
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-induced changes in the electrocatalytic activity of atomically precise Au25 nanoclusters†
Lipan
Luo‡
a,
Xia
Zhou‡
b,
Yuping
Chen
a,
Fang
Sun
a,
Likai
Wang
 *b and
Qing
Tang
*a
*b and
Qing
Tang
*a
aSchool of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Chemical Theory and Mechanism, Chongqing University, Chongqing 401331, China. E-mail: qingtang@cqu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, Shandong 255049, China. E-mail: lkwangchem@sdut.edu.cn
First published on 20th January 2025
Abstract
Atomically precise gold nanoclusters have shown great promise as model electrocatalysts in pivotal electrocatalytic processes such as the hydrogen evolution reaction (HER) and carbon dioxide reduction reaction (CO2RR). Although the influence of ligands on the electronic properties of these nanoclusters is well acknowledged, the ligand effects on their electrocatalytic performances have been rarely explored. Herein, using [Au25(SR)18]− nanoclusters as a prototype model, we demonstrated the importance of ligand hydrophilicity versus hydrophobicity in modulating the interface dynamics and electrocatalytic performance. Our first-principles calculations revealed that Au25 protected by hydrophilic –SCH2COOH ligands exhibits faster kinetics in stripping the thiolate ligand and better HER activity due to enhanced proton transfer facilitated by boosted interface hydrogen bonding. Conversely, Au25 protected by hydrophobic –SCH2CH3 ligands demonstrates enhanced CO2RR performance by minimizing water interference to stabilize the key *COOH intermediate and lower the barrier for CO formation. Experimental validation using synthesized hydrophilic and hydrophobic ligand-protected Au25 nanoclusters (NCs), such as [Au25(MPA)18]− (MPA = mercaptopropionic acid), [Au25(MHA)18]− (MHA = 6-mercaptohexanoic acid), and [Au25(SC6H13)18]−, confirms these findings, where the hydrophilic ligand-protected Au25 NCs exhibit better activity and stability in the HER, while the hydrophobic ligand-protected Au25 NCs achieve higher faradaic efficiency and current density in the CO2RR. The mechanistic insights in this study provide valuable guidance for the rational design of surface microenvironments in efficient nanocatalysts for sustainable energy applications.
1. Introduction
The electrocatalytic reactions such as the hydrogen evolution reaction (HER)1–4 and carbon dioxide reduction reaction (CO2RR) have been considered cornerstones in the pursuit of sustainable ways of meeting global energy demands.5–7 These reactions are critical for the production of clean energy and recycling of carbon, as they enable the conversion of CO2 into valuable chemicals and fuels and generate hydrogen as a clean energy carrier.8,9 Given the importance of these processes, the development of highly efficient and stable catalysts for both the CO2RR and HER is of paramount importance. Atomically precise metal nanoclusters (NCs), renowned for their unique structures and molecule-like properties, have shown exceptional catalytic capabilities due to their quantum confinement effects and a high density of active sites.10–18 Among these, [Au25(SR)18]− NCs have been particularly recognized for their significant electrocatalytic activities in both CO2 reduction12,19–22 and hydrogen evolution.23–27 The pioneering work by Kauffman et al.28 first highlighted the ability of [Au25(SR)18]− to efficiently reduce CO2 to CO with high selectivity and low overpotential at commercially viable current densities, establishing the potential of these nanoclusters in electrocatalysis. The bimetallic PtAu24(SR)18 NCs, explored by Kwak et al.,29 demonstrated remarkable catalytic activity for hydrogen production in the HER, surpassing previously reported molecular catalysts and even outperforming traditional platinum catalysts, setting a new benchmark in the field.In the context of Au25 NCs, the choice and nature of ligands are critical factors that significantly influence their stability and catalytic performance. Ligands act as the outer protective layers, not only preventing aggregation and stabilizing the nanocluster, but also modulating its electronic properties and surface chemistry.26,30–35 The thiol-based ligands, in particular, are widely used due to their strong S–metal bonds, which provide high stability and a straightforward preparation strategy.36–38 However, beyond the stabilization effect, the intrinsic properties of the ligands—such as their hydrophilicity or hydrophobicity—can profoundly affect the nanocluster's interaction with reactants and its overall electrocatalytic activity.39 For instance, Kwak et al.25 investigated the impact of different ligands on the HER performance and found that the 3-mercapto-1-propanesulfonic acid (MPS) protected Au25 NC achieved a rate constant of 121![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 at an overpotential of 0.7 V, which was 11 times higher than that of 1-hexanethiolate (C6S) protected Au25, highlighting the profound influence of ligand hydrophilicity on catalytic efficiency. Moreover, recent work by Yoo et al.40 has shown that the introduction of hydrophobic ligands in a silver nanocluster Ag25 can enhance the CO2 reduction activity, achieving a faradaic efficiency for CO (FECO) of over 90% and a partial current density (jCO) as high as −240 mA cm−2 in a gas-fed membrane electrode assembly device. Despite these experimental advancements, atomic-level understanding and elucidation of the specific effects of hydrophilic versus hydrophobic ligands on the electrocatalytic performance of atomically precise NCs have been lacking. This uncertainty underscores the urgent need for a systematic investigation into how the ligand properties influence these catalytic processes. In particular, understanding these interactions is crucial for further optimizing the performance of Au25 NCs and could also provide valuable insights into the rational design of nanocluster-based electrocatalysts for sustainable energy applications.
000 s−1 at an overpotential of 0.7 V, which was 11 times higher than that of 1-hexanethiolate (C6S) protected Au25, highlighting the profound influence of ligand hydrophilicity on catalytic efficiency. Moreover, recent work by Yoo et al.40 has shown that the introduction of hydrophobic ligands in a silver nanocluster Ag25 can enhance the CO2 reduction activity, achieving a faradaic efficiency for CO (FECO) of over 90% and a partial current density (jCO) as high as −240 mA cm−2 in a gas-fed membrane electrode assembly device. Despite these experimental advancements, atomic-level understanding and elucidation of the specific effects of hydrophilic versus hydrophobic ligands on the electrocatalytic performance of atomically precise NCs have been lacking. This uncertainty underscores the urgent need for a systematic investigation into how the ligand properties influence these catalytic processes. In particular, understanding these interactions is crucial for further optimizing the performance of Au25 NCs and could also provide valuable insights into the rational design of nanocluster-based electrocatalysts for sustainable energy applications.
In this work, we systematically investigated the ligand effect on the interface stability and the electrocatalytic performance of Au25 NCs in both acidic HER and alkaline CO2RR processes. Utilizing hydrophilic [Au25(SCH2COOH)17]− and hydrophobic [Au25(SCH2CH3)17]− NCs as theoretical models, we first explored how the ligand properties influence the Au–S interface dynamics, the electronic structures, and the electrocatalytic reaction kinetics via the constant potential calculations and ab initio molecular dynamics (AIMD) simulations. The results revealed that the Au–S interface is unstable at the applied electrochemical reduction potential, and hydrophilic [Au25(SCH2COOH)17]− exhibits faster kinetics for the stripping of the –SR ligand. Moreover, in the acidic environment, hydrophilic Au25 NCs exhibit superior HER performance compared to the hydrophobic one due to the enhanced proton transfer and hydrogen evolution facilitated by the hydrophilic environment. Conversely, under alkaline conditions, hydrophobic Au25 NCs show better CO2RR activity by promoting the adsorption and stabilization of CO2 intermediates while minimizing the water interference around the reaction interface. Our theoretical predictions are then validated through the experimental studies, where we synthesized mercaptopropionic acid-protected [Au25(MPA)18]−, 6-mercaptohexanoic acid-protected [Au25(MHA)18]−, and hexanethiol-protected [Au25(SC6H13)18]− NCs as model systems and performed the electrochemical tests. These insights provide a deeper understanding of how the ligand environments affect the electrocatalytic activities of Au25 nanoclusters and offer valuable guidance for the rational design of promising nanocatalysts for electrocatalytic applications.
