
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional cytochrome P450 orchestrates radical cleavage and non-radical cyclization in 5-oxaindolizidine biosynthesis†
Kaijin
Zhang‡
ab,
Jingxian
Sun‡
ab,
Wencai
Song
ab,
Junyu
Liu
ab,
Chuanteng
Ma
 ab,
Yinghan
Chen
ab,
Yan
Guan
ab,
Yuting
Liu
ab,
Zilin
Ren
ab,
Qian
Che
ab,
Guojian
Zhang
abc,
Yankai
Liu
abc,
Tianjiao
Zhu
ab and
Dehai
Li
*abc
ab,
Yinghan
Chen
ab,
Yan
Guan
ab,
Yuting
Liu
ab,
Zilin
Ren
ab,
Qian
Che
ab,
Guojian
Zhang
abc,
Yankai
Liu
abc,
Tianjiao
Zhu
ab and
Dehai
Li
*abc
aKey Laboratory of Marine Drugs, Ministry of Education, Ocean University of China, Qingdao 266003, China. E-mail: dehaili@ouc.edu.cn
bSanya Oceanographic Institute, School of Medicine and Pharmacy, Ocean University of China, Sanya 572025, China
cLaboratory for Marine Drugs and Bioproducts, Qingdao Marine Science and Technology Center, Qingdao 266237, People's Republic of China
First published on 10th May 2025
Abstract
Penicilactam A (1), a fungal alkaloid featuring a rare 5-oxaindolizidine scaffold, has long eluded biosynthetic characterization despite recent advances in microbial genomics. Through retro-biosynthetic analysis of Penicillium citrinum HDN11-186, we identified the pnlt gene cluster governing its production. This pathway ultilizes a hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) system to assemble the prolinol-containing precursor scalusamide A (2). The multifunctional cytochrome P450 enzyme PnltC then orchestrates two mechanistically distinct reactions: radical-mediated C–C bond cleavage followed by iminium-driven cyclization. Combined structural and computational analyses unveil PnltC's unprecedented catalytic logic, merging radical oxidation with non-radical cyclization within a single active site, which challenges existing paradigms of P450 enzymology. Our findings expand the functional repertoire of oxygenases in natural products (NPs) biosynthesis, revealing nature's sophisticated strategies for constructing complex nitrogen heterocycles.
Introduction
Fungal 5-oxaindolizidine alkaloids represent a distinctive class of heterocyclic natural products (NPs), with fewer than a dozen reported to date.1–5 Members of this family are classified into two principal structural subtypes (Fig. 1A). These compounds display diverse bioactivities, including anti-juvenile hormone effects,2 insecticidal activity against Oncopeltus fasciatus Dallas and Helicoverpa armigera Hubner,4,6 and broad-spectrum antimicrobial properties,7,8 making them attractive targets for synthetic exploration.1,7,9–11 The structural versatility of the 5-oxaindolizidine scaffold has enabled its application in rational drug design. For instance, strategic incorporation of this motif into phenolic DNA gyrase inhibitors via condensation of ortho-amino-hydroxymethyl-arene with salicylic aldehyde yields tetracyclic aminal derivatives exhibiting enhanced target binding affinity (Fig. 1B(i)).12 Dual inhibitors targeting prolyl oligopeptidase (POP) and fibroblast activation protein α (FAP) have been synthesized using these core structures through enantioselective Strecker reactions and copper-catalyzed borohydride reductions, achieving Ki values of 0.84 μM and 0.72 μM, respectively (Fig. 1B(ii)).13 These dual inhibitors demonstrate comparable efficacy to the clinical candidate Talabostat while showing improved metabolic stability.13 Penicilactam A (1), originally isolated from Penicillium brevicompactum alongide its biogenetically related congener brevioxime A (Fig. 1A),1,2 emerged as a focus of our investigation owing to their insecticidal potential.1 Despite their pharmacological potential, current synthetic routes to these compounds remain inefficient, yielding compound 1 in 3.2% over five steps and brevioxime A in 3.1% over eight steps.1,11,14 Consequently, deciphering the biosynthetic pathway of 1 could provide the foundation for further development of eco-friendly pesticides derived from 1. The N-acylated dihydropyrrole scaffold (3), postulated as a biosynthetic precursor to 1, is hypothesized through isotopic labeling studies to derive from acetate, methionine, and ornithine, though critical enzymatic steps remain uncharacterized (Fig. 1C).9 Building on prior co-isolation patterns and synthetic evidence,7,15 We propose a biogenetic relationship wherein compound 2 undergoes C2-hydroxymethyl cleavage to generate intermediate 3, followed by cyclization to form 1 (Fig. 1C). Recently, Chen et al. identified structurally analogous pyrrolizidinone alkaloids related to compound 1 (penicipyrrolizidinones A–C) from the mangrove-derived fungus Penicillium sp. DM27 (Fig. 1D).16 Their proposed biosynthetic network suggests the involvement of reactive aldehyde and carboxylic acid intermediates (Fig. 1D).16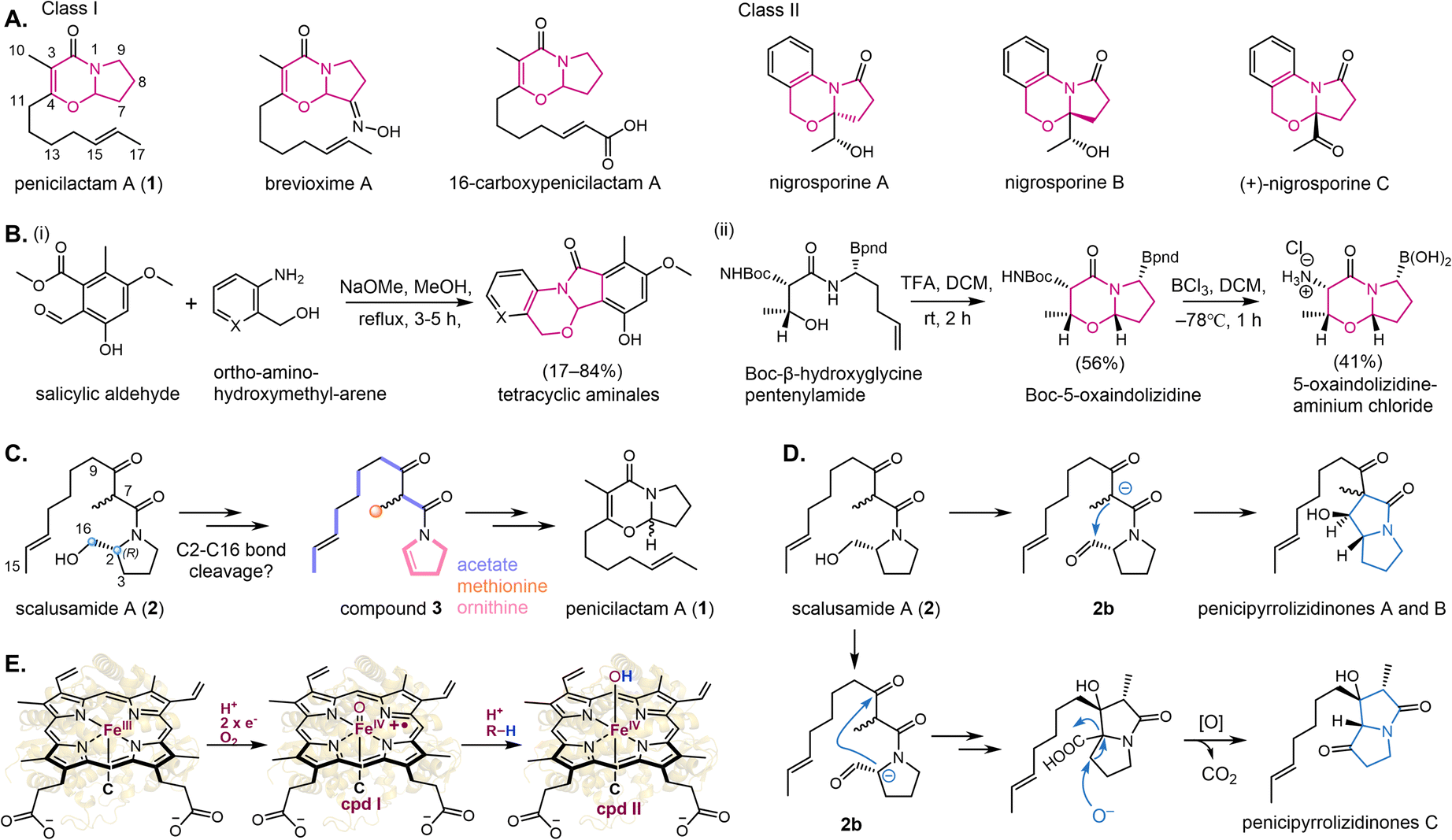 | ||
