
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDigitization of molecular complexity with machine learning†
Andrei S.
Tyrin‡
,
Daniil A.
Boiko‡
,
Nikita I.
Kolomoets
and
Valentine P.
Ananikov
 *
*
Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky prospekt 47, Moscow, 119991, Russia. Web: http://AnanikovLab.ruE-mail: val@ioc.ac.ru
First published on 19th March 2025
Abstract
Digitization of molecular complexity is of key importance in chemistry and life sciences to develop structure–activity relationships in chemical behavior and biological activity. The complexity of a given molecule compared to others is largely based on intuitive perception and lacks a standardized numerical measure. Quantifying molecular complexity remains a fundamental challenge, with key implications currently remaining controversial. In this study, we introduce a novel machine learning-based framework employing a Learning to Rank (LTR) approach to quantify molecular complexity on the basis of labeled data. As a result, we developed a ranking model utilizing the dataset that comprizes approximately 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 data points across diverse chemical structures, leveraging human expertise to capture complex decision rules that researchers intuitively use. Applications of our model in mapping the current organic chemistry landscape, analyzing FDA-approved drugs, guiding lead optimization processes, and interpreting total synthesis approaches reveal key trends in increasing molecular complexity and synthetic strategy evolution. Our study advances the methodologies available for quantifying molecular complexity, changing it from an elusive property to a numerical characteristic. With machine learning, we managed to digitize human perception of molecular complexity. Moreover, a corresponding large labeled dataset was produced for future research in this area.
000 data points across diverse chemical structures, leveraging human expertise to capture complex decision rules that researchers intuitively use. Applications of our model in mapping the current organic chemistry landscape, analyzing FDA-approved drugs, guiding lead optimization processes, and interpreting total synthesis approaches reveal key trends in increasing molecular complexity and synthetic strategy evolution. Our study advances the methodologies available for quantifying molecular complexity, changing it from an elusive property to a numerical characteristic. With machine learning, we managed to digitize human perception of molecular complexity. Moreover, a corresponding large labeled dataset was produced for future research in this area.
Introduction
Molecular complexity is a fundamental property of every molecule. However, quantifying the complexity of molecules is a long-standing challenge in the field of chemistry. The ability of the research community to capture human-assessed molecular complexity is quite limited. Having a numerically unbiased definition of molecular complexity would greatly benefit the field of chemistry and would provide a valuable research tool.Accessing molecular complexity is valuable in the field of medicinal chemistry, where it was experimentally validated that drug-like molecules tend to have more complex structures.1 Additionally, having a universal numerical definition of molecular complexity can provide insights into method development in the field of chemistry by quantifying the efficiency of synthetic approaches or discovering hidden trends in areas relying on organic chemistry.
A valuable group of methods for defining molecular complexity (MC) is based on substructure information. The first substructural approach for defining MC was proposed by Bertz in 1981.2 Bertz's method considers different substructures and estimates MC as the complexity of a molecular graph. Whitlock's approach models molecular complexity as a linear combination of the number of rings, unsaturations, heteroatoms, and chiral centers,3 but it lacks generality because it does not capture other important molecular features. Many new substructure-based MC estimation methods have been proposed recently. Among them are information theory-based approaches that include information content analysis,4 atom environment methods, MC estimation methods,5 and fractal-based approaches for calculating molecular complexity.6 Recent attempts also include the spatial score (SPS),7,8 which aims to evaluate the topological complexity of a molecule. Substructural approaches are attractive for their simplicity and usability. However, the problem with this group of approaches is the lack of universal acceptance in the community9 as well as challenges in identifying all the relevant molecular features.
Other methods include statistical approaches and machine learning. An approach by Sheridan et al.10 adopted crowdsourcing, which uses chemists' expertise to determine molecular complexity. However, modeling molecular complexity through the regression lens has significant disadvantages. Their model predicts the complexity of a molecule in a bounded 1–5 range, thereby potentially limiting the ability to distinguish molecules with similar or very high complexity. In addition, the training set of 2575 molecules for the model developed by Sheridan et al.10 is arguably insufficient to reflect the diversity of chemical space; therefore, the universality of the approach is lacking.
Assembly theory was proposed by Cronin et al.11 The molecular assembly index is grounded in the possible formation histories of a molecule from elementary building blocks. The advantage of the assembly index is the possibility of experimental identification, which distinguishes assembly theory from other approaches for defining molecular complexity.12 The challenges associated with assembly theory are the nontrivial choice of elementary building blocks and the difficulty in calculating the molecular assembly index.
Of course, it will be desirable to develop a general approach to deal with quantification of molecular complexity. Given a molecule, organic chemists usually characterize it as simple or complex or compare it with other molecules. It is challenging to formalize the decision an expert makes, but we believe that a definition that is based on scientists' expertise is the closest to that definition of molecular complexity in chemical methodology assessment. Therefore, machine learning methods can be particularly useful because they can capture complex patterns hidden in data.
In this work, we aim to achieve a foundational definition of molecular complexity by modeling it as a learning-to-rank problem and training a machine learning ranking model based on molecular data labeled by professional chemists. We believe that utilizing human expertise is the best way to quantify the complexity of molecules, as agreement on the definition is crucial. Furthermore, we provide an analysis across a wide range of application areas, demonstrating the fundamental nature of molecular complexity.
Molecular complexity is a fundamental concept of modern organic chemistry (Fig. 1a). Modern synthetic methods have been developed to rapidly increase molecular complexity, enabling more efficient navigation of the synthetic chemical space. Approaches developed by Nature show astounding examples of immense growth in molecular complexity.13
 | ||
| Fig. 1 Overview of the approach. (a) Illustration of the molecular complexity. (b) Visualization of PubChem samples annotated with molecular complexity values; examples of molecules from different complexity clusters. (c) Demonstration of existing methods for molecular complexity determination. (d) Workflow: the web application for experts to assess the complexity of molecules. Labeled data were then used to train a ranking model through an active learning process, where the experts reviewed the model's uncertain predictions. (e) Examples of different molecules were provided to an assessor during each stage. (f) Source of molecular data for each of the stages. (g) Quality control example. (h) Ternary diagram illustrating phase weights with the model performance measured in terms of pair accuracy. (i) Pairwise comparison between different molecular properties (including molecular complexity). | ||
To illustrate the fact that molecular complexity is an intrinsic property of every organic molecule, we randomly sampled 50000 molecules from the PubChem database.14 We then visualized the molecular complexity labels assigned by the developed machine learning model (Fig. 1b) using t-SNE to reduce the dimensionality of the molecular fingerprints. As can be seen, even such a small sample contains both very simple and immensely complex molecules.