2. Results and discussion
Ligand detachment dynamics
The removal of surface ligands from nanoclusters is a critical step in enhancing their electrocatalytic activity. Fully ligand-protected nanoclusters, such as Au25(SR)18, are typically considered electrochemically inactive and exhibit significant overpotential during the electrocatalytic process.20,41–44 This inactivity is primarily caused by the passivating effect of the ligands, which block the access of reactants to the metal core and hinder the efficient mass and charge transfer between the nanocluster and the reactants.45,46 Consequently, the selective removal of ligands from specific sites on the nanocluster surface is necessary to expose the active sites. Our recent studies have provided valuable insights into the mechanism of partial ligand removal in Au25(SCH3)18 NCs, where the applied reduction potential would induce the spontaneous desulfurization process via breaking the Au–S bonds regardless of the electrolyte environment (acidic, neutral or basic conditions).47Building on these prior findings, we first combined the constant potential calculations and AIMD simulations to explore how the presence of surface hydrophilic and hydrophobic ligands affects the stability of the Au–S interface under acidic electrochemical conditions. We modeled Au25 NCs with ligands aligned along the z-axis, simulating the system in both explicit water slabs and an implicit solvation environment to accurately capture the realistic electrochemical conditions. Our theoretical model includes hydrophilic [Au25(CH2COOH)18]− and hydrophobic [Au25(CH2CH3)18]− NCs (illustrated in Fig. S2†) placed in the simulation box filled with bulk water at an average density of approximately 1 g cm−3, which comprises 191 H2O molecules and one H3O+ ion to simulate the acidic environment.48 To explicitly consider the electrode potential, we manually adjusted the number of extra electrons to control the applied potential U (more details on the constant potential calculations can be found in the ESI†). Our AIMD simulations at room temperature (300 K) revealed that the –SR ligands on both the [Au25(CH2COOH)18]− and [Au25(CH2CH3)18]− NCs become unstable when subjected to the applied potential. In the dynamic process, the proton from the solvated H3O+ ion is attracted and adsorbed onto the sulfur atom, weakening the Au–S bonds. When the potential becomes sufficiently negative, this weakening leads to the complete breakage of the Au–S bonds and the formation of the HSR molecule, as illustrated in the AIMD snapshots (Fig. 1, left). Specifically, in the case of [Au25(CH2CH3)18]− NCs, the potential (URHE = −1.36 V) was not negative enough to facilitate proton adsorption onto the sulfur atom, leading instead to proton transfer and diffusion into the solvent (details provided in Fig. S3†). In contrast, when a lower potential (URHE = −1.54 V) was applied, the Au–S bonds in [Au25(CH2CH3)18]− NCs were completely broken (Fig. 1b).
 | ||
| Fig. 1 Relative distances between representative atoms (marked with numbers) during the equilibrated AIMD simulations of (a) [Au25(SCH2COOH)18]− and (b) [Au25(SCH2CH3)18]− NCs at around 300 K and at an applied potential of −0.98 V for [Au25(SCH2COOH)18]− and −1.54 V for [Au25(SCH2CH3)18]− in an acidic environment (pH = 0). The corresponding AIMD snapshots after 10 ps are shown on the left, while the insets (right) illustrate the local structures, highlighting the breaking of the Au(surface)–S and subsequent Au(staple)–S bonds. The water molecules in the water layer are shown in line mode. Color codes: yellow, Au; blue, S; grey, C; white, H from the ligand; green, the adsorbed H from the proton in solution. The same color scheme is used in the figures below. | ||
Fig. 1a illustrates the dynamic behavior of [Au25(SCH2COOH)18]− NC at an applied potential of URHE = −0.98 V at 300 K in an acidic environment (pH = 0). The Au1(surface)–S26 bond first rapidly breaks at around 0.12 ps with the spontaneous proton adsorption at the S site. The Au8(staple)–S26 bond then breaks at 4.54 ps, which oscillates, reattaches, and completely breaks again at 7.25 ps, leading to bond dissociation thereafter. This eventually results in the detachment of two Au–S bonds, and in the meantime, the –SR ligand combines with a proton to desorb and dissolve into the solution as a HSCH2COOH molecule. A similar –SR detachment process occurs for [Au25(SCH2CH3)18]− at URHE = −1.54 V (Fig. 1b). The proton attack is accompanied by the rapid breaking of the Au4(surface)–S28 bond at around 0.18 ps. Afterwards, the Au3(staple)–S28 bond breaks at 5.01 ps and again at 8.37 ps, after which the ligand with adsorbed H dissolves into the solution as a free HSCH2CH3 molecule. Interestingly, it seems that the hydrophilic [Au25(SCH2COOH)18]− NC exhibits faster etching dynamics in the process of ligand stripping. The faster dynamic process is further supported by the higher number of hydrogen bonds observed in the hydrophilic ligand-protected [Au25(SCH2COOH)18]− system. Specifically, about 113 hydrogen bonds were formed with the surrounding 192 H2O molecules, as shown in Fig. S4a,† compared to the 80 hydrogen bonds formed in the hydrophobic [Au25(SCH2CH3)18]− system. The increased hydrogen bonding in the hydrophilic system facilitates proton transfer to the sulfur atom, thus enhancing the kinetics of ligand detachment. To further investigate whether the carbon chain length of the ligands affects the interaction between the cluster and water molecules, we additionally conducted 10 ps dynamics simulations for Au25 clusters protected by longer hydrophilic and hydrophobic ligands, [Au25(MHA)18]− (MHA = 6-mercaptohexanoic acid) and [Au25(SC6H13)18]−, in the same water environment. The results showed that as the carbon chain length increases, the number of hydrogen bonds between the cluster and water molecules also increases. In the Au25(MHA)18 system, about 276 hydrogen bonds were formed, compared to 245 hydrogen bonds in Au25(SC6H13)18 (Fig. S4b†). Notably, the difference of 31 hydrogen bonds between the hydrophilic and hydrophobic [Au25(MHA)18]− and [Au25(SC6H13)18]− is nearly identical to the 33 hydrogen bond difference observed in [Au25(SCH2COOH)18]− and [Au25(SCH2CH3)18]−. This similarity indicates that, although the total number of hydrogen bonds increases with longer carbon chains, the differences in hydrogen bond counts between Au25 NCs with ligands of similar chain lengths are primarily attributed to the ligand hydrophilicity. Therefore, the accuracy of the simplified ligand calculations can be reliably ensured. These observations collectively underscore the critical role of applied potential, ligand environment, and hydrophilicity in driving the detachment of –SR ligands from the nanocluster surface—a necessary step for exposing active sites and enhancing the electrocatalytic performance of metal NCs.
HER performance in an acidic environment
Building on our above investigation into the dynamics of potential-induced ligand detachment, we now examine the electrocatalytic performance of the two dethiolated [Au25(SR)17]− structures, where one –SR ligand has been removed. To compare the HER performance of dethiolated [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]−, we constructed models that include both the explicit solvation with 50 water molecules and the implicit solvation effects.49 The HER testing was conducted under acidic conditions at pH = 0, providing an optimal environment for hydrogen evolution.Fig. 2a and b present the U–ΔG plots derived from the work function fitting for [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]−, respectively. These plots were obtained by calculating the Ne–U and U–G relationships (additional details are provided in Fig. S5†). The results indicate that the first step of the HER, the Volmer reaction, is thermodynamically favorable across the entire potential range, as evidenced by ΔG values consistently below zero. This suggests that the adsorption of protons onto the nanocluster surface with exposed Au sites occurs spontaneously. However, the second step of the HER, the Heyrovsky reaction—which involves the formation of H2—exhibits ΔG values greater than zero when the potential is not sufficiently negative. As the potential becomes more negative, the ΔG values gradually decrease, eventually falling below zero, indicating that the formation of H2 becomes thermodynamically favorable only at more negative potentials. Therefore, these constant potential thermodynamic calculations lead us to conclude that, for both [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]−, the formation of H2 in the second step of the HER (Heyrovsky reaction) is the rate-determining step.