| Fig. 1 (A) Chemical structures of represented 5-oxaindolizidine-containing NPs. Penicilactam A (1) and nigrosporine B are the only two types of NPs that contain the 5-oxaindolizidine core skeleton. (B) Application of 5-oxaindolizidine core in drug design. (C) Potential conversion between compounds 1, 2 and 3. Compound 2 is the precursor of 1, while the 3 may be the intermediate. (C) Proposed biogenetic relationship among compound 1, 2, 3. (D) Proposed biosynthetic pathway of pyrrolizidine using 2 as the precursor. (E) In the P450 catalytic cycle, upon substrate (R–H) binding to the active-site, the low-spin resting state shifts to a high-spin state of the ferric heme-iron (FeIII), which will be reduced and protonated with the participation of dioxygen to form the first reactive species heme-FeIII–O–OH (compound 0, Cpd 0) and then the high-energy porphyrin π radical cation ferryl intermediate [Por˙+–FeIV] = O] (Cpd I). Cpd I can readily initiate a reaction by hydrogen atom (H˙) abstraction (HAA) from the substrate to yield reactive radical species and is led to the ferryl-hydroxo heme-FeIV–OH species (Cpd II). | ||
We herein report the first identification of biosynthetic gene clusters (BGCs) responsible for compound 1 biosynthesis: a canonical polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) hybrid system synthesizes compound 2 through trans-acting enoyl reductase (trans-ER) activity. Remarkably, the single cytochrome P450 enzyme (PnltC) mediates the efficient conversion of 2 to 1, implying multifunctionality in coordinating hydroxymethyl oxidation and cyclization within a single catalytic cycle. As the most versatile oxygenase in nature, cytochrome P450s typically mediate monooxygenation through the well-established oxygen rebound mechanism involving key intermediates Compound I (Cpd I) and Compound II (Cpd II) (Fig. 1E).17,18 Most P450-catalyzed C–C bond cleavage events proceed through Cpd I-mediated radical mechanisms (Fig. S1†).19–22 In contrast, specific peroxygenases including CYP152 family members (e.g., OleTJE from Jeotgalicoccus sp. ATCC 8456, P450Spa from Sphingomonas paucimobilis, and P450BSβ from Bacillus subtilis) and human CYP19A1 utilize H2O2 activation to bypass the canonical reduction cycle, achieving C–C bond cleavage via peroxidase-like mechanisms.23–25 P450-mediated cyclization predominantly occurs through radical recombination mechanisms, as exemplified by HPQ melanin biosynthesis and the radical-dependent furapyrrole cyclization in (+)-azonazine A production.26,27 In communesin biosynthesis, P450 CnsC is hypothesized to catalyze radical-initiated coupling that generates iminium intermediates that may undergo enzymatic rearrangement.28 However, two critical knowledge gaps remain: (i) direct evidence for iminium intermediate formation is lacking; (ii) computational models neglect heme participation in reactions. Consequently, all well-characterized P450 cyclization mechanisms require radical initiation, with no confirmed cases of enzymatic non-radical cyclization reported. This strict radical dependence raises fundamental questions about P450's catalytic plasticity. Challenging such radical paradigms, our multidisciplinary approach employing synthetic analogues, in situ intermediates trapping, and computational methods reveal PnltC's unprecedented multifunctionality: initial radical-mediated C–C bond oxidative cleavage followed by discrete enzymatic non-radical iminium-driven cyclization. PnltC represents the first documented instance of P450-catalyzed cyclization independent of radical initiation, suggesting an evolutionary innovation in P450 catalytic chemistry.