Results and discussion
Approach for quantifying molecular complexity
Expert opinion is crucial in defining such a complicated quantity as molecular complexity. Utilizing human expertise with machine learning can efficiently capture all the corresponding features, making the definition as general and widely applicable as possible (Fig. 1c). Another important consideration that addresses the problems of the previously proposed regression methods is the formulation of molecular complexity as a Learning to Rank (LTR) problem (see the Methods for details). This decision allows further generalization of the developed method by capturing the difference in complexity among molecules. The overall data collection approach, driven by active learning, consists of three stages (Fig. 1d and e) to ensure the maximum training set diversity. We used PubChem14 as a source of molecules (Fig. 1f).This study involved over 50 chemists who made 294542 molecular complexity comparisons, with a total of 196083 data points involved in the training of the final model (164017 unique molecules). To ensure that the collected labels were of reliable quality, artificially created sets of molecules (we call them Quality Controls, QCs) were integrated for which it was straightforward to rank them according to the collective decision of this work's authors (see examples in Fig. 1g). The labels for molecules from the QCs were used to filter the assessors who did not pass their label quality check.
Learning-to-rank model development
Gradient Boosted Decision Trees (GDBT) architecture was selected as ranking model parameterization for several reasons. First, GDBT enables quick and intuitive analysis of feature importance, which is particularly useful in the molecular complexity problem for understanding the key features that define the complexity of molecules from the expert perspective. Second, GDBT achieves superior performance in LTR tasks.15 Finally, there are popular GBDT libraries, such as XGBoost or CatBoost, with available implementations of state-of-the-art ranking algorithms, GPU support, and common metrics.16,17As was mentioned, the proposed approach includes three different data collection stages, each of which contains a different number of data points as well as different sets of experts per phase. The default approach relies on assigning the same importance to all phases during training, which can lead to suboptimal performance due to the difference between the data stages. In the present study, phase weighting was included in the training pipeline. Phase weighting serves as an additional hyperparameter that allows simple model selection (Fig. 1h).
To choose the best-performing model, two criteria were considered: the performance of the ranking model on the test set in terms of pair accuracy, which captures the quality of the ranking, and the performance of the functional group test (FGT). The FGT proceeds as follows: for an arbitrary molecule with at least one hydrogen atom, a random hydrogen atom is replaced by a common organic functional group, such as a methyl, phenyl, or halogen group (see the ESI† for the full list of groups that were included in this test). After replacement, one obtains the molecule with a higher molecular complexity value, and the ranking model should be able to reflect this. The fraction of cases where the molecular complexity increases gives us the FGT value. For the selected model, we were able to achieve 77.5% PA averaged across three test phases and 98.1% FGT, with resulting weights of 70%, 10%, and 20% between the corresponding phases. The performance-averaged pair accuracy is depicted in the ternary diagrams in Fig. 1h.
Four features (SCScore, molecular weight, number of aromatic cycles, and TPSA) were analyzed to determine the relationship between them and the molecular complexity. The measured pairwise Pearson correlation is illustrated in Fig. 1i. The data used for Fig. 1i are a 1k sample from the ChEMBL18 database, accessed using the Datamol Python package (https://datamol.io/).19
Interpreting molecular complexity
The GBDT architecture offers the advantage of explainability. Feature importance analysis (Fig. 2a) based on the SHAP values20 was conducted to identify key molecular characteristics that guide experts in assigning molecular complexity scores to the molecules. | ||
| Fig. 2 Interpretability and analysis. (a) SHAP plot with the 7 most important molecular features and morphine molecules with two isomers along with the molecular complexity values. (b) Molecular complexity and different scores for synthetic or structural complexities (SAscore, SCScore, and SPS). (c) Distribution of the molecular complexity values in benchmarks common in molecular machine learning. | ||
From the plot presented in Fig. 2a, the feature that affects the experts' decisions the most is the molecular weight. The number of aromatic cycles in the structure also plays an important role in the complexity of molecules according to expert evaluation. The TPSA represents the topological information of the molecular graph and is the third most important feature that characterizes the importance of the topological characteristics of complex molecules. Fig. 2a additionally contains the (+)-ineleganolide molecule along with two molecules that have the same composition—all the compounds significantly differ in molecular complexity even though their molecular formula is the same. This highlights the importance of the topological molecular structure that guides both the experts during the complexity assessment and the developed ranking model. The latter is quite remarkable given that the model does not perform any explicit characterization of the 3D geometry.
Interestingly, the SCScore—a proxy for synthetic accessibility (SA)—is not among the most significant features for molecular complexity, which captures the difference between the SA and MC concepts.
To further investigate the relationships among molecular complexity, synthetic accessibility, and spatial complexity, a comparison between our approach and other methods was performed. The SAscore21 and SCScore22 are considered proxies for synthetic accessibility, whereas the recently developed Spatial Score (abbreviated as SPS)8 is considered a metric for the molecular complexity grounded in the topological complexity of a molecule. The results of the analysis for the subset of the ChEMBL18 database can be found in Fig. 2b. The MC, SAscore and SCScore are not strongly correlated. Notably, molecular complexity provides better differentiation among complex molecules in the case of the SCScore, while SAscore values are concentrated in the low to medium synthetic complexity region, suggesting that not all molecules that are easy to synthesize must be simple from the point of view of molecular complexity, which further supports the assumption regarding the difference between SA and MC. Finally, the ranking-based approach is correlated with the SPS, suggesting that the topology of the molecule is an important parameter in the context of molecular complexity estimation. A similar conclusion can be drawn in the section discussing the importance of molecular complexity for drug discovery.
Molecular complexity can serve as a valuable tool for dataset characterization, especially given the rapid development of machine learning methods. It is crucial to have reliable and comprehensive benchmarking datasets to understand the differences between different machine learning approaches. The QM9,23,24 Tox21,25 and HIV25 datasets were taken as widely used benchmarking datasets in the molecular machine learning community. Fig. 2c suggests that these datasets tend to contain molecules of low to medium molecular complexity and do not provide very comprehensive coverage of the chemical space, which might lead to biased results when machine learning models are evaluated on those benchmarks. This is an important issue because benchmarks should be as general and comprehensive as possible to correctly differentiate between different methods that rely on those benchmarks.
Reaction atlas: molecular complexity for reactions
Chemical reactions are fundamental in organic chemistry, and analyzing them through the lens of molecular complexity can enhance the characterization of chemical transformations.26 This is particularly useful for automated synthesis planning with limited resources, where selecting optimal reactions for given substrates and desired transformations is crucial.27Schwaller et al.28 proposed a method that enabled visualization of learned reaction embeddings as tree-like graphs, forming a “reaction atlas.” Fig. 3 contains such a tree based on the Schneider 50k29 dataset. Molecular complexity was integrated into this atlas by measuring the change in complexity between the main product and the most complex reagent (Fig. 3). Every node in the tree is colored according to the reaction class and contains the molecular complexity change value represented by the intensity (brighter points correspond to large complexity changes).