 | ||
| Fig. 2 Free energy changes (ΔG) for HER steps as a function of electrode potential for (a) [Au25(SCH2COOH)17]− and (b) [Au25(SCH2CH3)17]−. Energy profiles sampled by slow-growth (SG)-AIMD for the HER rate-determining step of (c) [Au25(SCH2COOH)17]− and (d) [Au25(SCH2CH3)17]−at similar potentials (vs. RHE) with representative initial state (IS) and final state (FS) structures. | ||
While thermodynamic calculations provide valuable insights into the feasibility of reaction steps, they often overlook the kinetic barriers that determine the rate at which these reactions proceed. To address this, we employed the slow-growth method within the framework of constrained kinetics to calculate the energy barriers associated with the rate-determining step of the HER for both [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]−. Fig. 2c and d depict the energy barriers as a function of the constrained variable (CV) (Fig. S1†) at an applied potential of −0.66 V. As the constraint increases, the energy barrier increases until it peaks at the point where two hydrogen atoms—one adsorbed at the Au bridge site and the other from H3O+ in the water environment—combine to form H2 and then desorb. The analysis of the reaction pathway shows that the [Au25(SCH2COOH)17]− system reaches this maximum energy barrier earlier, with a free energy of 0.42 eV, indicating a lower energy requirement for the rate-determining step. In contrast, at a similar potential, the energy barrier for the HER rate-determining step in the [Au25(SCH2CH3)17]− system is higher, at 0.54 eV. These results suggest that the hydrophilic [Au25(SCH2COOH)17]− nanocluster exhibits superior HER performance compared to the hydrophobic [Au25(SCH2COOH)17]− nanocluster. The lower kinetic barrier in the hydrophilic [Au25(SCH2COOH)17]− system facilitates a more efficient hydrogen evolution reaction. This improved performance can be attributed to the enhanced interaction between the hydrophilic ligands and the surrounding aqueous environment, which promotes the formation and desorption of H2, thereby making the HER process more favorable in [Au25(SCH2COOH)17]−.
CO2RR performance in an alkaline environment
We further evaluated the CO2RR performance of dethiolated [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]− by constant potential thermodynamic and kinetic calculations. The CO2RR performance testing was carried out under alkaline conditions at pH = 14. Note that the alkaline environments are favorable for CO2 reduction and significantly suppress the HER, minimizing the interference and allowing for a more targeted comparison of CO2RR activity between the two nanoclusters.Fig. 3a and b show the U–ΔG plots derived from work function fitting for [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]−, respectively (additional details are provided in Fig. S6†). These plots detail the ΔG values associated with the four key steps of the CO2RR. Notably, the reaction of *COOH → *CO + OH− exhibits the highest ΔG value when the potential is higher than −1.0 V, indicating that it is the thermodynamically most challenging step. In contrast, the other three steps show lower ΔG values, suggesting that they are either spontaneous or more easily facilitated under the same potential conditions. Therefore, we conclude that the conversion of *COOH to *CO is the rate-determining step for the CO2RR in both the nanocluster systems.
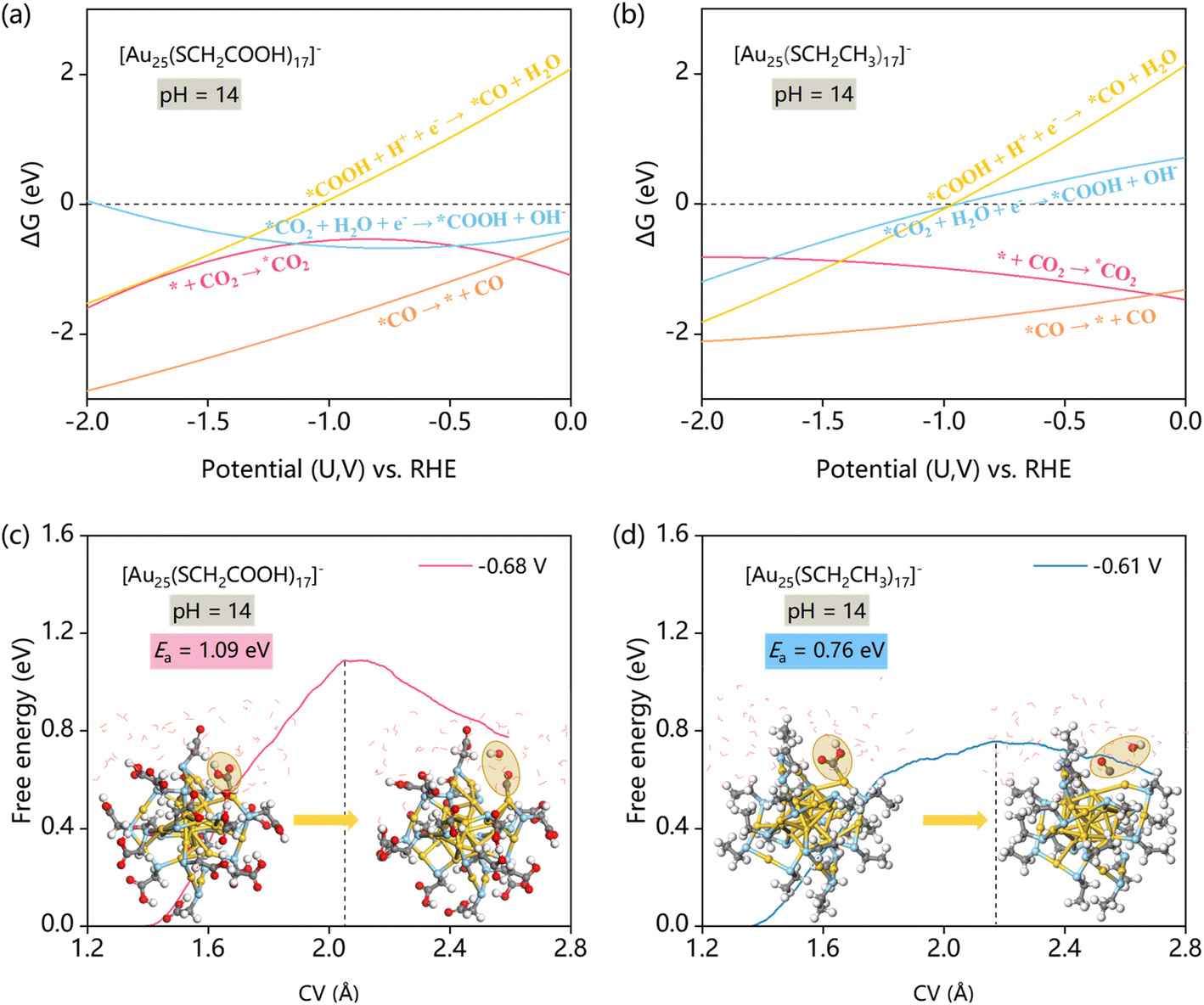 | ||
| Fig. 3 Free energy changes (ΔG) for CO2RR steps as a function of electrode potential for (a) [Au25(SCH2COOH)17]− and (b) [Au25(SCH2CH3)17]−. Energy profiles sampled by SG-AIMD for the rate-determining CO2RR step of (c) [Au25(SCH2COOH)17]− and (d) [Au25(SCH2CH3)17]− at similar potentials (vs. RHE) with representative IS and FS structures. | ||
Next, we simulated the constant potential kinetics of this rate-determining step for both nanoclusters at similar applied potentials. As depicted in Fig. 3c and d, we tracked the energy barrier as a function of the constrained variable, which corresponds to the C–O bond distance within the *COOH intermediate (Fig. S1†). The energy barrier increases as the CV constraint grows, reaching a peak when *COOH is converted into *CO, and in the meantime, the generated OH− is released into the water environment. The kinetic analysis revealed that the energy barrier for the [Au25(SCH2CH3)17]− NC is relatively lower, with a smoother curve and a peak barrier of 0.76 eV. In contrast, the [Au25(SCH2COOH)17]− NC exhibits a significantly higher energy barrier, with free energy peaking at 1.09 eV. This higher energy input indicates that the CO2 reduction process in [Au25(SCH2COOH)17]− is less efficient compared to that in [Au25(SCH2CH3)17]−. The superior CO2RR performance of [Au25(SCH2CH3)17]− can be attributed to the hydrophobic nature of the –SCH2CH3-protected nanocluster. The hydrophobic environment likely facilitates the desorption of the OH− species and the formation of *CO by minimizing interactions with the surrounding water molecules. This reduced interaction lowers the energy barrier for the rate-determining step, making the reaction pathway more favorable and thereby enhancing the overall CO2 reduction efficiency.