Results and discussion
Elucidating the biosynthetic pathway of penicilactam A (1)
Given the taxonomic specificity of penicilactam A (1) production in Penicillium species,1,3,4,8,29 we systematically screened 56 Penicillium strains under varied cultivation conditions. LC-MS profiling identified the halophyte-derived Penicillium citrinum HDN11-186 as a potental producer of 1 (m/z 250.1799 [M + H]+) (Fig. S2†). Large-scale fermentation coupled with 1H/13C NMR analysis unambiguously confirmed the structure of 1 (Fig. S29, S30 and Table S4†).4AntiSMASH 6.0 analysis of the HDN11-186 genome identified candidate hybrid PKS-NRPS biosynthetic gene clusters (BGCs),30 consistent with established nitrogen incorporation mechanisms in fungal polyketides.31–33 Fungal PKS-NRPS systems typically employ an N-terminal reducing PKS module to assemble polyketide chains, with C-terminal reductive (R/R*) domains facilitating product release through Dieckmann cyclization or reduction mechanisms (Fig. S3A†).32,34 Retro-biosynthetic analysis suggested proline-derived carbon elimination and subsequent cyclization, implying requisite post-assembly enzymatic modifications (Fig. S3B†). Consequently, we prioritized the uncharacterized pnlt cluster containing: (i) PKS-NRPS (PnltA), (ii) trans-ER (PnltB), (iii) cytochrome P450 (PnltC), and (iv) pathway-specific transcription factor (PnltD) (Fig. 2A). To circumvent low native production limitations of 1 (0.15 mg L−1), we used the engineering heterologous host Aspergillus nidulans A1145 for pathway reconstitution.35 Heterologous expression of pnltA alone did not yield detectable metabolites (Fig. 2A(iv)). Co-expression of pnltAB produced scalusamide A (2, m/z 282.2060 [M + H]+), existing as C7-methyl diastereomers (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) due to rapid epimerization of the acidic methine C7 (Fig. 2A(iii), S31, S32 and Table S5†).15,29 The D-prolinol moiety in 2—despite PnltA's lack of epimerization domains—implies selective recruitment of D-proline by the adenylation (A) domain (PnltA-A), consistent with some characterized NRPS systems.36–40 Despite inconclusive phylogenetic analysis (Fig. S4A†),41 molecular docking demonstrated preferential D-proline binding in PnltA-A's active site, with its carboxyl group oriented for efficient reaction with ATP's pyrophosphate moiety (Fig. S4B†).42–44 After unsuccessful purification efforts (Fig. S5A†), in vitro adenylation assays with A1145-PnltA-A crude enzymes confirmed D-proline selectivity (Fig. S5B†). The PnltA-R domain likely mediates reductive release of 2, as supported by functional analysis of its reductive domain (Fig. S6†).41 These findings establish a trans-ER-dependent PKS-NRPS pathway for 2 biosynthesis (Fig. 2A(iii)). Co-expression of pnltABC significantly accumulated the production of 1 (Fig. 2A(ii)), confirming the essential role of P450 PnltC.
:1 ratio) due to rapid epimerization of the acidic methine C7 (Fig. 2A(iii), S31, S32 and Table S5†).15,29 The D-prolinol moiety in 2—despite PnltA's lack of epimerization domains—implies selective recruitment of D-proline by the adenylation (A) domain (PnltA-A), consistent with some characterized NRPS systems.36–40 Despite inconclusive phylogenetic analysis (Fig. S4A†),41 molecular docking demonstrated preferential D-proline binding in PnltA-A's active site, with its carboxyl group oriented for efficient reaction with ATP's pyrophosphate moiety (Fig. S4B†).42–44 After unsuccessful purification efforts (Fig. S5A†), in vitro adenylation assays with A1145-PnltA-A crude enzymes confirmed D-proline selectivity (Fig. S5B†). The PnltA-R domain likely mediates reductive release of 2, as supported by functional analysis of its reductive domain (Fig. S6†).41 These findings establish a trans-ER-dependent PKS-NRPS pathway for 2 biosynthesis (Fig. 2A(iii)). Co-expression of pnltABC significantly accumulated the production of 1 (Fig. 2A(ii)), confirming the essential role of P450 PnltC.