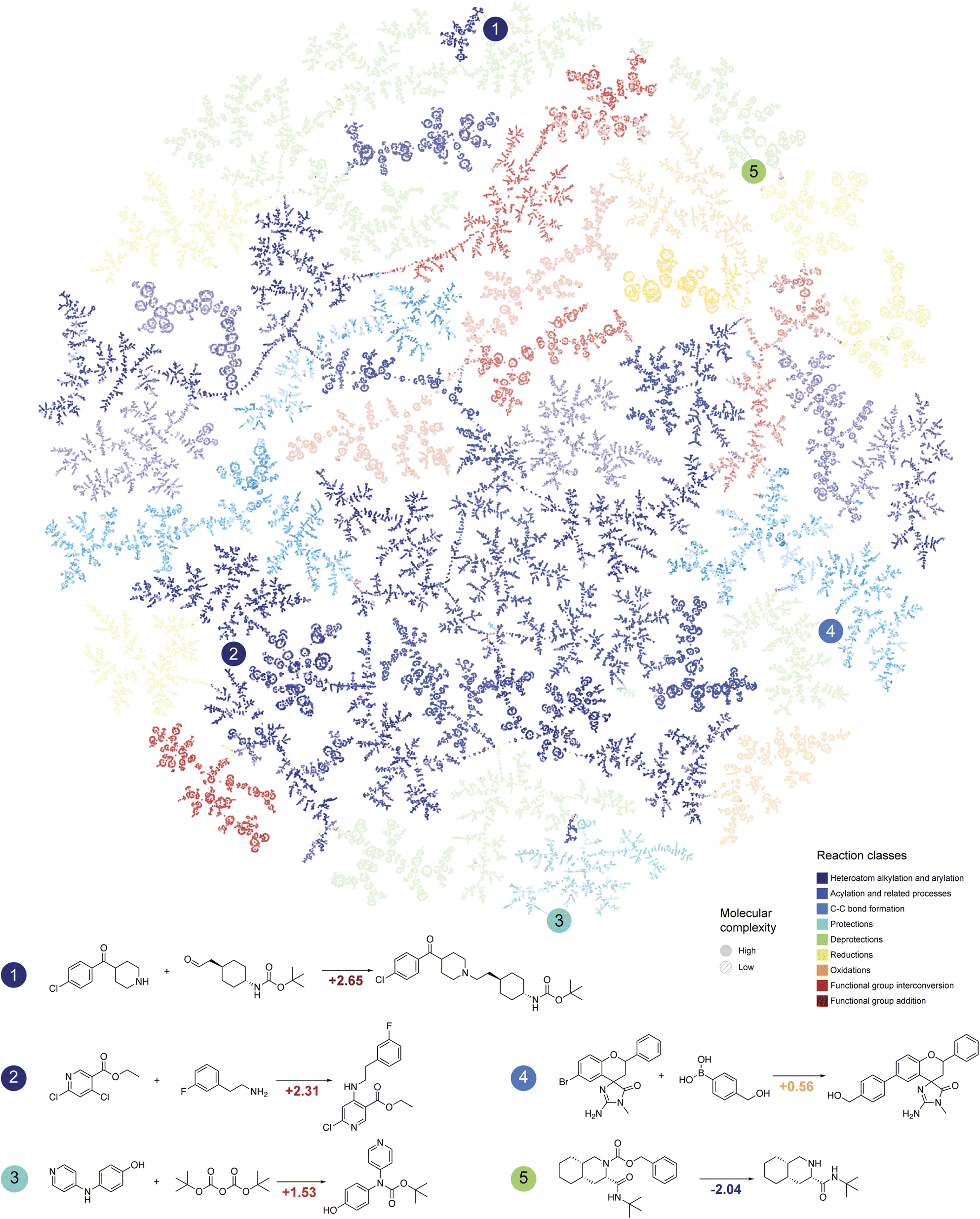 | ||
| Fig. 3 Reaction atlas annotated with molecular complexity values. Tree map of the reaction atlas colored by the reaction classes (the plot is obtained using the code developed in ref. 28). The brightness of every point in the atlas corresponds to the magnitude of molecular complexity change in chemical transformations. | ||
The results in Fig. 3 provide deep insight into the landscape of organic reactions. Most reaction classes either maintain or decrease the complexity of the most complex reagent, such as in deprotection reactions (Fig. 3, reaction 5). However, some classes, such as alkylation/arylation, acylation, and C–C bond formation (e.g., cross-coupling reactions), significantly increase molecular complexity, yielding more complex products.
Fig. 3 suggests that many current synthetic methods do not significantly change the molecular complexity. Protection/deprotection reactions or minor modifications constitute a significant part of the dataset illustrated in Fig. 3 rather than direct ways to increase intermediate molecule complexity. Nevertheless, synthetic methods are becoming increasingly atom efficient and tend to focus more on efficient molecular complexity growth,30–32 as evidenced by advancements in total synthesis and discussed in one of the following sections on total synthesis analysis.
Molecular complexity as a tool for drug discovery
The evolution of medicinal chemistry encapsulates significant molecular complexity trends. We analyzed FDA-approved small-molecule drugs and one of the lead optimization programs to highlight these trends.We examined a dataset of 623 FDA-approved small-molecule drugs from 1985 to 2022, as shown in Fig. 4a. The median molecular complexity has increased over the years. Notably, drugs from each period can be classified into simpler and more complex groups. Using Gaussian mixture model, we observed that the proportion of complex drugs has increased, reflecting advances in biological and synthetic methods.