To further substantiate the selectivity of the CO2RR over the HER under alkaline conditions, we performed the constrained kinetic simulations for the competitive HER process on [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]− NCs. The results revealed that while the Volmer reaction (water dissociation and proton adsorption) is feasible (with a barrier less than 0.7 eV) (Fig. S7†), the Heyrovsky reaction (H2 formation) encounters insurmountable barriers, where the splitting of the second H2O molecule and subsequent H2 generation display progressively increasing energy barriers (Fig. S8†). This indicates that the HER is kinetically hindered under alkaline conditions, thereby ensuring high selectivity for the CO2RR on these nanoclusters.
The catalytic performance difference in the HER and CO2RR
The optimized local structures of the Au(surface) and Au(staple) sites for both clusters are presented in Fig. 4a and b, while the adsorption configurations of the key *H and *COOH intermediates are provided in Fig. S9.†Fig. 4c presents the radial distribution function (RDF) between the Au active sites in [Au25(SCH2COOH)17]−/[Au25(SCH2CH3)17]− and the hydrogen atoms of water molecules during the rate-determining steps of the HER and CO2RR. The solid lines represent the RDF for both Au(surface) and Au(staple) during the HER, while the dashed lines depict the RDF for Au(staple) during the CO2RR. | ||
| Fig. 4 Local structures of the Au(surface) and Au(staple) sites in (a) [Au25(SCH2COOH)17]− and (b) [Au25(SCH2CH3)17]− NCs. (c) Radial distribution function (RDF) of Au active sites with hydrogen atoms from surrounding H2O during the kinetic process of the rate-determining steps in the HER (solid lines) and CO2RR (dashed lines) for [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]−. (d) Bader charge analysis of Au(surface) and Au(staple) sites in the NC catalysts before and after the adsorption of key *H and *COOH intermediates in acidic HER and alkaline CO2RR. | ||
In the HER rate-determining step, the RDF considers both Au(staple) and Au(surface) sites because the *H adsorption occurs at both the active Au sites. The first RDF peak corresponds to the closest interactions between the Au atoms and the hydrogen atoms of surrounding water molecules, occurring around 1.7 Å. For [Au25(SCH2COOH)17]−, the g(r) value at this peak is 3.25, whereas for [Au25(SCH2CH3)17]−, it is significantly lower at 0.98. The higher g(r) value for [Au25(SCH2COOH)17]− indicates stronger and more frequent interactions between the Au active sites and the hydrogen atoms. This can be attributed to the hydrophilic nature of the –SCH2COOH ligands, which draw water molecules closer to the Au surface, enhancing proton transfer and interaction with active sites, thereby improving the HER performance. In contrast, the lower g(r) value for [Au25(SCH2CH3)17]− reflects weaker interactions due to the hydrophobic nature of the –SCH2CH3 ligands, which repel water molecules and limit hydrogen bonding near the Au surface, thereby reducing the HER efficiency. The second RDF peak, occurring around 2.9 Å, reflects the next shell of water molecules interacting with the Au active sites. Here, the g(r) value is 5.66 for [Au25(SCH2COOH)17]− and 4.08 for [Au25(SCH2CH3)17]−. The higher and sharper peak for the hydrophilic cluster indicates a more ordered and denser water structure around the Au atoms, which facilitates proton transfer and H2 formation during the HER, further explaining its superior HER performance. Conversely, the less structured water network around the hydrophobic cluster hinders efficient proton transfer, limiting its HER activity.
For the CO2RR rate-determining step, the RDF focuses on the interaction between Au(staple) and the hydrogen atoms of water molecules because *CO2 and *COOH adsorption as well as the conversion to *CO occur exclusively at the Au(staple) site. Interestingly, [Au25(SCH2COOH)17]− also shows a higher and sharper peak compared to [Au25(SCH2CH3)17]−, indicating a more ordered and dense water structure around Au(staple). However, this structured water environment may hinder the adsorption and stabilization of CO2RR intermediates such as *COOH by increasing the energy barrier for *COOH conversion to *CO. In contrast, the less structured water environment in [Au25(SCH2CH3)17]− minimizes water interference with CO2RR intermediates, facilitating *COOH to *CO conversion with a lower barrier and enhancing CO2RR performance.
Fig. 4d presents the Bader charge analysis of the Au(surface) and Au(staple) sites for both nanoclusters before and after the adsorption of key intermediates (*H and *COOH) in the HER and CO2RR. These data reveal distinct differences in how the two systems interact with reactants, helping explain their differing catalytic performances. In the * state (without adsorption), both systems show similar Bader charges on the Au(surface) (0.13), but the Au(staple) in [Au25(SCH2COOH)17]− has a significantly lower charge (0.03) compared to [Au25(SCH2CH3)17]− (0.11). The lower charge on Au(staple) in the hydrophilic cluster suggests a lower electron density, which could promote better interaction with protons facilitating proton transfer and enhancing the hydrogen evolution. When hydrogen is adsorbed (*H state), both systems exhibit negative charges on the Au sites, indicating electron transfer from H to Au. However, the Au(staple) site in [Au25(SCH2CH3)17]− holds a higher negative charge (−0.04) compared to [Au25(SCH2COOH)17]− (−0.02), suggesting a stronger electron interaction with hydrogen. This stronger interaction likely hinders H2 desorption in the hydrophobic cluster, increasing the energy barrier for H2 release. In contrast, the more balanced electron distribution in the hydrophilic cluster allows for easier H2 desorption, contributing to its superior HER performance. In the *COOH state, a crucial intermediate for the CO2RR, the Au(staple) in [Au25(SCH2CH3)17]− shows a more negative charge (−0.12) than in [Au25(SCH2COOH)17]− (−0.08). This indicates that the hydrophobic cluster better stabilizes the *COOH intermediate by receiving more electrons from the adsorbed species. This enhanced stabilization lowers the energy barrier for *COOH reduction to *CO, explaining the superior CO2RR performance of [Au25(SCH2CH3)17]−. Conversely, the less negative charge on Au(staple) in the hydrophilic cluster results in weaker stabilization of *COOH, leading to a higher energy barrier for this reduction step and contributing to its lower CO2RR efficiency.
Fig. 5 illustrates the projected density of states (PDOS) and corresponding d-band centers (εd) of active Au sites for [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]− NCs, along with their key intermediates involved in the HER and CO2RR. The εd values offer valuable insights into the electronic properties of these catalysts and their interaction with reaction intermediates. In the pristine nanocluster (* state), [Au25(SCH2COOH)17]− exhibits an εd of −3.34 eV, while [Au25(SCH2CH3)17]− has a more negative εd of −3.61 eV. A more negative εd typically indicates a lower energy of the d-band center, which correlates with stronger binding of adsorbates. For the *H state, [Au25(SCH2COOH)17]− has an εd of −3.14 eV, whereas [Au25(SCH2CH3)17]− shows a more negative εd of −3.38 eV. This stronger interaction in [Au25(SCH2CH3)17]− reflects tighter hydrogen binding, which could make H2 desorption more difficult and increase the energy barrier for the HER. In contrast, the weaker interaction in [Au25(SCH2COOH)17]− facilitates easier H2 desorption, contributing to its superior HER activity. In the *COOH state, the εd for [Au25(SCH2COOH)17]− is −3.65 eV, compared to a more negative εd of −3.78 eV for [Au25(SCH2CH3)17]−. The more negative εd in [Au25(SCH2CH3)17]− indicates stronger stabilization of the *COOH intermediate, lowering the energy barrier for *COOH to *CO conversion and enhancing the CO2RR performance.
 | ||
| Fig. 5 PDOS and corresponding d-band centers (εd) for [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]−, and their key intermediates. (a) [Au25(SCH2COOH)17]−, (b) *H adsorbed on [Au25(SCH2COOH)17]−, (c) *COOH adsorbed on [Au25(SCH2COOH)17]−, (d) [Au25(SCH2CH3)17]−, (e) *H adsorbed on [Au25(SCH2CH3)17]−, and (f) *COOH adsorbed on [Au25(SCH2CH3)17]−. | ||
In summary, our above theoretical analysis revealed that the catalytic differences between [Au25(SCH2COOH)17]− and [Au25(SCH2CH3)17]− in the HER and CO2RR are contributed both by their surface interaction with the water environment and their electronic properties. The RDF analysis shows that [Au25(SCH2COOH)17]− with hydrophilic ligands enhances proton transfer and H2 desorption, leading to better HER performance. In contrast, the hydrophobic nature of [Au25(SCH2CH3)17]− reduces water interaction and lowers the HER efficiency. The Bader charge and d-band center analyses reveal that [Au25(SCH2CH3)17]− strongly stabilizes the *COOH intermediate, lowering the energy barrier for *CO formation, thereby excelling in the CO2RR, while [Au25(SCH2COOH)17]− shows weaker stabilization of *COOH, leading to higher CO2RR barriers.