 | ||
| Fig. 2 (A) The BGC pnlt responsible for the biosynthesis of 1 and the heterologous expression of various pnlt gene combinations in A1145. (B and C) Compound 3 only be converted to 1 under the PnltC catalysis. (D) Three potential pyrrole intermediates involved in the transformation from 2 to 1. (E) Analysis of AN-pnltC fed with 3, the products of which were further reduced to 3c-keto under the action of STAB. | ||
PnltC catalyzes sequential C–C bond cleavage and iminium-involved non-radical cyclization
To elucidate PnltC's catalytic function, we heterologously expressed pnltC in Saccharomyces cerevisiae RC01 (generating RC01-pnltC) and A1145 (AN-pnltC). While microsomal fractions exhibited no activity, whole-cell feeding of 2 produced detectable 1 (Fig. S7†), indicating membrane integrity is crucial for catalysis. This dichotomy suggests PnltC's catalytic competence requires intact cellular architecture, potentially through lipid interactions disrupted during microsome preparation (Fig. S8†).The biosynthetic conversion of 2 to 1 entails sequential C2–C16 bond cleavage followed by stereoselective cyclization (Fig. S9†). Retro-biosynthetic analysis and reported structural analogues (N-acetyldihydropyrrole monomers/heterodimers) suggested initial C2–OH oxidation could generate intermediate 3 (Fig. 1C).45–47 However, transient accumulation of 3 was undetectable in both co-expression and feeding experiments, likely due to rapid enzymatic processing in the optimized A. nidulans system. Chemical synthesis of 3 enabled feeding experiments demonstrating its conversion to 1 by AN-pnltC (Fig. 2B, S33, S34 and Table S6†), confirming its biosynthetic intermediacy. pH-controlled experiments (5–9) showed no abiotic conversion of 3 to 1 (Fig. 2C). Notably, the direct conversion of 3 to 1 contradicts classical radical cyclization mechanisms exemplified in (+)-azonazine A biosynthesis.27 No prior reports exist of P450-catalyzed cyclization independent of radical initiation mechanisms. The tetrahydropyrro moiety in 1 suggests C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 electron migration during cyclization, potentially involving enol tautomer 3a in electrophilic addition (Fig. 2D(vi)). However, lacking electron-withdrawing groups, this pathway remains thermodynamically unfavourable. Therefore, enzymatic stabilization of an activated intermediate from 3 is essential for efficient cyclization. Parallels exist in the cyclization of exo-1-acetamido-pyrrolizidine of loline biosynthesis where W279 stabilizes iminium intermediates via cation–π interactions (Fig. S10†), suggesting similar enzymatic strategies.48 Thus, a more plausible cyclization pathway involving iminium intermediate (3b) likely enhances nucleophilic driving force (Fig. 2D(vii)). In addition, while 3c—a reductive product of 3b or a direct product of C–C bond cleavage from 2 (low probability)—could theoretically undergo radical-mediated cyclization (Fig. 2D(viii)), its climbing energy barrier makes this pathway unlikely (Fig. S11†).
C3 electron migration during cyclization, potentially involving enol tautomer 3a in electrophilic addition (Fig. 2D(vi)). However, lacking electron-withdrawing groups, this pathway remains thermodynamically unfavourable. Therefore, enzymatic stabilization of an activated intermediate from 3 is essential for efficient cyclization. Parallels exist in the cyclization of exo-1-acetamido-pyrrolizidine of loline biosynthesis where W279 stabilizes iminium intermediates via cation–π interactions (Fig. S10†), suggesting similar enzymatic strategies.48 Thus, a more plausible cyclization pathway involving iminium intermediate (3b) likely enhances nucleophilic driving force (Fig. 2D(vii)). In addition, while 3c—a reductive product of 3b or a direct product of C–C bond cleavage from 2 (low probability)—could theoretically undergo radical-mediated cyclization (Fig. 2D(viii)), its climbing energy barrier makes this pathway unlikely (Fig. S11†).
Sodium triacetoxyborohydride (STAB) trapping is commonly employed to detect transient iminium species through in situ reduction.49 After quenching the fermentation system directly, STAB treatment confirmed iminium intermediacy through 3c-keto formation in: (i) 3-fed AN-pnltC cultures (Fig. 2E(i–iv)); and (ii) AN-pnltABC fermentations (Fig. S12, S35, S36 and Table S7†). Control experiments showed no 3c-keto formation from 3 in the absence of enzymatic activity (Fig. 2E(v–vi)). These results validate iminium-dependent cyclization while excluding direct enamine cyclization pathways.
We propose PnltC mediates sequential oxidation and cleavage: PnltC firstly oxidizes the C16–OH of 2 to carboxyl, followed by C–C bond lyase activity with concomitant decarboxylation (Fig. 2D). Attempted synthesis of carboxyl analogue 2c failed due to unexpected α-methylene substitution during proline derivatization (Fig. S13†). We instead synthesized cyclization-blocked analogues 4, 5, 7 and 8 (lacking β-carbonyl groups) to investigate reaction requirements (Fig. S14, S37–S48, Tables S8–S11†). LC-MS analysis revealed PnltC-mediated conversion of 4 → 5 (Fig. S14A†), with STAB trapping yielding 8 from both the feeding groups 4 and 5 (Fig. S14C†). Similar to the 3 detection (after feeding 2), compound 7 remained undetectable after feeding 4 and 5, suggesting transient enamine cationization (Fig. S14B†). However, STAB trapping showed no 8 generation after feeding analogue 7 (Fig. S14C†), likely due to impaired binding from simultaneous loss of polarity of the pyrrole modifying group and β-carbonyl recognition elements (see below). These findings collectively establish the 2 → 2c → 3 → 3b → 1 pathway with bifurcated oxidation and cyclization phases.