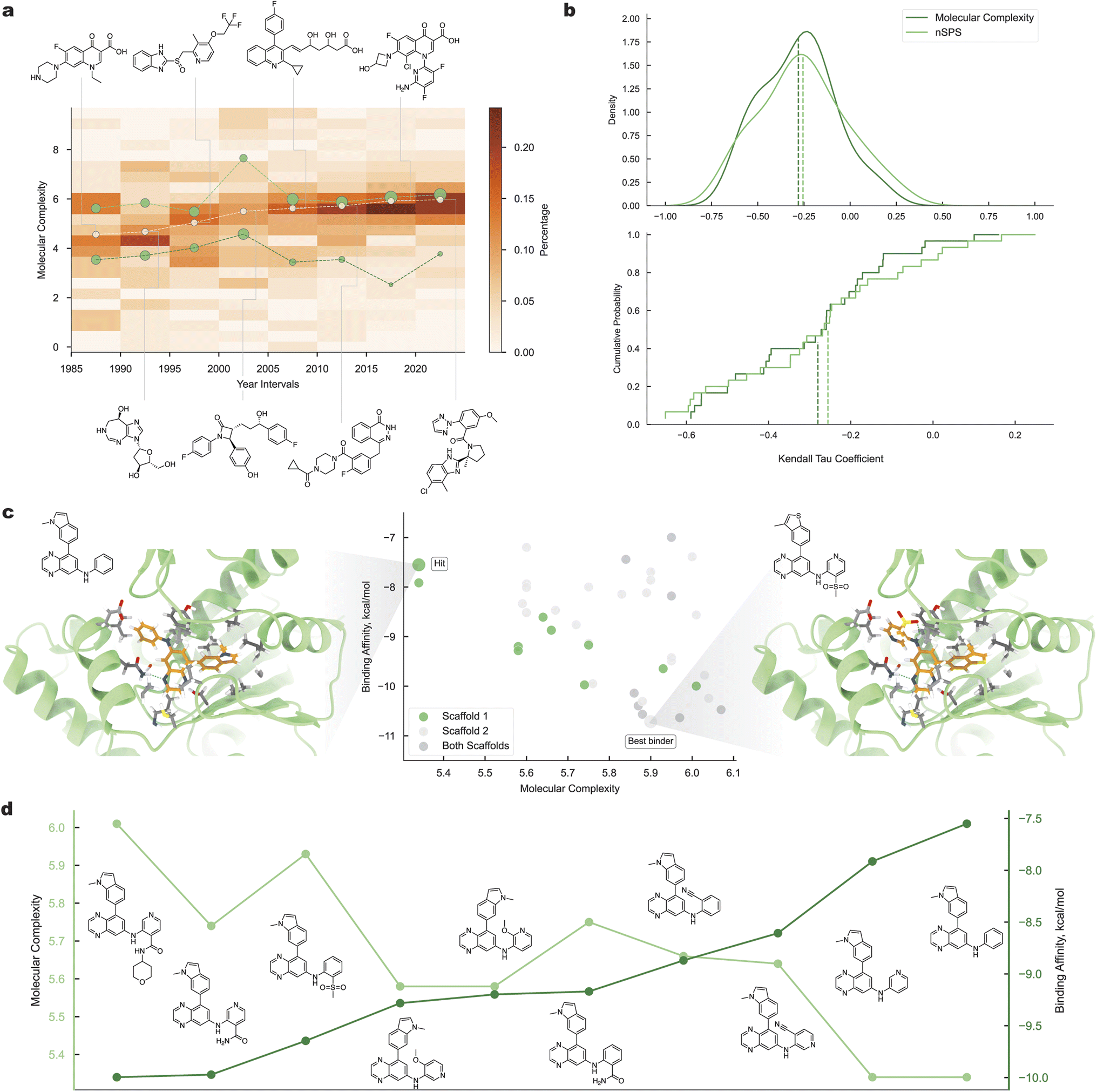 | ||
| Fig. 4 Drug molecules. (a) Molecular complexity evolution of small-molecule drugs over time: white points correspond to the molecules of median complexity, and green points correspond to each of the Gaussian mixture model components for “simple” and “complex” drugs. (b) Comparison of the proposed method with the nSPS: Kendall Tau correlation coefficient distribution among proteins (upper) and the cumulative empirical distribution of the Kendall Tau coefficient (lower). (c) Structure-based analysis of the hit molecule with the identified lead for PFKFB3 kinase. (d) Analysis of Scaffold 1, showing the relationships between the molecular structures, molecular complexity, and binding affinity. | ||
To explore the relationship between molecular complexity and binding affinity, we analyzed data aggregated by Ross et al.33Fig. 4b compares our method with the topology-based space-normalized spatial score (nSPS). For each protein target, we calculated the Kendall Tau correlation between the molecular complexity and binding free energy. The results show that our method and nSPS exhibit similar correlations, with our approach having a slightly higher mean. This underscores our method's universality, as it captures topological information implicitly, aligning with the notion that chemists consider 3D molecular information when assessing complexity.
Further analysis focused on PFKFB3 kinase,34 a key target in cancer drug discovery programs.35 Modifications of an identified hit compound were considered (Fig. 4c, left). The hit molecule showed one of the worst affinities among the explored molecules. Two different scaffolds and a combined scaffold were analyzed, as shown in the scatterplot in Fig. 4c (middle), which displays the relationship between molecular complexity and binding affinity. Compared with simpler molecules, more complex molecules generally exhibit stronger binding. The right part of Fig. 4c shows the structure of the protein and the best binder-docked complex, indicating tighter ligand packing in the PFKFB3 pocket and additional noncovalent interactions between the binder and pocket residues compared with the hit molecule.
In addition to structure-based analysis, Scaffold 1 exhibited a near-linear relationship between molecular complexity and binding affinity. Fig. 4d illustrates the molecular structures, along with the corresponding MC and binding affinity values. The hit molecule had the worst binding in the selected scaffold, with subsequent consistent improvement in binding as the molecular complexity increased.
Exploring synthetic strategies with molecular complexity
To further demonstrate the potential applications of molecular complexity, the field of total synthesis was explored, where molecular complexity is crucial research driving force, as was concluded in the work of Wright and Sarpong.36 Unlike the reaction atlas, total synthesis provides a temporal structure, detailing how particular synthetic steps are planned and executed with foresight in later stages. A trained ranking model was applied to analyze synthetic routes, yielding several profound observations on the evolution of human-developed methods and their comparison to natural approaches.Importantly, molecular complexity is a numerical estimate reflecting expert opinions on how complex a molecule is. It is not synonymous with synthetic accessibility, as was determined throughout the analysis in Fig. 2b. Indeed, some molecules with high molecular complexity can be easy to synthesize and vice versa. Molecular complexity remains invariant with the development of new synthetic methods, allowing for the comparison and analysis of synthetic sequences involving many intermediates.
For this analysis, the natural product and popular total synthesis target molecule strychnine37 was selected, due to its well-documented biosynthesis38 and numerous total syntheses performed by organic chemists.39–42 This comparison provides insights into the differences between human and natural synthetic approaches and traces the development and improvement of synthetic methods in organic chemistry from a quantitative perspective.
In the biosynthesis route (Fig. 5a, biosynthesis), Nature uses specially designed enzymes for each transformation, allowing chemical transformations to be carried out with excellent selectivity and efficiency. The non-monotonic behavior of molecular complexity during biosynthesis is evident. For instance, in the biosynthesis of strychnine, one step includes the glycosylation of the alcohol group (stage 6), which later disappears (stage 11). Although this approach does not align with the atom economy paradigm, the glucose molecule is reused for other biosystem purposes.
 | ||
| Fig. 5 Total synthesis analysis. (a) Changes in molecular complexity in different syntheses of strychnine; selected reactions illustrate some changes in molecular complexity. The crosses denote the starting reagents; the star corresponds to the final product. (b) Molecular complexity descent directions for strictosidine. (c) Comparison of human-developed (top, left) and Nature-utilized (top, right) approaches for the synthesis of molecules in terms of molecular complexity and other characteristics as well as the evolution of human-developed approaches with time. | ||
Initially developed synthetic approaches also exhibit non-monotonic molecular complexity changes, often due to steps such as protection/deprotection stages. For example, in 1954, the primary objective of Woodward's39 or Overman's40 syntheses of strychnine was to synthesize the target molecule (Fig. 5a, Woodward; Overman). Extensive use of protective groups and functional fragments that are not part of the final product's structure was common. These previously developed approaches resemble Nature's methods in terms of changing molecular complexity, driven by limited synthetic techniques. In contrast, modern approaches allow easier manipulation of molecular complexity.
For example, Vanderwal's 2011 total synthesis41 aimed to synthesize the target molecule efficiently. Vanderwal's synthesis demonstrates the atom economy paradigm, which uses no protection groups and focuses on the monotonic and rapid growth of molecular complexity (Fig. 5a, Vanderwal). This approach minimizes the number of steps between the starting reagents and the target molecule.
Interestingly, Nature's approach efficiently allocates “molecular” resources to multiple targets.43–45 For example, strictosidine plays a crucial role in the biosynthesis of strychnine and other alkaloids. After stage 10 of strychnine biosynthesis, strictosidine, which is more complex than strychnine, serves as an intermediate in at least three other biosynthetic pathways (Fig. 5b). This approach by Nature, termed “ascent–descent” synthesis (Fig. 5c), involves the creation of a high-order intermediate with a relatively high molecular complexity value, which can then be used for various syntheses. This results in a variety of molecules at the desired complexity level.
Overall, the “ascent–descent” synthesis approach is a clever design for synthesizing a family of complex molecules rather than a single molecule. This strategy is not only relevant in natural settings but also addresses major issues in the preparation of compound libraries for the pharmaceutical industry.46 Therefore, large variations in molecular complexity are justified in Nature's methods because highly complex intermediates undergo further modification to form other natural components. This finding suggests that while synthetic methods are improving, they differ significantly from natural pathways.
It is not obvious which approach is better because both have advantages and disadvantages. The benefits of the human approach include atom economy and higher yields due to the smaller number of steps. However, this approach does not address cross-synthesis problems, such as late-stage functionalization, because the paradigm of monotonically increasing molecular complexity lacks flexibility and aims to design a unique synthetic path for each molecule. One of the potential solutions is multicomponent reactions47 that can be highly universal and potentially match Nature's philosophy. In contrast, the natural approach involves transforming intermediate molecules with high molecular complexity into other products, facilitating the modification of already complex molecules during later stages.
Conclusions
This study introduced a novel machine learning-based framework utilizing a Learning to Rank (LTR) approach to quantify molecular complexity, a concept that has traditionally relied on subjective human judgment. By modeling molecular complexity as a ranking problem, we transformed it into a quantifiable property, capturing the intricate decision-making process used by chemists. Over 50 chemists labeled approximately 164000 molecules contributing nearly 200000 molecular complexity comparisons that were used to develop the final model. This large dataset and the active learning process ensured diversity and accuracy, enabling the model to generalize across various chemical structures.
The Gradient Boosted Decision Trees (GBDT) model was selected due to its interpretability and superior performance in ranking tasks. The model achieved 77.5% pair accuracy and 98.1% accuracy in functional group tests, confirming its reliability in distinguishing molecules with different levels of complexity. Key molecular features, such as molecular weight, number of aromatic cycles, and topological polar surface area (TPSA), were identified as the most influential in determining molecular complexity. Interestingly, synthetic accessibility scores, such as SAscore and SCScore, showed significant differences from molecular complexity scores, emphasizing that ease of synthesis does not always correlate with structural complexity.
The developed model has broad potential applications in medicinal chemistry and synthetic methodology. Analysis of FDA-approved drugs from 1985 to 2022 revealed a clear trend toward increasing molecular complexity, reflecting advances in both synthetic techniques and biological targets. The study also explored the use of molecular complexity as a tool to assess reaction strategies, identifying how certain reaction classes increase complexity more efficiently than others. Additionally, the analysis of total synthesis routes compared human-developed methods with natural biosynthetic approaches, highlighting differences in how molecular complexity evolves through synthetic processes. Modern synthetic strategies prioritize atom economy and efficiency, while natural biosynthesis often takes a more flexible “ascent–descent” approach, generating complex intermediates that serve multiple biosynthetic pathways.
Overall, this work provides a step forward in the quantification of molecular complexity, offering a versatile and data-driven tool for analyzing chemical space. By transforming molecular complexity from an intuitive concept into a measurable characteristic, the study opens new possibilities for optimizing synthetic methods, guiding drug discovery, and enhancing our understanding of the molecular landscape.
Although the developed numeric score provides a powerful tool for quantifying an inherently subjective concept, it is important to acknowledge some limitations. Despite rigorous model development, extensive data collection, and validation with expert input, the score may not fully capture every aspect of molecular complexity, particularly in cases involving rare or unconventional molecular structures. Additionally, the reliance of the model on labeled data and specific molecular features means that its predictions are inherently influenced by the available dataset and the expertise of participating experts. Users should exercise caution and avoid treating the score as an absolute measure of complexity, but rather as a useful reference point within the broader context of chemical analysis. Continuous refinement and validation with new data will be essential for improving the accuracy and applicability of the model in the future.
Methods
Molecular complexity: ranking approach
Learning to Rank (LTR) is one of the classical machine learning tasks that is often overlooked by researchers, which is nevertheless widely used in critical areas such as information retrieval, recommender systems, machine translation, and many other fields. LTR is a unique task that partially resembles both regression and classification but also has its own features. The formulation of the problem for LTR is as follows: one needs to construct a real-valued function:where X is a set of all objects that would satisfy a given order on pairs of objects, i.e., for a given pair of objects with indices i and j, such that

 (which means that object xj is more relevant than object xi) function a should satisfy the condition a(xi) < a(xj).
(which means that object xj is more relevant than object xi) function a should satisfy the condition a(xi) < a(xj).
Multiple problems can be formulated as ranking tasks in the context of chemistry. In particular, medicinal chemists have attempted to replace the drug likeness score with the intuition of medicinal chemists. In the work by Choung et al.,48 medicinal chemists were offered to pick better-looking molecules for a particular task in the proposed pair of molecules. Compared with previous methods, this very simple LTR approach has several advantages.
LTR algorithms are usually divided into pointwise, pairwise, and listwise.49 The first group of algorithms calculates the relevance based on one object. The second or pairwise group of LTR algorithms considers pairs of objects and assigns relevance to one of the documents in the pair. In other words, it tries to answer the question: which document is more relevant? Finally, the last group of algorithms (listwise approach) attempts to analyze the whole list of documents to predict the order of ranked documents correctly. Recent research conducted by Prokhorenkova et al. has shown that pairwise algorithms, particularly the YetiRank algorithm, demonstrate state-of-the-art performance according to ranking metrics, so we also used this algorithm in our work.15 YetiRank is optimizing the following objective function:

Many quality metrics have been invented specifically for the task of ranking. In our work, we used scores from 1 to 5, so we consider only the metrics that fit our requirements to assessors.
where p is the position of the document on the page and where relevance is the assessor's score. The DCG allows one to take into account the position of the document in the output.

where IDCG is a DCG value for perfect ranking. NDCG has all the features of DCG but takes values between 0 and 1 and is, therefore, more interpretable.
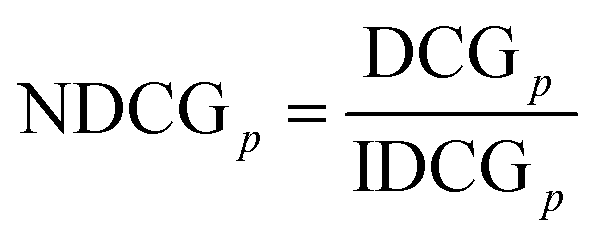
where i,j are pairs of indices and where nq is the number of documents relevant to the entered query. Thus, the denominator of the fraction above is the number of all possible pairs of documents relevant to the entered query.

Given the specificity of the molecular complexity problem, we can reformulate it in terms of LTR in the following way: the query in this setting would be a list of N molecules provided to an expert (in our case, we chose N to be 5). The ranking model learns from the expert scores given to each sample of molecules, and the scores themselves act as a measure of relevance if we reduce our problem to an example of the search engine ranking task.
How is our approach better than any other approach? This approach is more flexible than the regression approach for the following reasons:
• It is possible to integrate active learning into the labeling pipeline (see Fig. 1d).
• LTR allows for different molecular set selection strategies (see Fig. 1e).
These advantages make the exploration of the chemical space significantly more efficient and thus allow the labeling of molecules of high relevance.
Data collection
To collect the data, we split the process into several stages, as illustrated in Fig. 1e:(1) Absolute values phase. In this data collection stage, the experts were asked to assign a score from 1 to 5 to each presented molecule on the web page (labels could repeat for the given set of 5 molecules). The votes did not need to be unique. The initial pool of molecules was selected as a random subset of PubChem50 by splitting by the molecular weights (Fig. 1f) so that the initial training data sample would contain a set that is diverse enough and, at the same time, would not be too large so that each molecule in the set would receive enough labels.
(2) Active learning phase. For this stage, we utilized the labeled data from the first stage to select the molecules of similar complexity; this way, during the training, the model would be able to distinguish the subtle complexity difference between the provided molecules. The experts were asked to provide unique labels.
(3) Ranking phase. This phase combined some features of the first and second stages. The experts were given randomly selected molecules and were asked to provide unique labels. By including the data from this phase, the model would be able to learn the information related to the ordering of molecules based on molecular complexity.
Most off-the-shelf platforms for data labeling are not customizable enough. One of our goals was to incorporate the active learning cycle into the data collection and model training. Additionally, we needed full control over the labeled data and timely correction of minor flaws. Therefore, the decision to build a custom web application was made.
For this purpose, we developed a web interface based on the Python programming language using the Flask web framework. The advantages of Flask, compared to other web frameworks, are its simplicity, speed of operation, and easy integration for any need.
We also created a Docker container for the deployment of our website. This allowed us to create additional protection for the data stored on our server by isolating the contents of the container from the host machine. The container was composed of 8 GB of RAM and 256 GB of persistent memory.
Within the constraints of time and resources, we decided to use a local database, which would not require additional capacity. The database used was SQLite3, which was integrated with Python. The data were written to a file stored in the container, which all project developers had access to. Storing the database as files allowed us to quickly respond to any errors that occurred in the process of labeling the molecules and to find artifacts in the data.
Approximately 1.6 million compounds, which included C, H, O, N, S, Si, P, B, As, I, Br, Cl, F, and other elements, were obtained from the PubChem dataset. Molecules were displayed in the web interface using the RDKit chemistry framework: using this framework, SMILES strings from PubChem were converted to SVG format and integrated into the HTML markup.
At the time of registration, all users were classified according to their level of education (DSc in Chemistry, PhD in Chemistry, MSc/BA in Chemistry, and currently an MSc/BA Chemistry program student).
A total of 294542 labels were collected by acquiring expert opinions from the researchers. In total, we had 127 participants. After processing the quality controls, the number of participants whose data were used for the training of the final model was narrowed to 50 experts. Each participant was asked to rank molecules from 1 to 5 within one page based on their molecular complexity. A gamification technique was used to make the labeling process more enjoyable. Each person was assigned a score that depended on the number of labeled molecules. This score was used to assign virtual rewards. In addition, there was a secret prize for the first person to score 5000 labeled molecules.
To ensure that each of the participants accurately labeled the data, we created a small set of 50 molecules for which the correct ranking was trivial. We gave this subset of molecules to the participants to detect people who did not label the molecules correctly. The labels of these participants were filtered and not used for model training.
To represent molecules numerically, we selected the following set of features:
RDKit-generated features 51—encoding the structural feature of a molecule.
Among the features of this group are molecular weight, number of atoms, number of heavy atoms (heavier than hydrogen), topological surface polar area, number of heteroatoms, number of atoms in spirocycles, number of rotatable bonds, number of aliphatic and aromatic cycles, number of bridgehead atoms, number of stereocenters, and number of stereoisomers of a molecule.
SCScore22is a score developed to evaluate the synthetic complexity of the molecules.
For a summary of the other datasets used throughout this work, see Section 7 of the ESI.†
Machine learning model development
All the data we used were processed using the Pandas library, and all the machine learning models were trained using the CatBoost library.17 The phase weighting can be specified by providing the group_weight values to the CatBoost Pool object.We used the SMILES representation of molecules to perform the analysis and calculate descriptors. To relate the molecular descriptors to the molecule representation, the RDKit library was used.51 The descriptors were then used to train the model. Python was the main programming language of this project.
Synthesis data mining
All syntheses were redrawn in ChemDraw, and the SMILES representation of each chemical compound was used for the analysis (the molecular complexity value can be calculated provided a SMILES representation of the molecule): strychnine38–42 and artemisinine.52–55Code availability
Code for molecular complexity prediction and analysis as well as information about the data can be found at https://github.com/Ananikov-Lab/digitizing_molecular_complexity/.Data availability
The data for this study are described in the article and the ESI.† Data downloading instructions can be found on the Github page of the project.Author contributions
A. T. co-developed the web service, developed the models, designed, executed and analyzed experiments. D. B. co-developed the web service, developed the models, analyzed the experiments, conceptualized the work. N. K. co-developed and maintained the web service. V. A. developed the project, co-analyzed the results, supervised and conceptualized the work. All authors participated in the discussion and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the people who were involved in the data labeling process.References
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, The Maximal Affinity of Ligands, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(18), 9997–10002, DOI:10.1073/pnas.96.18.9997.
- S. H. Bertz, The First General Index of Molecular Complexity, J. Am. Chem. Soc., 1981, 103(12), 3599–3601, DOI:10.1021/ja00402a071.
- J. B. Hendrickson, P. Huang and A. G. Toczko, Molecular Complexity: A Simplified Formula Adapted to Individual Atoms, J. Chem. Inf. Comput. Sci., 1987, 27(2), 63–67, DOI:10.1021/ci00054a004.
- T. Böttcher, An Additive Definition of Molecular Complexity, J. Chem. Inf. Model., 2016, 56(3), 462–470, DOI:10.1021/acs.jcim.5b00723.
- J. R. A. Proudfoot, Path Based Approach to Assessing Molecular Complexity, Bioorg. Med. Chem. Lett., 2017, 27(9), 2014–2017, DOI:10.1016/j.bmcl.2017.03.008.
- M. von Korff and T. Sander, Molecular Complexity Calculated by Fractal Dimension, Sci. Rep., 2019, 9(1), 967, DOI:10.1038/s41598-018-37253-8.
- T. I. Oprea and C. Bologa, Molecular Complexity: You Know It When You See It, J. Med. Chem., 2023, 66(18), 12710–12714, DOI:10.1021/acs.jmedchem.3c01507.
- A. Krzyzanowski, A. Pahl, M. Grigalunas and H. Waldmann, Spacial Score-A Comprehensive Topological Indicator for Small-Molecule Complexity, J. Med. Chem., 2023, 66(18), 12739–12750, DOI:10.1021/acs.jmedchem.3c00689.
- O. Méndez-Lucio and J. L. Medina-Franco, The Many Roles of Molecular Complexity in Drug Discovery, Drug Discov. Today, 2017, 22(1), 120–126, DOI:10.1016/j.drudis.2016.08.009.
- R. P. Sheridan, N. Zorn, E. C. Sherer, L.-C. Campeau, C. Chang, J. Cumming, M. L. Maddess, P. G. Nantermet, C. J. Sinz and P. D. O'Shea, Modeling a Crowdsourced Definition of Molecular Complexity, J. Chem. Inf. Model., 2014, 54(6), 1604–1616, DOI:10.1021/ci5001778.
- A. Sharma, D. Czégel, M. Lachmann, C. P. Kempes, S. I. Walker and L. Cronin, Assembly Theory Explains and Quantifies Selection and Evolution, Nature, 2023, 622(7982), 321–328, DOI:10.1038/s41586-023-06600-9.
- M. Jirasek, A. Sharma, J. R. Bame, S. H. M. Mehr, N. Bell, S. M. Marshall, C. Mathis, A. MacLeod, G. J. T. Cooper, M. Swart, R. Mollfulleda and L. Cronin, Investigating and Quantifying Molecular Complexity Using Assembly Theory and Spectroscopy, ACS Cent. Sci., 2024, 10(5), 1054–1064, DOI:10.1021/acscentsci.4c00120.
- M. F. Freeman, M. J. Helf, A. Bhushan, B. I. Morinaka and J. Piel, Seven Enzymes Create Extraordinary Molecular Complexity in an Uncultivated Bacterium, Nat. Chem., 2017, 9(4), 387–395, DOI:10.1038/nchem.2666.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem 2023 Update, Nucleic Acids Res., 2023, 51(D1), D1373–D1380, DOI:10.1093/nar/gkac956.
- I. Lyzhin, A. Ustimenko, A. Gulin and L. Prokhorenkova, Which Tricks Are Important for Learning to Rank?, 2022.
- T. Chen and C. Guestrin, XGBoost, in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, ACM, New York, NY, USA, 2016, pp 785–794, DOI:10.1145/2939672.2939785.
- L. Prokhorenkova, G. Gusev, A. Vorobev, A. V. Dorogush and A. C. Gulin, Unbiased Boosting with Categorical Features, Adv. Neural Inf. Process. Syst., 2018, 6638–6648 Search PubMed.
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. P. Magariños, J. F. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. J. Radoux, A. Segura-Cabrera, A. Hersey and A. R. Leach, ChEMBL: Towards Direct Deposition of Bioassay Data, Nucleic Acids Res., 2019, 47(D1), D930–D940, DOI:10.1093/nar/gky1075.
- H. Mary, E. Noutahi, DomInvivo, L. Zhu, M. Moreau, S. Pak, D. Gilmour, S. Whitfield; J. H. Valence, H. Hounwanou, I. Kumar, S. Maheshkar, S. Nakata, K. M. Kovary, C. Wognum, M. Craig, D. Bot, Datamol-Io/Datamol: 0.12.3, Zenodo, 2024, DOI:10.5281/zenodo.10535844.
- S. M. Lundberg and S. I. Lee, A Unified Approach to Interpreting Model Predictions, Adv. Neural Inf. Process. Syst., 2017, 4766–4775 Search PubMed.
- P. Ertl and A. Schuffenhauer, Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions, J. Cheminf., 2009, 1(1), 8, DOI:10.1186/1758-2946-1-8.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, SCScore: Synthetic Complexity Learned from a Reaction Corpus, J. Chem. Inf. Model., 2018, 58(2), 252–261, DOI:10.1021/acs.jcim.7b00622.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, MoleculeNet: A Benchmark for Molecular Machine Learning, Chem. Sci., 2018, 9(2), 513–530, 10.1039/C7SC02664A.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Quantum Chemistry Structures and Properties of 134 Kilo Molecules, Sci. Data, 2014, 1(1), 140022, DOI:10.1038/sdata.2014.22.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun and M. Zitnik, Artificial Intelligence Foundation for Therapeutic Science, Nat. Chem. Biol., 2022, 18(10), 1033–1036, DOI:10.1038/s41589-022-01131-2.
- X. Verdaguer, Building Molecular Complexity from Scratch, Science, 2016, 353(6302), 866–867, DOI:10.1126/science.aah5871.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, A Robotic Platform for Flow Synthesis of Organic Compounds Informed by AI Planning, Science, 2019, 365(6453), eaax1566, DOI:10.1126/science.aax1566.
- P. Schwaller, D. Probst, A. C. Vaucher, V. H. Nair, D. Kreutter, T. Laino and J. L. Reymond, Mapping the Space of Chemical Reactions Using Attention-Based Neural Networks, Nat. Mach. Intell., 2021, 3(2), 144–152, DOI:10.1038/s42256-020-00284-w.
- N. Schneider, D. M. Lowe, R. A. Sayle and G. A. Landrum, Development of a Novel Fingerprint for Chemical Reactions and Its Application to Large-Scale Reaction Classification and Similarity, J. Chem. Inf. Model., 2015, 55(1), 39–53, DOI:10.1021/ci5006614.
- M. Murakami and N. Ishida, A Shortcut to Molecular Complexity, Nat. Chem., 2017, 9(4), 298–299, DOI:10.1038/nchem.2750.
- J. Ye, Z. Shi, T. Sperger, Y. Yasukawa, C. Kingston, F. Schoenebeck and M. Lautens, Remote C−H Alkylation and C−C Bond Cleavage Enabled by an in Situ Generated Palladacycle, Nat. Chem., 2017, 9(4), 361–368, DOI:10.1038/nchem.2631.
- D. S. Reddy, I. M. Novitskiy and A. G. Kutateladze, Maximizing Step-Normalized Increases in Molecular Complexity: Formal [4+2+2+2] Photoinduced Cyclization Cascade to Access Polyheterocycles Possessing Privileged Substructures, Angew. Chem., Int. Ed., 2022, 61(4), e202112573, DOI:10.1002/anie.202112573.
- G. A. Ross, C. Lu, G. Scarabelli, S. K. Albanese, E. Houang, R. Abel, E. D. Harder and L. Wang, The Maximal and Current Accuracy of Rigorous Protein-Ligand Binding Free Energy Calculations, Commun. Chem., 2023, 6(1), 222, DOI:10.1038/s42004-023-01019-9.
- L. Shi, H. Pan, Z. Liu, J. Xie and W. Han, Roles of PFKFB3 in Cancer, Signal Transduct. Targeted Ther., 2017, 2(1), 17044, DOI:10.1038/sigtrans.2017.44.
- N. Boutard, A. Białas, A. Sabiniarz, P. Guzik, K. Banaszak, A. Biela, M. Bień, A. Buda, B. Bugaj, E. Cieluch, A. Cierpich, Ł. Dudek, H. Eggenweiler, J. Fogt, M. Gaik, A. Gondela, K. Jakubiec, M. Jurzak, A. Kitlińska, P. Kowalczyk, M. Kujawa, K. Kwiecińska, M. Leś, R. Lindemann, M. Maciuszek, M. Mikulski, P. Niedziejko, A. Obara, H. Pawlik, T. Rzymski, M. Sieprawska-Lupa, M. Sowińska, J. Szeremeta-Spisak, A. Stachowicz, M. M. Tomczyk, K. Wiklik, Ł. Włoszczak, S. Ziemiańska, A. Zarębski, K. Brzózka, M. Nowak and C. Fabritius, Discovery and Structure–Activity Relationships of N-Aryl 6-Aminoquinoxalines as Potent PFKFB3 Kinase Inhibitors, ChemMedChem, 2019, 14(1), 169–181, DOI:10.1002/cmdc.201800569.
- B. A. Wright and R. Sarpong, Molecular Complexity as a Driving Force for the Advancement of Organic Synthesis, Nat. Rev. Chem, 2024, 8(10), 776–792, DOI:10.1038/s41570-024-00645-8.
- J. Bonjoch and D. Solé, Synthesis of Strychnine, Chem. Rev., 2000, 100(9), 3455–3482, DOI:10.1021/cr9902547.
- B. Hong, D. Grzech, L. Caputi, P. Sonawane, C. E. R. López, M. O. Kamileen, N. J. Hernández Lozada, V. Grabe and S. E. O'Connor, Biosynthesis of Strychnine, Nature, 2022, 607(7919), 617–622, DOI:10.1038/s41586-022-04950-4.
- R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, The Total Synthesis Of Strychnine, J. Am. Chem. Soc., 1954, 76(18), 4749–4751, DOI:10.1021/ja01647a088.
- S. D. Knight, L. E. Overman and G. Pairaudeau, Asymmetric Total Syntheses of (-)- and (+)-Strychnine and the Wieland-Gumlich Aldehyde, J. Am. Chem. Soc., 1995, 117(21), 5776–5788, DOI:10.1021/ja00126a017.
- D. B. C. Martin and C. D. Vanderwal, A Synthesis of Strychnine by a Longest Linear Sequence of Six Steps, Chem. Sci., 2011, 2(4), 649, 10.1039/c1sc00009h.
- M. Nakanishi and M. Mori, Total Synthesis of (−)-Strychnine, Angew. Chem., Int. Ed., 2002, 41(11), 1934, DOI:10.1002/1521-3773(20020603)41:11<1934::AID-ANIE1934>3.0.CO;2-F.
- J. Gao, Y. Zuo, F. Xiao, Y. Wang, D. Li, J. Xu, C. Ye, L. Feng, L. Jiang, T. Liu, D. Gao, B. Ma, L. Huang, Z. Xu and J. Lian, Biosynthesis of Catharanthine in Engineered Pichia Pastoris, Nat. Synth., 2023, 2, 231–242, DOI:10.1038/s44160-022-00205-2.
- M. Kang, R. Fu, P. Zhang, S. Lou, X. Yang, Y. Chen, T. Ma, Y. Zhang, Z. Xi and J. Liu, A Chromosome-Level Camptotheca Acuminata Genome Assembly Provides Insights into the Evolutionary Origin of Camptothecin Biosynthesis, Nat. Commun., 2021, 12(1), 3531, DOI:10.1038/s41467-021-23872-9.
- F. Trenti, K. Yamamoto, B. Hong, C. Paetz, Y. Nakamura and S. E. O'Connor, Early and Late Steps of Quinine Biosynthesis, Org. Lett., 2021, 23(5), 1793–1797, DOI:10.1021/acs.orglett.1c00206.
- M. Moir, J. J. Danon, T. A. Reekie and M. Kassiou, An Overview of Late-Stage Functionalization in Today's Drug Discovery, Expet Opin. Drug Discov., 2019, 14(11), 1137–1149, DOI:10.1080/17460441.2019.1653850.
- E. Ruijter, R. Scheffelaar and R. V. A. Orru, Multicomponent Reaction Design in the Quest for Molecular Complexity and Diversity, Angew. Chem., Int. Ed., 2011, 50(28), 6234–6246, DOI:10.1002/anie.201006515.
- O.-H. Choung, R. Vianello, M. Segler, N. Stiefl and J. Jiménez-Luna, Extracting Medicinal Chemistry Intuition via Preference Machine Learning, Nat. Commun., 2023, 14(1), 6651, DOI:10.1038/s41467-023-42242-1.
- T. Y. Liu, Learning to Rank for Information Retrieval, Found. Trends Inf. Retr., 2009, 3(3), 225–331, DOI:10.1561/1500000016.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem in 2021: New Data Content and Improved Web Interfaces, Nucleic Acids Res., 2021, 49(D1), D1388–D1395, DOI:10.1093/nar/gkaa971.
- G. Landrum, RDKit: Open-source Cheminformatics, http://www.rdkit.org/.
- N. K. B. K. Ikram and H. T. Simonsen, A Review of Biotechnological Artemisinin Production in Plants, Front. Plant Sci., 2017, 8, 1966, DOI:10.3389/fpls.2017.01966.
- M. A. Avery, W. K. M. Chong and C. Jennings-White, Stereoselective Total Synthesis of (+)-Artemisinin, the Antimalarial Constituent of Artemisia Annua L, J. Am. Chem. Soc., 1992, 114(3), 974–979, DOI:10.1021/ja00029a028.
- C. Zhu and S. P. Cook, A Concise Synthesis of (+)-Artemisinin, J. Am. Chem. Soc., 2012, 134(33), 13577–13579, DOI:10.1021/ja3061479.
- J. Krieger, T. Smeilus, M. Kaiser, E. Seo, T. Efferth and A. Giannis, Total Synthesis and Biological Investigation of (−)-Artemisinin: The Antimalarial Activity of Artemisinin Is Not Stereospecific, Angew. Chem., Int. Ed., 2018, 57(27), 8293–8296, DOI:10.1002/anie.201802015.
Footnotes |
| † Electronic supplementary information (ESI) available: Information about machine learning development; calculated molecular complexity at each step of the analyzed synthetic procedures; information on the analyzed synthetic pathways; summary of all the datasets used in this work. See DOI: https://doi.org/10.1039/d4sc07320g |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2025 |