Experimental validation
To validate the computational findings, which indicate that the hydrophilic ligand-protected Au25 cluster exhibits superior HER performance in an acidic environment, whereas the hydrophobic ligand-protected Au25 cluster excels in the CO2RR in an alkaline environment, we conducted further experimental investigations. Specifically, we synthesized [Au25(MPA)18]− as the hydrophilic model and [Au25(SC6H13)18]− as its hydrophobic counterpart. To confirm the successful synthesis of [Au25(MPA)18]− and [Au25(SC6H13)18]− NCs, we conducted the UV-Vis absorption spectroscopy measurements. The UV-Vis spectra of the two Au25 NCs, provided in Fig. S10a and b,† exhibit distinct absorption peaks at 400 nm, 450 nm, and 670 nm, which are consistent with the characteristic optical behavior of Au25 NCs reported in previous studies.22,25In the HER experiments conducted in 0.5 M H2SO4 (Fig. 6a), [Au25(MPA)18]− demonstrated a lower overpotential of 445 mV at a current density of 10 mA cm−2 compared to 541 mV for [Au25(SC6H13)18]−, indicating an enhanced hydrogen evolution activity of the [Au25(MPA)18]− NC. Additionally, the Tafel slope for [Au25(MPA)18]− was slightly lower (106 mV dec−1) than that of [Au25(SC6H13)18]− (111 mV dec−1), suggesting faster reaction kinetics (Fig. 6b). The stability tests over 8.5 hours of continuous operation (Fig. 6c) revealed that [Au25(MPA)18]− maintained a stable current density, whereas [Au25(SC6H13)18]− exhibited a reduction of about 10% in the current density, indicating that the hydrophilic [Au25(MPA)18]− is more stable under acidic HER conditions.
 | ||
| Fig. 6 Electrocatalytic HER performance in 0.5 M H2SO4. (a) LSV curves of [Au25(MPA)18]− and [Au25(SC6H13)18]−. (b) Tafel slopes of the catalysts. (c) Chronoamperometric plots at 10 mA cm−2 for the HER stability testing. | ||
For the CO2RR performance evaluation, linear sweep voltammetry (LSV) was performed using a CO2 flow cell with 1.0 M KOH electrolyte. As shown in Fig. 7a, the current density difference between CO2-saturated and N2-saturated electrolytes for [Au25(SC6H13)18]− was larger than that for [Au25(MPA)18]−, indicating that [Au25(SC6H13)18]− is more favorable for CO2 reduction to CO. The faradaic efficiency for CO (FECO) at various potentials (Fig. 7b) showed that [Au25(SC6H13)18]− achieved a higher FECO, reaching 97.64% at −0.48 V. In terms of the partial current density for CO production (jCO) (Fig. 7c), [Au25(MPA)18]− exhibited slightly higher values in the potential range of −0.18 V to −0.48 V, whereas [Au25(SC6H13)18]− outperformed in the lower potential range of −0.58 V to −0.98 V. The Tafel slopes (Fig. S11†) further indicated the faster reaction kinetics for [Au25(SC6H13)18]− (199 mV dec−1) than that for [Au25(MPA)18]− (242 mV dec−1). Additionally, the turnover frequency (TOF) values (Fig. 7d) showed that [Au25(MPA)18]− has slightly higher TOF between −0.18 V and −0.48 V, while [Au25(SC6H13)18]− exhibits significantly higher TOF values from −0.58 V to −0.98 V, indicating the much enhanced activity in lower potential ranges. The long-term stability tests over 8.5 hours of continuous operation demonstrated that [Au25(MPA)18]− showed a slight decrease of around 4% in FECO and current density (Fig. 7e), whereas [Au25(SC6H13)18]− maintained the stable FECO and current density (Fig. 7f), suggesting that the hydrophobic nanocluster is more stable under alkaline CO2RR conditions.
 | ||
| Fig. 7 Electrocatalytic CO2RR performance in a CO2 flow cell with 1 M KOH electrolyte. (a) LSV curves of [Au25(MPA)18]− and [Au25(SC6H13)18]− in N2 and CO2 environments. (b) Faradaic efficiency for CO (FECO) of [Au25(MPA)18]− and [Au25(SC6H13)18]− at different potentials. (c) Fractional current density of CO at various potentials. (d) Turnover frequency (TOF) of CO at different potentials. (e and f) Long-term stability tests of [Au25(MPA)18]− and [Au25(SC6H13)18]− for the CO2RR at −0.48 V. | ||
Recognizing that the difference in the ligand chain length could also influence the electrocatalytic activity, we further synthesized and tested Au25 clusters protected by ligands with similar chain lengths. Specifically, [Au25(MHA)18]− (MHA = 6-mercaptohexanoic acid) was chosen as a hydrophilic analog with a chain length comparable to that of [Au25(SC6H13)18]−. These additional experiments, detailed in the ESI,† provide a comprehensive analysis of how both ligand hydrophilicity and chain length affect the catalytic performance of Au25 NCs. The UV-Vis spectra of the synthesized [Au25(MHA)18]− NCs exhibit distinct absorption peaks at 400 nm, 450 nm, and 670 nm (Fig. S10c†), confirming the successful synthesis of Au25 NCs.21,22,25
The LSV curves were measured in a flow cell with 1.0 M KOH electrolyte (Fig. S12a†), revealing that [Au25(SC6H13)18]− NCs are more favorable for CO2 reduction to CO. In the comparison of FECO at various potentials (Fig. S12b†), [Au25(SC6H13)18]− NCs demonstrate higher CO2 reduction selectivity than [Au25(MHA)18]− NCs, with the maximum FECO for [Au25(MHA)18]− reaching only 88%. As shown in Fig. S12c,† the jCO of [Au25(SC6H13)18]− NCs is up to twice as high as that of [Au25(MHA)18]− NCs. The Tafel slopes (Fig. S13†) further highlight the faster reaction kinetics for [Au25(SC6H13)18]− NCs (199 mV dec−1) compared to [Au25(MHA)18]− (498 mV dec−1). Additionally, the TOF values (Fig. S12d†) indicate that [Au25(SC6H13)18]− exhibits significantly higher TOF than [Au25(MHA)18]− across various potentials.
These experimental results have well corroborated our computational predictions, confirming that the hydrophilic ligand-protected Au25 NCs are more effective for the HER in acidic environments due to enhanced proton transfer facilitated by the hydrophilic ligands. Conversely, the hydrophobic ligand-protected Au25 NCs demonstrate superior CO2RR activity under alkaline conditions by promoting adsorption of CO2RR intermediates and minimizing the water interference, thereby enhancing CO2 reduction efficiency. Further experiments with ligands of similar chain lengths to the hydrophilic and hydrophobic Au25 NCs reveal that both ligand hydrophilicity and chain length influence catalytic performance. However, the hydrophilicity of the ligands plays a dominant role in determining the overall catalytic behavior. This alignment between computational and experimental findings underscores the critical role of ligand properties in tuning the electrocatalytic performance of atomically precise metal NCs, providing valuable insights for the rational ligand design of nanocatalysts in sustainable energy applications.