Proposed catalytic mechanism of PnltC
Building on the established 2 → 2c → 3 → 3b → 1 pathway, we propose two distinct C–C bond cleavage mechanisms for PnltC based on t P450's single-electron transfer processes (Fig. 3A): Path A: (i) C16–OH oxidation to aldehyde 2bvia gem-diol intermediate, analogous to CYP51A1 catalysis (Fig. 3A);21 (ii) 2b oxidation to 2c; (iii) Cpd I-mediated HAA generates C3 radical; (iv) β-scission via C2 radical (induced though HAA of carboxyl group) recombination, resembling DuxD's mechanism;50 (v) enzyme-guided iminium formation and stereoselective cyclization. While enamine → iminium → 1 pathway remains plausible, iminium-enamine interconversion aligns with established organic equilibrium principles (Fig. S12A†). Therefore, Path B proposes direct C2 carbocation formation via radical transfer to heme iron, proceeding along non-radical iminium formation (Fig. 3A). | ||
| Fig. 3 (A) Two proposed mechanisms of C–C bond cleavage catalyzed by PnltC. The main node is the formation of a carboxyl group. After that, the pathway is considered to have two possible C–C bond cleavages: Path A and B. Note: in Path B, after the carboxyl proton is extracted, a cascade of free radical transfers is induced to form one carbon free radical, which then spontaneously transfers to form a carbon cation (sp3 hybridization) and then spontaneously to an iminium (sp2 hybridization). (B) DFT calculation for the oxidative C–C bond cleavage of 2 (Path A and Path B). Structural optimization was calculated by opt = GDIIS/freq/b3lyp/def2svp/empiricaldispersion = gd3bj//opt/freq/b3lyp/def2svp/empiricaldispersion = gd3bj/scf = xqc; Transition states (TSs) calculations were conducted by freq/ub3lyp/def2svp/empiricaldispersion = gd3bj/scf = xqc//freq/b3lyp/def2svp/empiricaldispersion = gd3bj scf = xqc; the energies of key species involved in different pathways at were calculated by b3lyp/def2tzvp/empiricaldispersion = gd3bj and are given in kcal mol−1. The optimized geometries of these key species along the reaction. | ||
Density functional theory (DFT) calculations employed a residue-free heme-substrate model approximates the conserved catalytic core while acknowledging natural systems may exhibit insignificant energy barrier reductions through scaffold effects.51–53 Calculations commenced with aldehyde 2b, the bifurcation point for both pathways. Cpd I-mediated HAA from 2b's aldehyde group forms carbonyl radical 2e (8.0 kcal mol−1 barrier) (Fig. S15†). Subsequent Cpd II-mediated hydroxylation (2.9 kcal mol−1 barrier) produces carboxylic acid 2c (Fig. 3B). This low-barrier oxidized carboxylation process establishes 2c as the pathway branch point. Path A's rate-limiting step involves C3–H abstraction (TS3, 15.2 kcal mol−1) generating diradical 2f (Fig. 3B). Cpd II facilitates decarboxylation via carboxyl dehydrogenation (TS4, 2.2 kcal mol−1), driving β-scission to form stable low-energy enamine 3 (−130 kcal mol−1) through conjugated diradical 3. Path B initiates through Cpd I-mediated carboxyl HAA from 2c, generating radical 2h. However, single-radical decarboxylation encounters a prohibitive barrier (TS5, 45.6 kcal mol−1), contrasting with Path A's diradical efficiency. Although enzymatic environments can lower barriers (e.g., Pyrl4's 12.7 kcal mol−1 reduction),54 similar effects remain unreported for heme-dependent P450 C–C bond lyases. Despite thermodynamic favorability (ΔG = −62.6 kcal mol−1) between 2h and TS5, Path B's energy barrier renders 2h formation improbable, even considering barrierless 2h → 2i conversion via a single electron transfer without an energy barrier (3.9 kcal mol−1) (Fig. 3B). Thus, Path A represents the exclusive feasible route for C–C bond cleavage and enamine production. These computations corroborate experimental observations: (1) STAB-trapped iminium in 3-fed cultures; (2) No 3 → 1 conversion without enzymatic activity. The enamine-iminium equilibrium requires protonation likely mediated by active-site residues, motivating subsequent mutagenesis studies.
AlphaFold2 modeling generated high-confidence PnltC structures with well-defined active site architecture (Fig. S16A†).42,43 Molecular docking positioned the heme cofactor within an active pocket featuring dual entry channels (Fig. S16B, C and S17A†).55 Channel 1 contains strict categorized polar and hydrophobic residues that enforce substrate orientation through complementary interactions (Fig. S17†). This explains analogue 7's failed conversion due to disrupted polar interactions. Evolutionary conservation analysis coupled with FoldX 5.0 predictions identified 31 residues for alanine scanning across the complete substrate access pathway (Fig. S18 and S19†).56 As shown in Fig. S20,† mutations at K50, S51, M53, T56, L63, G158, T162, K164, H235, F238, Y305, L307, K309, and W424 substantially or completely abolished the production of 1 (>80% reduction), indicating essential roles in catalysis or substrate channel. Fifteen mutants (H52, D55, P64, S65, L150, V157, H165, S231, D239, E242, S245, N302, S308, L420, and V423) showed partial activity (30–70%), suggesting roles in transition state stabilization or microenvironment modulation. The remaining mutations showed negligible effects.
Docking simulations with pathway intermediates identified six critical residues (H235, F238, D239, E242, N302, and Y305) through interaction analysis (RMSD = 0, Fig. 4A and S21†).42,44 Y305 forms a critical 3.0 Å hydrogen bond with substrate 2's C6O (Fig. 4B), essential for catalysis (Y305A: 0% activity, Fig. 4D). [Por˙+–FeIV] = O (Cpd I) is positioned 3.1 Å from C16–OH versus 4.8-5.0 Å from C16–H (Fig. 4E(a)). Then FeIV–OH (Cpd II)-mediated C16–H abstraction would necessitate water-bridged proton transfer for aldehyde formation through diradical coupling (Fig. 4E(b)).57 The 4.8 Å Fe–C16 distance disfavors gem-diol mechanisms requiring continuous water-mediated hydrogen extraction, making this pathway energetically unfavourable versus direct oxidation (Fig. 4E(c)). Thus, we propose C16–OH → C16OH occurs via dual HAA-mediated diradical coupling, bypassing the gem-diol pathway (Fig. 4E(b)). F238 stabilizes iminium 3bvia π–cation interactions (4.0 Å centroid distance), while H235's imidazole ring (2.8 Å from C8–OH) likely facilitates proton transfer (Fig. 4C). F238Y retains partial activity (50%) through preserved π-conjugation, demonstrating aromaticity's critical role over hydrophobicity (Fig. 4D and S20†). F238W showed complete activity loss, likely from tryptophan's steric bulk impeding substrate entry (Fig. 4D and S20†). In situ quenching revealed Y305A and F238W abolished 3c-keto production, while F238A retained it, confirming their roles pre-iminium formation (Fig. S22†). H235A's 85% activity loss may be related to its imidazole-N(δ1) abstracting proton (2.8 Å H-bond from C8–OH of 3b, Fig. 4C).58 H235A retained 3c-keto production in STAB trapping assays, indicating post-iminium formation role (Fig. S22†) Furthermore, D239A (35%), E242A (55%), and N302A (45%) implicate hydrogen bonding and pH modulation roles (Fig. 4D). Chirality analysis showed negligible energy differences between R/S-1 precursors (ΔΔG < 0.3 kcal mol−1), indicating minimal inherent stereochemical preference in spontaneous processes (Fig. 5A). Therefore, the observed 37:63 R/S ratio of 1 under enzymatic catalysis demonstrated stereochemical control by the active site (Fig. S9†). Based on the above results, we propose the complete catalytic process (Fig. 5B): (i) C16–OH → CHO (direct oxidization) → COOH; (ii) diradical-mediated β-scission producing enamine 3; (iii) acid-catalyzed iminium formation; (iv) stereoselective cyclization. Notably, non-enzymatic cyclization of activated 3b_act would produce racemic products, emphasizing enzymatic control's necessity for stereoselectivity.
 | ||
| Fig. 4 (A) View of the predicted substrates-binding sites of PnltC. (B) Predicted interaction between residue Y305 and substrate 2. (C) Predicted interaction between residues F238 and H235 and substrate 3b. (D) Analysis of the relative production ratio of 1 and 2via expressing pnltC and its mutants together with pnltAB in A1145. Error bars represent the standard deviation of three independent results. Analysis of the relative conversion rate (mean ± SD) of scanning mutagenesis of these residues combined with in vivo assays in A1145. (E). The mechanism discussion of producing aldehyde by direct oxidation of the hydroxyl group at C16 and by the gem-diol pathway. | ||
 | ||
| Fig. 5 (A) Proposed cyclization process from enamine which will be converted to iminium intermediate for generating 1 with no energy barrier (heme-free). (B) Biosynthetic strategy to produce N-acyl-D-prolinol and 5-oxaindolizidine core in the biosynthesis pathway of penicilactam A. | ||
Evolutionary and functional analysis of PnltC homologs
Having established the biosynthetic pathway of compound 1 (Fig. 5B), we investigated the phylogenetic distribution of PnltC-containing BGCs. Comparative genomics revealed strong conservation of the such BGC, especially in Penicillium species (Fig. S23†). Sequence similarity network (SSN) analysis identified PnltC as phylogenetically distinct, forming a singleton cluster with only Penicillium steckii P450Ps (Fig. S24†). Expanded homology searches in NCBI database identified limited homologs (14 total) predominantly in Penicillium, confirming phylogenetic distribution (Fig. S25†). Multiple sequence alignment showed partial conservation of F238 among homologs, with other catalytic residues being widely conserved (Fig. S26†). Functional analysis of two homologs – P450Ph (Penicillium hetheringtonii IBT 29057, F238, 95% BGC similarity) and P450Cg (Colletotrichum godetiae CBS 193.32, L238, 45% similarity) – revealed strict evolutionary conservation: P450Ph exhibited full catalytic activity, whereas P450Cj was inactive (Fig. S27†). This evolution constraint enables targeted BGC mining for industrial strain development. Phylogenetic clustering with multi-oxidase P450 s suggests unexplored catalytic potential for complex oxidations (Fig. S28†).41Conclusions
This study elucidates the biosynthetic logic behind 5-oxaindolizidine alkaloids through functional characterization of PnltC, a paradigm-altering cytochrome P450 that redefines enzymatic complexity in natural product assembly. We demonstrate how this metalloenzyme orchestrates mechanistically distinct reactions—sequential C–OH/H oxidations, radical-mediated C–C bond cleavage, and non-radical iminium cyclization—through an elegant “energy landscape editing” strategy. Intriguingly, the enzyme exhibits metabolic circularity through prolinol substrate reoxidation (derived from proline reduction by the R domain), showcasing nature's strategic use of redox plasticity. Structural dissection identifies conserved residues (especially H235/F238/Y305) enabling both radical initiation and active transition state stabilization. Phylogenetic analysis traces this innovation to insect-pathogenic and plant-endophytic Penicillium species.2,46,59 Ecologically, these findings reveal fungal-plant coevolution as a driver of metabolic innovation, with biosynthetic clusters serving as molecular armaments in symbiotic defense systems. This work expands the catalytic repertoire of oxygenases and provide an evolutionary roadmap for discovering bioactive nitrogen heterocycles.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. Z. and J. S: investigation, data curation, validation, writing – original draft. W. S., J. L., C. M., Y. C. and Y. G.: investigation. Y. L, Z. R., Q. C., G. Z., Y. L., and T. Z.: formal analysis, visualization. D. L.: supervision, formal analysis, funding acquisition, resources, project administration. All the authors contribute to writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Qingdao Marine Science and Technology Center (2022QNLM030003-1, 2022QNLM030003-2), the Fundamental Research Funds for the Central Universities (202172002, 202262015), the National Natural Science Foundation of China (82473837), Taishan Scholar Distinguished Expert Program in Shandong Province (tstp20240504), Taishan Scholar Youth Expert Program in Shandong Province (tsqn202103153), Fundamental Research Funds for the Central Universities and China Scholarship Council (202461057). The authors also acknowledge the support of the High-Performance Biological Supercomputing Center at the Ocean University of China for this research and thank Prof. Yi Tang (UCLA) for providing A. nidulans A1145 and expression vectors.Notes and references
- Á. Cantín, P. Moya, M.-A. Castillo, J. Primo, M. A. Miranda and E. Primo-Yúfera, Eur. J. Org Chem., 1999, 1999, 221–226 CrossRef
.
- P. Moya, M. Castillo, E. Primo-Yúfera, F. Couillaud, R. Martínez-Máñez, M.-D. Garcerá, M. A. Miranda, J. Primo and R. Martínez-Pardo, J. Org. Chem., 1997, 62, 8544–8545 CrossRef CAS PubMed
- D. Lai, H. Brötz-Oesterhelt, W. E. G. Müller, V. Wray and P. Proksch, Fitoterapia, 2013, 91, 100–106 Search PubMed
- W. Li, X. Zhou, S. Ji, Y. Wang, Z. Sun, Z. Huang, Z. Zhou, Y. Hui and W. Chen, Nat. Prod. Res., 2022, 38, 372–378 CrossRef PubMed
- X. Chen, S. Chen, H. Guo, X. Lu, H. Shen, L. Liu, L. Wang, B. Chen, Y. Zhang and Y. Liu, Mar. Drugs, 2024, 22, 214 CrossRef CAS PubMed
- M.-A. Castillo, P. Moya, A. Cantín, M. A. Miranda, J. Primo, E. Hernández and E. Primo-Yúfera, J. Agric. Food Chem., 1999, 47, 2120–2124 CrossRef CAS PubMed
- K. Sadorn, S. Saepua, N. Boonyuen, P. Laksanacharoen, P. Rachtawee and P. Pittayakhajonwut, RSC Adv., 2016, 6, 94510–94523 RSC
- J.-K. Yang, B. Zhang, T. Gao, M.-Y. Yang, G.-Z. Zhao, H.-J. Zhu, L. Liu and F. Cao, Nat. Prod. Res., 2018, 32, 2366–2369 CrossRef CAS PubMed
- S. Romminger, E. F. Pimenta, E. S. Nascimento, A. G. Ferreira and R. G. S. Berlinck, J. Braz. Chem. Soc., 2012, 23, 1783–1788 CrossRef CAS
- B. Karadogan and P. J. Parsons, Tetrahedron, 2001, 57, 8699–8703 CrossRef CAS
- D. L. J. Clive and S. Hisaindee, Chem. Commun., 1999, 2251–2252 RSC
- T. Lübbers, P. Angehrn, H. Gmünder and S. Herzig, Bioorg. Med. Chem. Lett., 2007, 17, 4708–4714 CrossRef PubMed
- J. Plescia, D. Hédou, M. E. Pousse, A. Labarre, C. Dufresne, A. Mittermaier and N. Moitessier, Eur. J. Med. Chem., 2022, 240, 114543 CrossRef CAS PubMed
- Á. Cantín, M. P. López-Gresa, M. C. González, P. Moya, M. Á. Miranda, J. Primo, V. Romero, E. Peris and E. Estornell, J. Agric. Food Chem., 2005, 53, 8296–8301 CrossRef PubMed
- M. Tsuda, M. Sasaki, T. Mugishima, K. Komatsu, T. Sone, M. Tanaka, Y. Mikami and J. Kobayashi, J. Nat. Prod., 2005, 68, 273–276 CrossRef CAS PubMed
- Z. Chen, J.-J. Zhang, C.-Y. Huang, W.-C. Chen, L.-M. He, Q.-Q. Tang, K.-K. Zhu, J. Li, P. Gao, M.-K. Zhang and Y.-S. Cai, Phytochemistry, 2025, 229, 114273 CrossRef CAS PubMed
- X. Zhang, J. Guo, F. Cheng and S. Li, Nat. Prod. Rep., 2021, 38, 1072–1099 RSC
- E. Yang, Y. Yao, H. Su, Z. Sun, S.-S. Gao, S. Sureram, P. Kittakoop, K. Fan, Y. Pan, X. Xu, Z.-H. Sun, G. Ma and G. Liu, J. Am. Chem. Soc., 2024, 146, 11457–11464 CAS
- Z.-Q. Cao, G.-Q. Wang, R. Luo, Y.-H. Gao, J.-M. Lv, S.-Y. Qin, G.-D. Chen, T. Awakawa, X.-F. Bao, Q.-H. Mei, X.-S. Yao, D. Hu, I. Abe and H. Gao, J. Am. Chem. Soc., 2024, 146, 12723–12733 CrossRef CAS PubMed
- S.-S. Gao, T. Zhang, M. Garcia-Borràs, Y.-S. Hung, J. M. Billingsley, K. N. Houk, Y. Hu and Y. Tang, J. Am. Chem. Soc., 2018, 140, 6991–6997 CrossRef CAS PubMed
- S. Kalita, S. Shaik and K. D. Dubey, ACS Catal., 2022, 12, 5673–5683 CrossRef CAS
- F. P. Guengerich and F. K. Yoshimoto, Chem. Rev., 2018, 118, 6573–6655 CrossRef CAS PubMed
- Y. Jiang, W. Peng, Z. Li, C. You, Y. Zhao, D. Tang, B. Wang and S. Li, Angew. Chem., Int. Ed., 2021, 60, 24694–24701 CrossRef CAS PubMed
- I. M. Mokhosoev, D. V. Astakhov, A. A. Terentiev and N. T. Moldogazieva, Cells, 2024, 13, 1958 CrossRef CAS PubMed
- Y. Tateishi, K. D. McCarty, M. V. Martin, F. K. Yoshimoto and F. P. Guengerich, Angew. Chem., Int. Ed., 2024, 63, e202406542 CrossRef CAS PubMed
- J. Liu, A. Liu and Y. Hu, Nat. Prod. Rep., 2021, 38, 1469–1505 RSC
- M. Liu, M. Ohashi, Q. Zhou, J. N. Sanders, E. P. McCauley, P. Crews, K. N. Houk and Y. Tang, Angew. Chem., Int. Ed., 2023, 62, e202311266 CrossRef CAS PubMed
- H.-C. Lin, T. C. McMahon, A. Patel, M. Corsello, A. Simon, W. Xu, M. Zhao, K. N. Houk, N. K. Garg and Y. Tang, J. Am. Chem. Soc., 2016, 138, 4002–4005 CrossRef CAS PubMed
- Y. Gu, P. Ding, Z. Liang, Y. Song, Y. Liu, G. Chen and J. L. Li, Fitoterapia, 2018, 127, 207–211 CrossRef CAS PubMed
- K. Blin, S. Shaw, A. M. Kloosterman, Z. Charlop-Powers, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2021, 49, W29–W35 CrossRef CAS PubMed
- D. Boettger and C. Hertweck, ChemBioChem, 2013, 14, 28–42 CrossRef CAS PubMed
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185–1209 RSC
- K. Zhang, J. Liu, Y. Jiang, S. Sun, R. Wang, J. Sun, C. Ma, Y. Chen, W. Wang, X. Hou, T. Zhu, G. Zhang, Q. Che, R. A. Keyzers, M. Liu and D. Li, J. Am. Chem. Soc., 2024, 146, 18172–18183 CrossRef CAS PubMed
- L. Chen, X. Wang, Y. Zou and M.-C. Tang, Org. Lett., 2024, 26, 3597–3601 CrossRef CAS PubMed
- C.-Y. Chiang, M. Ohashi and Y. Tang, Nat. Prod. Rep., 2023, 40, 89–127 RSC
- G.-L. Tang, Y.-Q. Cheng and B. Shen, J. Biol. Chem., 2007, 282, 20273–20282 CrossRef CAS PubMed
- J. Yu, J. Song, C. Chi, T. Liu, T. Geng, Z. Cai, W. Dong, C. Shi, X. Ma, Z. Zhang, X. Ma, B. Xing, H. Jin, L. Zhang, S. Dong, D. Yang and M. Ma, ACS Catal., 2021, 11, 11733–11741 CrossRef CAS
- C. J. Balibar, F. H. Vaillancourt and C. T. Walsh, Chem. Biol., 2005, 12, 1189–1200 CrossRef CAS PubMed
- L. Luo, M. D. Burkart, T. Stachelhaus and C. T. Walsh, J. Am. Chem. Soc., 2001, 123, 11208–11218 CrossRef CAS PubMed
- K. Hoffmann, E. Schneider-Scherzer, H. Kleinkauf and R. Zocher, J. Biol. Chem., 1994, 269, 12710–12714 CrossRef CAS PubMed
- K. Tamura, G. Stecher and S. Kumar, Mol. Biol. Evol., 2021, 38, 3022–3027 CrossRef CAS PubMed
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed
- Z. Yang, X. Zeng, Y. Zhao and R. Chen, Signal Transduct. Target. Ther., 2023, 8, 1–14 CrossRef PubMed
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed
- E. F. Pimenta, A. M. Vita-Marques, A. Tininis, M. H. R. Seleghim, L. D. Sette, K. Veloso, A. G. Ferreira, D. E. Williams, B. O. Patrick, D. S. Dalisay, R. J. Andersen and R. G. S. Berlinck, J. Nat. Prod., 2010, 73, 1821–1832 CrossRef CAS PubMed
- D. Lai, H. Brötz-Oesterhelt, W. E. G. Müller, V. Wray and P. Proksch, Fitoterapia, 2013, 91, 100–106 CrossRef CAS PubMed
- J. Wei, X. Chen, Y. Ge, Q. Yin, X. Wu, J. Tang, Z. Zhang and B. Wu, J. Org. Chem., 2022, 87, 13270–13279 CrossRef CAS PubMed
- J. Gao, S. Liu, C. Zhou, D. Lara, Y. Zou and Y. Hai, Nat. Catal., 2023, 6, 476–486 CrossRef CAS
- S. J. Oliphant and R. H. Morris, ACS Omega, 2022, 7, 30554–30564 CrossRef CAS PubMed
- S.-S. Gao, T. Zhang, M. Garcia-Borràs, Y.-S. Hung, J. M. Billingsley, K. N. Houk, Y. Hu and Y. Tang, J. Am. Chem. Soc., 2018, 140, 6991–6997 CrossRef CAS PubMed
- G. Ma, H. Yu, T. Xu, X. Wei, J. Chen, H. Lin and G. Schüürmann, Environ. Sci. Technol., 2018, 52, 11838–11847 CAS
- F. Guo, L. Chai, S. Zhang, H. Yu, W. Liu, K. P. Kepp and L. Ji, Environ. Sci. Technol., 2020, 54, 2902–2912 CrossRef CAS PubMed
- R. Yang, Y. Ye, Y. Chen, Y. Yang, L. Yang, Y. Yao, W. Zhong and L. Zhu, Environ. Sci. Technol., 2023, 57, 451–462 CrossRef CAS PubMed
- Y. Zou, S. Yang, J. N. Sanders, W. Li, P. Yu, H. Wang, Z. Tang, W. Liu and K. N. Houk, J. Am. Chem. Soc., 2020, 142, 20232–20239 CrossRef CAS PubMed
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed
- J. Delgado, L. G. Radusky, D. Cianferoni and L. Serrano, Bioinformatics, 2019, 35, 4168–4169 CrossRef CAS PubMed
- S.-H. Li, X. Zhang, Z.-L. Mei, Y. Liu, J.-A. Ma and F.-G. Zhang, J. Am. Chem. Soc., 2024, 146, 19962–19973 CrossRef CAS PubMed
- H. N. Raum, K. Modig, M. Akke and U. Weininger, J. Am. Chem. Soc., 2024, 146, 22284–22294 CrossRef CAS PubMed
- S. P. Putri, K. Ishido, H. Kinoshita, S. Kitani, F. Ihara, Y. Sakihama, Y. Igarashi and T. Nihira, J. Biosci. Bioeng., 2014, 117, 557–562 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07174c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |