance of the LFP/Li battery at 100 °C and 1C. (g) Cycling performance of the LFP/Li battery at 30 °C and 0.1C. (h) LED-lighting security test of the pouch cell under extreme circumstances. (i) The distribution of the magnetic field mapping at Bx + By and its corresponding current flow diagram of the multilayer pouch cells. (j) Cycling performance of the pouch-type cell under extreme conditions. Inset: multilayer microstructure characterization of the post-tested battery. (k) Comparison of comprehensive performance with that of related reported CSEs.35,42–48 | ||
In addition, the charge–discharge behaviors of LFP/CPE-3-PAP/Li, LFP/CPE-3-PHD/Li, and LFP/PEO10/Li were measured at 100 °C. As shown in Fig. 7f, the initial discharge capacity of LFP/CPE-3-PAP/Li, LFP/CPE-3-PHD/Li, and LFP/PEO10/Li is 159.7, 138.4, and 101.3 mA h g−1 at 1C. The discharge capacity of LFP/CPE-3-PAP/Li is 103 mA h g−1 after 500 cycles. In contrast, LFP/CPE-3-PHD/Li and LFP/PEO10/Li failed after 301 and 47 cycles. These excellent electrochemical properties indicate that CPE-3-PAP has good interfacial stability and cycling stability at high temperature.
To evaluate the practical value of polymer electrolytes, it is necessary to consider the cycle performance of the battery at room temperature. Here, the LFP/CPE-3-PAP/Li battery is subjected to a cycle test at 30 °C. As shown in Fig. 7g, the initial discharge capacity is 146.1 mA h g−1 at 0.1C, and the discharge capacity after 177 cycles is 100.6 mA h g−1. The initial capacity of LFP/CPE-3-PHD/Li is only 93 mA h g−1. The results show that the LFP/CPE-3-PAP/Li battery can cycle stably at room temperature while the LFP/CPE-3-PHD/Li battery is invalid at room temperature because of its low initial capacity.
To evaluate the safety performance of CPE-3-PAP, a series of flexibility and safety tests were performed on LFP/CPE-3-PAP/Li solid state package tanks at 30 °C. As shown in Fig. 7h, the LFP/CPE-3-PAP/Li pouch battery has a voltage of 3.16V. After folding, extruding, and cutting, the battery can still light up the LED lamp and is safer than typical liquid bag batteries. The distribution of the magnetic field and the flow of current in Bx + By are depicted in Fig. 7i, indicating a uniform magnetic field and a closed-loop current. This implies that there is an even distribution of current within the pouch cell and efficient conduction internally, suggesting a compact and uniform internal structure as well as excellent stability between the electrode and electrolyte. It is worth noting that the CPE-3-PAP pouch cell can maintain a consistent output voltage above 3.18V for more than 30 cycles, while exhibiting a normal charge–discharge capacity performance of up to 150 mA h g−1 (Fig. 7j). Besides, we cut and characterized the multilayer microstructure of the pouch cell that was cycled to death. Obviously, the CPE-3-PAP pouch cell exhibits a tighter Li/electrolyte interface after cycling. These results highlight its significant potential for practical applications compared with other SLMBs (Fig. 7k and Table S2†).
To further emphasize the excellent electrochemical performance from the porous polymer filler, we increased the LFP cathode mass loading and prepared a pouch cell to test the cycling performance. The results are shown in Fig. S12,† where CPE-3-PAP was assembled into a pouch cell with 18.5 mg loading of LFP, and the cell still maintains a high cycling performance. Meanwhile, we assembled CPE-3-PAP with an NCM811 high-capacity cathode into a button cell. The results are shown in Fig. S13,† and the battery still maintains a high cycling performance. This highlights the excellent telephony performance of CPE-3-PAP.
Conclusion
In this work, POPs with aromatic and non-aromatic side groups were successfully synthesized and combined with PEO to prepare CPE-x-PHD and CPE-x-PAP (x = 1, 3, 5). The effects of aromatic groups in POPs on the properties of CPE were investigated. It was found that the aromatic groups in PAP could stimulate the decomposition of LiTFSI and form a LiF-rich SEI layer, which could effectively prevent more side reactions and inhibit the growth of lithium dendrites. Based on the LiF-rich SEI layer, the Li/CPE-3-PAP/Li symmetrical battery is cycled for more than 2000 h at 60 °C. The initial discharge capacity of the LFP/CPE-3-PAP/Li battery is 139.8 mA h g−1 and the discharge capacity is 108 mA h g−1 after 600 cycles at 1C (60 °C). In particular, the network structure of the triamine ring and benzene ring makes the CPEs have high thermal stability. The LFP/CPE-3-PAP/Li battery can also cycle for more than 500 cycles at 1C (100 °C). It provides a promising method to improve the stability of SEI layers and ASSLBs.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. Dai performed the chemical research and prepared the manuscript; M. Cai helped with pouch cell assembly and performance testing; X. Zhou, W. Liang and Z. Zhao helped with SEM, and TOF-SIMS characterization; Z. Xia helped with FTIR and DSC; B. Zhang conceived the idea and designed the research; B. Zhang, J. Jiang and W. Jiang conceived the experimental work and participated in the discussion of the data; F. Huang and Z. Ma contributed to the DFT calculation. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (No. 2024M763135), Outstanding Youth Project of Education Bureau of Hunan Province (23B0161), Natural Science Foundation of Hunan Province (No. 2024JJ5376), National Natural Science Foundation of China (No. 12472100) and College Students Innovation and Entrepreneurship Training Program of Hunan Province (No. S202410530061 and S202310530174).References
- N. Abdi, Electrical energy storage for the grid: applications and benefits, in Conference: International Conference on Energy Management and Technology, At Tehran, Iran, 2017 Search PubMed.
- Y. Liu and Y. Cui, Lithium Metal Anodes: A Recipe for Protection, Joule, 2017, 1(4), 649–650 CrossRef.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Toward Safe Lithium Metal Anode in Rechargeable Batteries: A Review, Chem. Rev., 2017, 117, 10403 CrossRef CAS PubMed.
- Y. Xu, R. Zhao, L. Gao, T. Gao, W. Wang, J. Bian and Y. Zhao, A fiber-reinforced solid polymer electrolyte by in situ polymerization for stable lithium metal batteries, Nano Res., 2023, 12, 452–1157 Search PubMed.
- Z. Li, A. Li, H. Zhang, R. Lin and Y. Yang, Interfacial Engineering for Stabilizing Polymer Electrolytes with 4V Cathodes in Lithium Metal Batteries at Elevated Temperature, Nano Energy, 2020, 72, 104655 CrossRef CAS.
- J. Liang, D. Chen, K. Adair, Q. Sun, N. G. Holmes, Y. Zhao, Y. Sun, J. Luo, R. Li and L. Zhang, Insight into Prolonged Cycling Life of 4 V All-Solid-State Polymer Batteries by a High-Voltage Stable Binder, Adv. Energy Mater., 2020, 11, 2002455 CrossRef.
- N. Meng, F. Lian and G. Cui, Macromolecular Design of Lithium Conductive Polymer as Electrolyte for Solid-State Lithium Batteries, Small, 2021, 17, 202005762 Search PubMed.
- H. Chen, D. Adekoya, L. Hencz, J. Ma and S. Zhang, Stable seamless interfaces and rapid ionic conductivity of Ca-CeO2/LiTFSI/PEO composite electrolyte for highrate and high-voltage all-solid-state battery, Adv. Energy Mater., 2020, 10, 2000049 CrossRef CAS.
- X. Wu, K. Chen, Z. Yao, J. Hu, M. Huang, J. Meng, S. Ma, T. Wu, Y. Cui and C. Li, Metal organic framework reinforced polymer electrolyte with high cation transference number to enable dendrite-free solid state Li metal conversion batteries, J. Power Sources, 2021, 501, 229946 CrossRef CAS.
- H. Li, Y. Du, X. Wu, J. Xie and F. Lian, Developing “Polymer-in-Salt” High Voltage Electrolyte Based on Composite Lithium Salts for Solid-State Li Metal Batteries, Adv. Funct. Mater., 2021, 31, 2103049 CrossRef CAS.
- Q. Zhang, K. Liu, F. Ding and X. Liu, Recent advances in solid polymer electrolytes for lithium batteries, Nano Res., 2017, 10, 4139–4174 CrossRef.
- W. Zhang, Z. Su, S. Yi, H. Chen, M. Wang, B. Niu, Y. Zhang and D. Long, Sandwich Structured Metal oxide/Reduced Graphene Oxide/Metal Oxide-Based Polymer Electrolyte Enables Continuous Inorganic–Organic Interphase for Fast Lithium-Ion Transportation, Small, 2023, 2207536 CrossRef CAS PubMed.
- D. Wu, J. He, J. Liu, M. Wu, S. Qi, H. Wang, J. Huang, F. Li, D. Tang and J. Ma, Li2CO3/LiF-Rich Heterostructured Solid Electrolyte Interphase with Superior Lithiophilic and Li+-Transferred Characteristics via Adjusting Electrolyte Additives, Adv. Energy Mater., 2022, 12, 2200337 CrossRef CAS.
- Z. Zhao, X. Zhou, B. Zhang, Y. Huang, Z. Ma and J. Liu, Regulating Steric Hindrance of Porous Organic Polymers in Composite Solid-State Electrolytes to Induce the Formation of LiF-Rich SEI in Li-Ion Batteries, Angew. Chem., Int. Ed., 2023, 62, e202308738 CrossRef CAS PubMed.
- Z. Zhang, J. Wang, S. Zhang, H. Ying, Z. Zhuang, F. Ma, P. Huang, T. Yang, G. Han and W. Q. Han, Stable all-solid-state lithium metal batteries with Li3N-LiF-enriched interface induced by lithium nitrate addition, Energy Storage Mater., 2021, 43, 229–237 CrossRef.
- C. Fang, Y. Huang and Y. Sun, Revealing and reconstructing the 3D Li-ion transportation network for superionic poly(ethylene) oxide conductor, Nat. Commun., 2024, 15, 6781 CrossRef CAS PubMed.
- W. Lyu, H. Fu, A. M. Rao, X. Lu, X. Yu, Y. Lin, J. Zhou and B. Lu, Permeable void-free interface for all-solid-state alkali-ion polymer batteries, Sci. Adv., 2024, 42, 9602 CrossRef CAS PubMed; J. Xu, J. Li, Y. Li, M. Yang, L. Chen, H. Li and F. Wu, Long-Life Lithium-Metal All-Solid-State Batteries and Stable Li Plating Enabled by In Situ Formation of Li3PS4 in the SEI Layer, Adv. Mater., 2022, 34, 2203281 CrossRef PubMed.
- H. Bildirir, V. G. Gregoriou, A. Avgeropoulos, U. Scherfd and C. L. Chochos, Porous organic polymers as emerging new materials for organic photovoltaic applications: current status and future challenges, Mater. Horiz., 2017, 4, 546–556 RSC.
- R. Dawson, A. I. Cooper and D. J. Adams, Nanoporous organic polymer networks, Prog. Polym. Sci., 2011, 37, 530–563 CrossRef.
- X. Zhan, Z. Chen and Q. Zhang, Recent progress in two-dimensional COFs for energy-related applications, J. Mater. Chem. A, 2017, 5, 14463–14479 RSC.
- N. Chaoui, M. Trunk, R. Dawson, J. Schmidt and A. Thomas, Trends and challenges for microporous polymers, Chem. Soc. Rev., 2017, 46, 3302–3321 RSC.
- Q. Sun, Z. Dai, X. Meng and F. S. Xiao, Porous polymer catalysts with hierarchical structures, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC.
- S. B. Peh, Y. Wang and D. Zhao, Scalable and Sustainable Synthesis of Advanced Porous Materials, ACS Sustainable Chem. Eng., 2019, 7, 3647–3670 CrossRef CAS.
- K. Jie, Y. Zhou, E. Li and F. Huang, Nonporous Adaptive Crystals of Pillararenes, Acc. Chem. Res., 2018, 51, 2064–2072 CrossRef CAS PubMed.
- J. Liang, Z. Xiao, Y. Gao, X. Xu, D. Kong, M. Wagner and L. Zhi, Ionothermal strategy towards template-free hierarchical porous carbons for supercapacitive energy storage, Carbon, 2019, 143, 487–493 CrossRef CAS.
- N. Angutakshmi, R. B. Dhanalakshmi, M. Kathiresan, Y. Zhou and A. M. Stephan, The suppression of lithium dendrites by a triazine-based porous organic polymer-laden PEO-based electrolyte and its application for all-solid-state lithium batteries, Mater. Chem. Front., 2020, 4, 933–940 RSC.
- C. Li, S. Zhou, L. Dai, X. Zhou, B. Zhang, L. Chen, T. Zeng, Y. Liu, Y. Tang, J. Jiang and J. Huang, Porous polyamine/PEO composite solid electrolyte for high performance solid-state lithium metal batteries, J. Mater. Chem. A, 2021, 9, 24661–24669 RSC.
- Z. Zhang, Y. Zhou, P. Chen, S. Zeng, W. Nie and Y. Xu, Investigation of Capacity Increase in Schiff-Base Networks as the Organic Anode for Lithium-Ion Batteries, ACS Appl. Energy Mater., 2021, 4, 12882–12891 CrossRef CAS.
- D. Luo, M. Li, Q. Ma, G. Wen, H. Dou, B. Ren, Y. Liu, X. Wang, L. Shui and Z. Chen, Porous organic polymers for Li-chemistry-based batteries: functionalities and characterization studies, Chem. Soc. Rev., 2022, 51, 2917–2938 RSC.
- W. Liang, X. Zhou, Z. Zhao, X. Song, Z. Wang, Z. Ma and J. Liu, The Versatile Establishment of Charge Storage in Polymer Solid Electrolyte with Enhanced Charge Transfer for LiF-Rich SEI Generation in Lithium Metal Batteries, Angew. Chem., Int. Ed., 2024, 63, e202320149 CrossRef CAS PubMed.
- W. Huang, Y. Wang, L. Lv, G. Zhu, Q. Qu and H. Zheng, A versatile LiTFSI-like anchor for constructing robust interfacial layers with tailored structures for silicon anodes, Energy Storage Mater., 2022, 52, 646–654 CrossRef.
- L. Qiao, S. R. Pena, M. Martinez-Ibanez, A. Santiago, I. Aldalur, E. Lobato, E. Sanchez-Diez, Y. Zhang, H. Manzano and H. Zhu, Anion π–π Stacking for Improved Lithium Transport in Polymer Electrolytes, J. Am. Chem. Soc., 2022, 22, 9806–9816 CrossRef PubMed.
- J. Wang, A. E. Lakraychi, X. Liu, L. Sieuw, C. Morari, P. Poizot and A. Vlad, Conjugated sulfonamides as a class of organic lithium-ion positive electrodes, Nat. Mater., 2021, 20, 665–673 CrossRef CAS PubMed.
- F. Wu, K. Zhang, Y. Liu, H. Gao, Y. Bai, X. Wang and C. Wu, Polymer electrolytes and interfaces toward solid-state batteries: Recent advances and prospects, Energy Storage Mater., 2020, 33, 26–54 CrossRef.
- C. Tao, M. H. Gao, B. H. Yin, B. Li, Y. P. Huang, G. Xu and J. J. Bao, A promising TPU/PEO blend polymer electrolyte for all-solid-state lithium ion batteries, Electrochim. Acta, 2017, 257, 31–39 CrossRef CAS.
- J. Pickelmann and J. Plank, A mechanistic study explaining the synergistic viscosity increase obtained from polyethylene oxide (PEO) and β-naphthalene sulfonate (BNS) in shotcrete, Cem. Concr. Res., 2012, 42, 1409–1416 CrossRef CAS.
- L. Yang, Y. Nie, Y. Liu, Y. Zheng, D. Luo, N. Yang, Q. Ma, M. Xu, X. Ma, A. Yu, L. Shui, X. Wang and Z. Chen, The plasticizer-free composite block copolymer electrolytes for ultralong lifespan all-solid-state lithium-metal batteries, Nano Energy, 2022, 100, 107499 CrossRef CAS.
- S. S. Chi, Q. Wang, B. Han, C. Luo, Y. Jiang, J. Wang, C. Wang, Y. Yu and Y. Deng, Lithiophilic Zn Sites in Porous CuZn Alloy Induced Uniform Li Nucleation and Dendrite-free Li Metal Deposition, Nano Lett., 2020, 20, 2724–2732 CrossRef CAS PubMed.
- Q. Wang, J. Wan, X. Cao, R. Wen, Y. Guo, W. Liu and H. Zhou, Organophosphorus Hybrid Solid Electrolyte Interphase Layer Based on LixPO4 Enables Uniform Lithium Deposition for High-Performance Lithium Metal Batteries, Adv. Funct. Mater., 2022, 32, 2107923 CrossRef CAS.
- C. Wang, T. Wang, L. Wang, Z. Hu, Z. Cui, J. Li, S. Dong, X. Zhou and G. Cui, Differentiated Lithium Salt Design for Multilayered PEO Electrolyte Enables a High-Voltage Solid-State Lithium Metal Battery, Adv. Sci., 2019, 6, 1901036 CrossRef CAS PubMed.
- Y. Chen, W. Li, C. Sun, J. Jin, Q. Wang, X. Chen, W. Zha and Z. Wen, Sustained Release-Driven Formation of Ultrastable SEI between Li6PS5Cl and Lithium Anode for Sulfide-Based Solid-State Batteries, Adv. Energy Mater., 2021, 11, 2002545 CrossRef CAS.
- F. Zeng, Y. Sun, B. Hui, Y. Xia, Y. Zou, X. Zhang and D. Yang, Three-Dimensional Porous Alginate Fiber Membrane Reinforced PEO-Based Solid Polymer Electrolyte for Safe and High-Performance Lithium Ion Batteries, ACS Appl. Mater. Interfaces, 2020, 12, 43805–43812 CrossRef CAS PubMed.
- J. Y. Lee, T. Y. Yu, P. H. Chung, W. Y. Lee and R. J. Jeng, Semi-Interpenetrating Polymer Network Electrolytes Based on a Spiro-Twisted Benzoxazine for All-Solid-State Lithium-Ion Batteries, ACS Appl. Energy Mater., 2021, 4, 2663–2671 CrossRef CAS.
- J. Tan, X. Ao, A. Dai, Y. Yuan, H. Zhuo, H. Lu, L. Zhuang, Y. Ke, C. Su, X. Peng, B. Tian and J. Lu, Polycation ionic liquid tailored PEO-based solid polymer electrolytes for high temperature lithium metal batteries, Energy Storage Mater., 2020, 33, 173–180 CrossRef.
- T. Y. Yu, S. C. Yeh, J. Y. Lee, N. L. Wu and R. J. Jeng, Epoxy-Based Interlocking Membranes for All Solid-State Lithium Ion Batteries: The Effects of Amine Curing Agents on Electrochemical Properties, Polymers, 2021, 13, 3244 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, W. Feng, Y. Zhen, P. Zhao, Z. Cai and L. Li, Effects of the shapes of BaTiO3 nanofillers on PEO-based electrolytes for all-solid-state lithium-ion batteries, Ionics, 2019, 25, 1471–1480 CrossRef CAS.
- Y. Ma, J. Wan and Y. Yang, et al., Scalable, ultrathin, and high-temperature-resistant solid polymer electrolytes for energy-dense lithium metal batteries, Adv. Energy Mater., 2022, 12, 2103720 CrossRef CAS.
- Z. Hu, Y. Zhang and Y. Zhang, et al., In-situ construction of high-temperature-resistant 3D composite polymer electrolyte membranes towards high-performance all-solid-state lithium metal batteries, J. Power Sources, 2022, 548, 232052 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07449a |
| This journal is © The Royal Society of Chemistry 2025 |