3. Conclusion
In this study, we have systematically explored how the ligand hydrophilicity and hydrophobicity influence the interface dynamics and the electrocatalytic performance of Au25 NCs in important reactions such as the HER and CO2RR. Our computations revealed that the hydrophilic [Au25(SCH2COOH)17]− NC exhibits faster breakage of the Au–S bond and desorption of the thiolate ligand into solution at the applied reduction potential. The dethiolated [Au25(SCH2COOH)17]− NC is predicted to exhibit superior acid HER activity due to enhanced proton transfer and efficient hydrogen evolution facilitated by stronger hydrogen bond interactions with water molecules. Conversely, the hydrophobic [Au25(SCH2CH3)17]− NC demonstrates enhanced alkaline CO2RR performance by stabilizing key CO2RR intermediates and minimizing the water interference to lower the barrier for CO formation. The electrochemical experiments using synthesized [Au25(MPA)18]− and [Au25(SC6H13)18]− as models, which have similar ligand molecular weights, have further validated our predictions. Hydrophilic [Au25(MPA)18]− showed better activity, lower overpotential, and greater stability in the acidic HER testing, while hydrophobic [Au25(SC6H13)18]− achieved higher faradaic efficiency, current density, and stability in alkaline CO2RR. Moreover, the results from clusters with similar chain lengths, such as [Au25(MHA)18]− and [Au25(SC6H13)18]−, further corroborated our computations, highlighting that both ligand hydrophilicity and chain length influence the catalytic performance, with hydrophilicity being the dominant factor in determining the overall catalytic behavior. These results not only provide an atomic level understanding of the ligand effects on the interface dynamics and electrocatalytic performance of metal NCs, but also stimulate ligand engineering as a facile strategy to optimize the catalytic activity of nanocluster catalysts for specific catalytic targets in future electrocatalysis research.4. Computational methods and experimental section
Ligand removal dynamics
The dynamics of ligand removal from the nanocluster were investigated using the CP2K package (version 2023.1).50 The simulations employed the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) in combination with the hybrid Gaussian/Plane-Wave (GPW) scheme.51 The computational domain was defined with a periodic lattice size of 23 × 23 × 28 Å3, including 192 water molecules to simulate the solvent environment. Molecular dynamics (MD) simulations were performed in the canonical (NVT) ensemble using Nose–Hoover thermostats to maintain a constant temperature of 300 K.52,53 A time step of 1.0 fs was used throughout the simulations, which were run for a total of 10 ps. Dispersion corrections were applied using the DFT-D3 method to account for van der Waals interactions.54,55 The electronic structure was described by density functional theory (DFT) with spin-polarization and a mixed double-ζ Gaussian and plane-wave basis set, with an energy cutoff of 400 Ry.56Thermodynamic calculations for the HER and CO2RR
Spin-polarized DFT calculations were carried out using the Vienna Ab initio Simulation Package (VASP, version 5.4.4).57 The exchange–correlation effects were described using the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA).51 The projector-augmented wave (PAW) method was employed to account for electron–ion interactions with a cutoff energy of 400 eV.58 For these calculations, the computational cell was set with a lattice size of 23 × 23 × 28 Å3, including 50 water molecules to adapt to the specific requirements of the reaction environments. The empirical DFT-D3 dispersion correction was applied to include van der Waals interactions.59 Brillouin zone integration was performed using a gamma centered 1 × 1 × 1 k-point grid, with convergence criteria set at 0.05 eV Å−1 for forces and 10−4 eV per cell for energy. The electrode potential was determined via tuning the work function (additional details are provided in the ESI†).Dynamic calculations for the HER and CO2RR
Ab initio molecular dynamics (AIMD) simulations were conducted using the Nose–Hoover thermostat in the canonical (NVT) ensemble at 300 K with a time step of 1 fs.52,60 Thermodynamic integrations and the “slow-growth” method were employed to derive the free energy profiles for the rate-determining steps in the HER and CO2RR. The collective variable (CV) increment was set to 0.0008 Å, and the simulation time was 3 ps. Reaction barriers and reaction energies were determined by integrating the free-energy gradients to generate the free energy profiles.61,62Chemicals and materials
Tetrachloroauric(III) acid (HAuCl4·3H2O, >99.99%), tetraoctylammonium bromide (TOAB, 98%), 1-hexanethiol (97%), 3-mercaptopropionic acid (MPA, 98%), 6-mercaptohexanoic acid (MHA, 98%), sodium borohydride (NaBH4), sodium hydroxide (NaOH), potassium hydroxide (KOH), Nafion solution (5 wt%), and solvents: reverse osmosis water, methanol, ethanol, acetonitrile (CH3CN), dichloromethane (CH2Cl2) and tetrahydrofuran (THF) were used. High purity (>99.999%) Ar and CO2 gases were used.Preparation of the [Au25(MPA)18]− NCs
In a typical synthesis of [Au25(MPA)18]− clusters, 4 mg of HAuCl4·3H2O (0.05 mmol) and 2.1 mg of MPA (0.09 mmol) were mixed in 4.7 mL of ultrapure water under mild stirring. Subsequently, 3 mL of NaOH solution (1.0 M) was added, causing the solution color to change from yellow to pale yellow. Then, 1 mL of NaBH4 solution (0.1 M) was added dropwise, turning the solution yellowish-brown. After 3 hours of vigorous stirring, unreacted MPA and other impurities were removed using organic solvents and spin distillation, yielding highly pure MPA-protected Au25 nanoclusters.Preparation of the [Au25(MHA)18]− NCs
The procedure of the [Au25(MHA)18]− NCs is similar to the synthesis of [Au25(MPA)18]−. 4.0 mg of HAuCl4·3H2O (0.05 mmol) and 4.0 mL of MHA (50 mM) were mixed in 4.7 mL of ultrapure water under mild stirring. Subsequently, 0.5 mL of NaOH solution (1.0 M) was added, causing the solution color to change from yellow to pale yellow. Then, 1 mL of NaBH4 solution (0.1 M) was added dropwise, turning the solution yellowish-brown. After 2 h of vigorous stirring, the obtained product was purified using organic solvents to remove unreacted MHA and other impurities, yielding [Au25(MHA)18]− nanoclusters.Preparation of the [Au25(SC6H13)18]− NCs
HAuCl4·3H2O (0.125 mmol) and TOAB (0.145 mmol) were dissolved in 14 mL of THF in a 50 mL vial. After vigorous stirring for 15 minutes, the solution color changed from orange to red. Then, 1-hexanethiol (0.625 mmol) was slowly added, and the mixture was stirred for 1 hour until the red solution turned colorless. Next, freshly prepared NaBH4 (1.25 mmol) in 2 mL of cold water was quickly added, producing bubbles as the solution turned black, indicating the formation of gold clusters. The reaction mixture was stirred for an additional 24 hours. The product was then transferred to a 25 mL round-bottom flask and dried by rotary evaporation. The dried product was redissolved in CH2Cl2, and the supernatant was transferred to another round-bottom flask and dried again by rotary evaporation. Finally, the product was washed with ethanol, collected by centrifugation, and the washing process was repeated at least 10 times to ensure the removal of impurities.Electrochemical measurements
The electrochemical tests were conducted using a CHI 760E electrochemical workstation (CH Instruments Inc.) at room temperature. For HER testing, cyclic voltammetry (CV) and LSV curves were obtained in a three-electrode cell setup, consisting of a glassy carbon disk electrode (GCE, diameter 5 mm, surface area 0.07 cm2) as the working electrode, polished with alumina slurry and cleaned with ethanol and deionized water. A carbon rod served as the counter electrode, and an Ag/AgCl electrode in 0.5 M H2SO4 was used as the reference electrode. The potential of the reference electrode was calibrated to the reversible hydrogen electrode (RHE) using the equation: ERHE = EHg/HgCl2 + (0.244 + 0.0591 × pH) V in 0.5 M H2SO4 solution. The working electrode for the HER was prepared by dissolving 1 mg of carbon nanotube and 1 mg of Au25 NCs in 0.5 mL of CH2Cl2, creating an ink with 10 μL of 5 wt% Nafion. Before testing, 3.5 μL of the catalyst and 3 μL of Nafion (5 wt%) were cast onto the GCE.The catalytic activity of Au25 NCs in the CO2RR was determined using an electrochemical workstation (CHI 600E) with a three-electrode system coupled to a CO2 flow cell. The electrolyte solution was 1 M KOH, and the reference electrode was an Ag/AgCl electrode immersed in saturated KCl solution. An anion-exchange membrane and a platinum plate were used as the ion mobility channel and counter electrode, respectively. The working electrode was prepared by dissolving 1 mg of carbon nanotube and 1 mg of Au25 NCs in 0.5 mL of CH2Cl2, creating a uniform dispersion ink containing 10 μL of 5 wt% Nafion solution. This solution was sprayed onto a 1 cm2 gas diffusion layer (GDL) with a mass loading of 2 mg cm−2. The potentials were converted to the RHE using the following equation:
| E(RHE) = E(Ag/AgCl) + 0.197 V + 0.0591 × pH |
Before the electrochemical CO2 reduction reaction, the cathodic electrolyte was saturated with CO2 for 30 minutes. The cathodic and anodic reaction chambers were separated using an anion exchange membrane. During the CO2RR process, each electrolyte cell contained 30 mL of electrolyte, circulated at 40 rpm using a peristaltic pump. The gas products were analyzed quantitatively with a gas chromatograph (GC, Huaai 9560). The faradaic efficiency (FE) of the gas products was calculated using the formula:
 485 C mol−1).
485 C mol−1).
The turnover frequency (TOF) was calculated as follows:
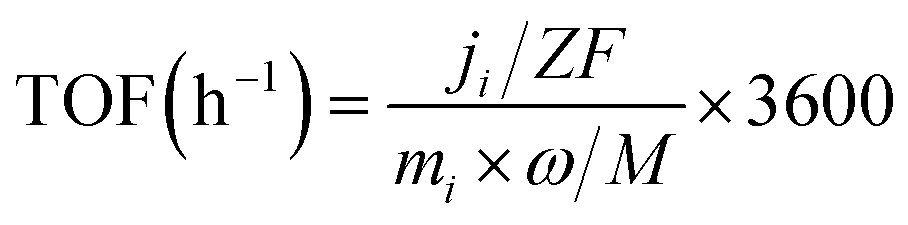 485 C mol−1), mi is the mass of the loaded catalyst, ω is the relative mass fraction of Au in the catalyst, and M is the relative atomic mass of Au. Linear sweep voltammetry (LSV) was conducted in a 1 M KOH solution saturated with either N2 or CO2, using a scan rate of 50 mV s−1.
485 C mol−1), mi is the mass of the loaded catalyst, ω is the relative mass fraction of Au in the catalyst, and M is the relative atomic mass of Au. Linear sweep voltammetry (LSV) was conducted in a 1 M KOH solution saturated with either N2 or CO2, using a scan rate of 50 mV s−1.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Q. T. conceived the idea. L. L. performed the theoretical calculations with help from Y. C. and F. S. X. Z. performed the experiments and analyzed the data under the guidance of L. W. L. L. wrote the manuscript with inputs from all authors. Q. T. and L. W. finalized the manuscript. All the authors approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22473017) and the Chongqing Science and Technology Commission (CSTB2024NSCQ-MSX0250).References
- Z. Zhou, Z. Pei, L. Wei, S. Zhao, X. Jian and Y. Chen, Electrocatalytic hydrogen evolution under neutral pH conditions: current understandings, recent advances, and future prospects, Energy Environ. Sci., 2020, 13, 3185–3206 RSC.
- W. Zhai, Y. Ma, D. Chen, J. C. Ho, Z. Dai and Y. Qu, Recent progress on the long-term stability of hydrogen evolution reaction electrocatalysts, InfoMat, 2022, 4, e12357 CrossRef CAS.
- L. Xiong, Y. Qiu, X. Peng, Z. Liu and P. K. Chu, Electronic structural engineering of transition metal-based electrocatalysts for the hydrogen evolution reaction, Nano Energy, 2022, 104, 107882 CrossRef CAS.
- M. Ďurovič, J. Hnát and K. Bouzek, Electrocatalysts for the hydrogen evolution reaction in alkaline and neutral media. A comparative review, J. Power Sources, 2021, 493, 229708 CrossRef.
- X. Zhang, S.-X. Guo, K. A. Gandionco, A. M. Bond and J. Zhang, Electrocatalytic carbon dioxide reduction: from fundamental principles to catalyst design, Mater. Today Adv., 2020, 7, 100074 CrossRef.
- T. Ahmad, S. Liu, M. Sajid, K. Li, M. Ali, L. Liu and W. Chen, Electrochemical CO2 reduction to C2+ products using Cu-based electrocatalysts: a review, Nano Res. Energy, 2022, 1, e9120021 CrossRef.
- D. Xu, K. Li, B. Jia, W. Sun, W. Zhang, X. Liu and T. Ma, Electrocatalytic CO2 reduction towards industrial applications, Carbon Energy, 2023, 5, e230 CrossRef CAS.
- X. An, S. Li, X. Hao, Z. Xie, X. Du, Z. Wang, X. Hao, A. Abudula and G. Guan, Common strategies for improving the performances of tin and bismuth-based catalysts in the electrocatalytic reduction of CO2 to formic acid/formate, Renewable Sustainable Energy Rev., 2021, 143, 110952 CrossRef CAS.
- X. Mao, W. Gong, Y. Fu, J. Li, X. Wang, A. P. O'Mullane, Y. Xiong and A. Du, Computational Design and Experimental Validation of Enzyme Mimicking Cu-Based Metal–Organic Frameworks for the Reduction of CO2 into C2 Products: C–C Coupling Promoted by Ligand Modulation and the Optimal Cu–Cu Distance, J. Am. Chem. Soc., 2023, 145, 21442–21453 CrossRef CAS PubMed.
- K. Sokołowska, S. Malola, M. Lahtinen, V. Saarnio, P. Permi, K. Koskinen, M. Jalasvuori, H. Häkkinen, L. Lehtovaara and T. Lahtinen, Towards controlled synthesis of water-soluble gold nanoclusters: synthesis and analysis, J. Phys. Chem. C, 2019, 123, 2602–2612 CrossRef.
- P. Maity, S. Xie, M. Yamauchi and T. Tsukuda, Stabilized gold clusters: from isolation toward controlled synthesis, Nanoscale, 2012, 4, 4027–4037 RSC.
- K. Kwak and D. Lee, Electrochemistry of atomically precise metal nanoclusters, Acc. Chem. Res., 2018, 52, 12–22 CrossRef PubMed.
- Y. Jo, M. Choi, M. Kim, J. S. Yoo, W. Choi and D. Lee, Promotion of alkaline hydrogen production via Ni-doping of atomically precise Ag nanoclusters, Bull. Korean Chem. Soc., 2021, 42, 1672–1677 CrossRef CAS.
- S. Zhao, R. Jin and R. Jin, Opportunities and challenges in CO2 reduction by gold-and silver-based electrocatalysts: from bulk metals to nanoparticles and atomically precise nanoclusters, ACS Energy Lett., 2018, 3, 452–462 CrossRef CAS.
- Q. Tang, G. Hu, V. Fung and D.-e. Jiang, Insights into interfaces, stability, electronic properties, and catalytic activities of atomically precise metal nanoclusters from first principles, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS PubMed.
- F. Sun, C. Deng, S. Tian and Q. Tang, Oxygen electrocatalysis by [Au25(SR)18]: charge, doping, and ligand removal effect, ACS Catal., 2021, 11, 7957–7969 CrossRef CAS.
- F. Sun, Q. Tang and D.-e. Jiang, Theoretical advances in understanding and designing the active sites for hydrogen evolution reaction, ACS Catal., 2022, 12, 8404–8433 CrossRef CAS.
- T. Kawawaki, T. Okada, D. Hirayama and Y. Negishi, Atomically precise metal nanoclusters as catalysts for electrocatalytic CO2 reduction, Green Chem., 2024, 26, 122–163 RSC.
- W. Choi, H. Seong, V. Efremov, Y. Lee, S. Im, D.-H. Lim, J. S. Yoo and D. Lee, Controlled syngas production by electrocatalytic CO2 reduction on formulated Au25(SR)18 and PtAu24(SR)18 nanoclusters, J. Chem. Phys., 2021, 155, 014305 CrossRef CAS PubMed.
- D. R. Alfonso, D. Kauffman and C. Matranga, Active sites of ligand-protected Au25 nanoparticle catalysts for CO2 electroreduction to CO, J. Chem. Phys., 2016, 144, 184705 CrossRef PubMed.
- H. Seong, V. Efremov, G. Park, H. Kim, J. S. Yoo and D. Lee, Atomically precise gold nanoclusters as model catalysts for identifying active sites for electroreduction of CO2, Angew. Chem., 2021, 133, 14684–14691 CrossRef.
- S. M. Han, M. Park, J. Kim and D. Lee, Boosting the Electroreduction of CO2 to CO by Ligand Engineering of Gold Nanoclusters, Angew. Chem., 2024, 63, e202404387 CrossRef CAS PubMed.
- O. López-Estrada, N. Mammen, L. Laverdure, M. M. Melander, H. Häkkinen and K. Honkala, Computational Criteria for Hydrogen Evolution Activity on Ligand-Protected Au25-Based Nanoclusters, ACS Catal., 2023, 13, 8997–9006 CrossRef.
- Y. Li, S. Li, A. V. Nagarajan, Z. Liu, S. Nevins, Y. Song, G. Mpourmpakis and R. Jin, Hydrogen evolution electrocatalyst design: turning inert gold into active catalyst by atomically precise nanochemistry, J. Am. Chem. Soc., 2021, 143, 11102–11108 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, D.-e. Jiang and D. Lee, Rationally designed metal nanocluster for electrocatalytic hydrogen production from water, J. Mater. Chem. A, 2018, 6, 19495–19501 RSC.
- G. Hu, Q. Tang, D. Lee, Z. Wu and D.-e. Jiang, Metallic hydrogen in atomically precise gold nanoclusters, Chem. Mater., 2017, 29, 4840–4847 CrossRef CAS.
- F. Sun, L. Qin, Z. Tang and Q. Tang, Revisiting the activity origin of the PtAu24(SR)18 nanocluster for enhanced electrocatalytic hydrogen evolution by combining first-principles simulations with the experimental in situ FTIR technique, Chem. Sci., 2024, 15, 16142–16155 RSC.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, Experimental and computational investigation of Au25 clusters and CO2: a unique interaction and enhanced electrocatalytic activity, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-e. Jiang and D. Lee, A molecule-like PtAu24(SC6H13)18 nanocluster as an electrocatalyst for hydrogen production, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- E. Pensa, L. M. Azofra, R. C. Salvarezza and P. Carro, Effect of ligands on the stability of gold nanoclusters, J. Phys. Chem. Lett., 2022, 13, 6475–6480 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, The role of ligands in atomically precise nanocluster-catalyzed CO2 electrochemical reduction, Nanoscale, 2021, 13, 2333–2337 RSC.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Gold nanoclusters as electrocatalysts: size, ligands, heteroatom doping, and charge dependences, Nanoscale, 2020, 12, 9969–9979 RSC.
- S. F. Yuan, R. L. He, X. S. Han, J. Q. Wang, Z. J. Guan and Q. M. Wang, Robust gold nanocluster protected with amidinates for electrocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2021, 60, 14345–14349 CrossRef CAS PubMed.
- J. Wang, F. Xu, Z. Y. Wang, S. Q. Zang and T. C. Mak, Ligand-Shell Engineering of a Au28 Nanocluster Boosts Electrocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2022, 61, e202207492 CrossRef CAS PubMed.
- X. Li, S. Takano and T. Tsukuda, Ligand effects on the hydrogen evolution reaction catalyzed by Au13 and Pt@Au12: alkynyl vs. thiolate, J. Phys. Chem. C, 2021, 125, 23226–23230 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-assembled monolayers of thiolates on metals as a form of nanotechnology, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- C. Vericat, M. Vela, G. Benitez, P. Carro and R. Salvarezza, Self-assembled monolayers of thiols and dithiols on gold: new challenges for a well-known system, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- R. G. Nuzzo and D. L. Allara, Adsorption of bifunctional organic disulfides on gold surfaces, J. Am. Chem. Soc., 1983, 105, 4481–4483 CrossRef CAS.
- Z. Liu, H. Tan, B. Li, Z. Hu, D.-e. Jiang, Q. Yao, L. Wang and J. Xie, Ligand effect on switching the rate-determining step of water oxidation in atomically precise metal nanoclusters, Nat. Commun., 2023, 14, 3374 CrossRef CAS PubMed.
- S. Yoo, S. Yoo, G. Deng, F. Sun, K. Lee, H. Jang, C. W. Lee, X. Liu, J. Jang, Q. Tang, Y. J. Hwang, T. Hyeon and M. S. Bootharaju, Nanocluster surface microenvironment modulates electrocatalytic CO2 reduction, Adv. Mater., 2024, 36, 2313032 CrossRef CAS PubMed.
- S. Zhao, N. Austin, M. Li, Y. Song, S. D. House, S. Bernhard, J. C. Yang, G. Mpourmpakis and R. Jin, Influence of atomic-level morphology on catalysis: the case of sphere and rod-like gold nanoclusters for CO2 electroreduction, ACS Catal., 2018, 8, 4996–5001 CrossRef CAS.
- N. Austin, S. Zhao, J. R. McKone, R. Jin and G. Mpourmpakis, Elucidating the active sites for CO2 electroreduction on ligand-protected Au25 nanoclusters, Catal. Sci. Technol., 2018, 8, 3795–3805 RSC.
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, Monopalladium substitution in gold nanoclusters enhances CO2 electroreduction activity and selectivity, ACS Catal., 2020, 10, 12011–12016 CrossRef CAS.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Boosting CO2 electrochemical reduction with atomically precise surface modification on gold nanoclusters, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef CAS PubMed.
- R. Jin, Quantum sized, thiolate-protected gold nanoclusters, Nanoscale, 2010, 2, 343–362 RSC.
- P. Hu, L. Chen, X. Kang and S. Chen, Surface functionalization of metal nanoparticles by conjugated metal–ligand interfacial bonds: impacts on intraparticle charge transfer, Acc. Chem. Res., 2016, 49, 2251–2260 CrossRef CAS PubMed.
- F. Sun, L. Qin, Z. Tang, G. Deng, M. S. Bootharaju, Z. Wei, Q. Tang and T. Hyeon, SR removal or –R removal? A mechanistic revisit on the puzzle of ligand etching of Au25(SR)18 nanoclusters during electrocatalysis, Chem. Sci., 2023, 14, 10532–10546 RSC.
- T. Giorgino, Computing 1-D atomic densities in macromolecular simulations: the density profile tool for VMD, Comput. Phys. Commun., 2014, 185, 317–322 CrossRef CAS.
- X. Zhao and Y. Liu, Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction, J. Am. Chem. Soc., 2021, 143, 9423–9428 CrossRef CAS PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Quickstep: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- W. G. Hoover, Canonical dynamics: equilibrium phase-space distributions, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, Nosé–Hoover chains: the canonical ensemble via continuous dynamics, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- I. L. Garzon and A. Posada-Amarillas, Structural and vibrational analysis of amorphous Au55 clusters, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11796 CrossRef CAS PubMed.
- R. W. Hall and B. J. Berne, Nonergodicity in path integral molecular dynamics, J. Chem. Phys., 1984, 81, 3641–3643 CrossRef CAS.
- M. Sprik and G. Ciccotti, Free energy from constrained molecular dynamics, J. Chem. Phys., 1998, 109, 7737–7744 CrossRef CAS.
- A. A. Hassanali, J. Cuny, V. Verdolino and M. Parrinello, Aqueous solutions: state of the art in ab initio molecular dynamics, Philos. Trans. R. Soc., A, 2014, 372, 20120482 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07181f |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |