
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLi1.6AlCl3.4S0.6: a low-cost and high-performance solid electrolyte for solid-state batteries†
Tej P.
Poudel‡
 abe,
Ifeoluwa P.
Oyekunle‡
be,
Michael J.
Deck
be,
Yudan
Chen
be,
Dewen
Hou
c,
Pawan K.
Ojha
be,
Bright O.
Ogbolu
be,
Chen
Huang
ab,
Hui
Xiong
d and
Yan-Yan
Hu
*abe
abe,
Ifeoluwa P.
Oyekunle‡
be,
Michael J.
Deck
be,
Yudan
Chen
be,
Dewen
Hou
c,
Pawan K.
Ojha
be,
Bright O.
Ogbolu
be,
Chen
Huang
ab,
Hui
Xiong
d and
Yan-Yan
Hu
*abe
aMaterials Science and Engineering Program, The Graduate School, Florida State University, 2005 Levy Ave., Tallahassee, FL 32310, USA. E-mail: yhu@fsu.edu
bDepartment of Chemistry and Biochemistry, Florida State University, 95 Chieftan Way, Tallahassee, FL 32306, USA
cCenter for Nanoscale Materials, Argonne National Laboratory, 9700 S Cass Ave, Lemont, IL 60439, USA
dMicron School of Materials Science and Engineering, Boise State University, Boise, ID 83725, USA
eCenter of Interdisciplinary Magnetic Resonance, National High Magnetic Field Laboratory, 1800 East Paul Dirac Drive, Tallahassee, FL 32310, USA
First published on 8th January 2025
Abstract
Solid electrolytes (SEs) are crucial for advancing next-generation rechargeable battery technologies, but their commercial viability is partially limited by expensive precursors, unscalable synthesis, or low ionic conductivity. Lithium tetrahaloaluminates offer an economical option but exhibit low Li+ conductivities with high activation energy barriers. This study reports the synthesis of lithium aluminum chalcohalide (Li1.6AlCl3.4S0.6) using inexpensive precursors via one-step mechanochemical milling. The resulting Cl–S mixed-anion sublattice significantly improves the ionic conductivity from 0.008 mS cm−1 for LiAlCl4 to 0.18 mS cm−1 for Li1.6AlCl3.4S0.6 at 25 °C. Structural refinement of the high-resolution XRD patterns and 6Li magic-angle-spinning (MAS) NMR quantitative analysis reveals the formation of tetrahedrally-coordinated, face- and edge-shared LiClxSy octahedra that facilitate 3D Li+ transport. Ab initio molecular dynamics (AIMD) simulations on Li1.6AlCl3.4S0.6 support an enhanced 3D network for Li+ migration with increased diffusivity. All-solid-state battery (ASSB) half-cells using Li1.6AlCl3.4S0.6 exhibit high-rate and long-term stable cycling performance. This work highlights the potential of Li1.6AlCl3.4S0.6 as a cost-effective and high-performance SE for ASSBs.
Introduction
All-solid-state batteries (ASSBs) are rapidly gaining traction due to their high energy density and enhanced safety features, positioning them as strong contenders to replace traditional liquid-electrolyte-based lithium-ion batteries (LIBs) in portable electronic devices and electric vehicles.1,2 The practical commercialization of ASSBs hinges on discovering SEs that satisfy the cost and performance metrics. In recent years, ternary halides such as LiAlCl4 and NaAlCl4 have received significant attention owing to their low cost and fast ion transport when in solution or as a melt.3–6Aluminum is the third most abundant element in the earth's crust,7 which makes it a desirable lost-cost component in SEs for ASSBs. However, despite recent advancements in enhancing Li+ dynamics within the lithium tetrahaloaluminate class of solid electrolytes, their ionic conductivity remains notably low, thereby restricting their practical applications in ASSBs. Recent computational studies have demonstrated that stabilizing a metastable phase of LiAlCl4 can force Li-ions to occupy interstitial sites, thereby reducing the activation energy barrier for ion transport. Specifically, mechanochemical synthesis has induced structural disorder in LiAlCl4, resulting in a small fraction (2.5%) of Li-ions occupying interstitial tetrahedral sites.8 This has significantly improved the ionic conductivity of LiAlCl4 to 0.02 mS cm−1 at 25 °C, compared to the previous value of 0.001 mS cm−1.9 However, this improvement is still insufficient for commercial applications in ASSBs, which require conductivities greater than 0.1 mS cm−1 at 25 °C.
A synthesis approach that does not require high-temperature heating is highly attractive for fabricating commercially viable SEs. For example, by subjecting LiAlBr4 to mechanical stress, a moderate ionic conductivity of 0.033 mS cm−1 at room temperature has been reported.3 Li+ transport within this material is facilitated via octahedral and tetrahedral interstitial sites, which are intrinsic to lithium tetrahaloaluminates.3,8 Previous studies have shown that for some Li-SEs, e.g., Li6PS5Cl, Li3PS4, and LiAlCl4, ionic conductivity can be enhanced by introducing local disorder within the anion sublattice.10–17 Moreover, studies have reported connecting non-accessible ion-conducting channels by a random distribution of cations in a host structure via local disorder.18 To further improve the ionic conductivity, local disorder on the atomic scale can be implemented.19
Herein, we demonstrate an effective approach for improving Li+ conduction in LiAlCl4 by mixing inexpensive and poor-conducting Li2S and AlCl3. An ionic conductivity of 0.18 mS cm−1 is achieved at 25 °C for Li1.6AlCl3.4S0.6 compared to 0.008 mS cm−1 for LiAlCl4. Powder X-ray diffraction and solid-state nuclear magnetic resonance (NMR) spectroscopy, combined with Ab initio molecular dynamics (AIMD) simulations, were utilized to determine the structural origin of the improved ionic conductivity in Li1.6AlCl3.4S0.6. Galvanostatic measurements of Li1.6AlCl3.4S0.6-containing ASSB half-cells show an improved rate performance and long-term cycling stability compared with the control cell utilizing LiAlCl4.
Experimental
Material synthesis
LiCl (Sigma-Aldrich) was dried at 200 °C for 12 hours under a dynamic vacuum before being stored in an argon-filled glovebox. Anhydrous Li2S (Alfa Aesar) and ultra-dry AlCl3 (Alfa Aesar) were used as received and handled under argon. Stoichiometric amounts of precursors were ground using a mortar/pestle for 5 minutes inside an argon-filled glovebox. The hand-milled powder was transferred into a ZrO2 jar containing two 10 mm balls as milling media. After the jar was vacuum sealed, mechanochemical mixing was performed using a SPEX 8000M MIXER/MILL (SPEX®SamplePrep, USA) for 20 hours. Afterward, the ball-milled powder, typically ∼130 mg, was pressed into a pellet of 8 mm in diameter under ∼400 MPa inside an argon-filled Mbraun glovebox. The resulting pellet had a thickness of ∼1.2 mm and appeared shiny white.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 562 Å). The data was collected in the 2θ range of 10–80° at a step size of 0.03° for 30 minutes.
562 Å). The data was collected in the 2θ range of 10–80° at a step size of 0.03° for 30 minutes.
Variable-temperature 7Li T1 relaxation NMR experiments were performed using a Bruker Avance-I 300 MHz Spectrometer from 25 to 70 °C. An inversion recovery pulse sequence with a π/2 pulse length of 2.63 μs was utilized. The 7Li Larmor frequency was 116.6 MHz. Sample powders were packed into 4 mm ZrO2 rotors under Argon and spun at a MAS rate of 10 kHz.
A 6Li|SE|6Li symmetrical cell was assembled in an argon-filled glovebox for the 6Li → 7Li tracer exchange experiments. The cell was then subjected to galvanostatic cycling for 3 days at a current density of 5 μA cm−2 to drive the diffusion of 6Li+ ions from the 6Li foil into the Li2AlCl3S pellet. Following the galvanostatic cycling, 6Li MAS NMR experiments were performed on the cycled Li2AlCl3S pellet using the Bruker Advance-III 500 spectrometer at a spinning rate of 24 kHz, using the same parameters described above.
:carbon black (C: Super P) composite was evenly spread and pressed at 300 MPa for 10 s. On the opposite side of the LiAlCl4 (or Li2AlCl3S) pellet, a piece of indium (In) foil measuring 5/16 inch in diameter and 0.1 mm in thickness, with an approximate weight of 32 mg was attached. Subsequently, lithium foil with a 3/16-inch diameter and weighing around 1 mg was pressed onto the In foil to form Li–In and used as the counter electrode, giving a final cell setup of Li–In|SE|3SE:C. With the cell sealed using vacuum grease, it was subjected to electrochemical cycling under an estimated stack pressure of approximately 30 MPa at 22 °C. CV and LSV measurements were conducted with a scan rate of 0.2 mV s−1 from 0 to 4 V vs. Li–In and 0 to 4.5 V respectively.
To prepare the composite cathode (or catholyte) for galvanostatic cycling, spherical TiS2 particles of ∼74 microns were purchased from Sigma-Aldrich. It was dried at 200 °C for 12 hours, then subjected to ball milling for 5 hours at 300 rpm to reduce particle size. Subsequently, LiAlCl4 or Li2AlCl3S was combined with TiS2 at a TiS2:SE mass ratio of 1:2 and ground together using a mortar and pestle for 10 minutes. Li6PS5Cl, synthesized following the established method by Patel et al.,13 was pressed into pellets at 300 MPa for 10 seconds as the separator. For the half-cell assembly, 12 mg of the catholyte was evenly spread onto one side of the Li6PS5Cl pellet, achieving an aerial loading of approximately 1.25 mA h cm−2, followed by further pressing at 300 MPa for 10 seconds. A Li–In alloy foil was affixed to the opposite side of the Li6PS5Cl pellet to assemble the Li–In|Li6PS5Cl|2SE:TiS2 (SE: LiAlCl4 or Li2AlCl3S) half cells. Finally, the cells were sealed with vacuum grease and subjected to controlled cycling conditions at 22 °C with a stack pressure of ∼30 MPa, within a voltage window of 1–2.5 V vs. Li–In. For rate performance evaluations, the cells underwent cycling for 5 cycles at each of the following rates: 0.1C, 0.2C, 0.5C, 1C, and 2C, with C representing the charge–discharge rate. Correspondingly, these rates translate to current densities of 0.14 mA cm−2, 0.28 mA cm−2, 0.70 mA cm−2, 1.40 mA cm−2, and 2.80 mA cm−2, respectively. Subsequently, long-term stability testing was conducted over 175 cycles at 0.2C.
Results and discussion
X-ray diffraction and structure
X-ray diffraction (XRD) was employed to investigate the long-range structure of the synthesized solid electrolytes. The powder XRD confirms the presence of a monoclinic LiAlCl4 phase (Fig. 1). With sulfur substitution, the long-range monoclinic structure is maintained for Li2AlCl3S. However, the weak and diffuse diffraction pattern of the as-milled Li2AlCl3S suggests a decrease in the crystallinity of the material. Rietveld refinement was performed against high-resolution XRD data using GSAS-II. Fig. 1(c)–(f) shows the diffraction patterns and the refined structures. For LiAlCl4, the crystalline phase was refined with a monoclinic structure type in the P21/c space group. The structure is built from distorted LiX6 octahedra and AlCl4− tetrahedra. Two LiCl65− octahedra are edge-shared to form Li2Cl108− dimers.1 AlCl4− tetrahedra are isolated from one another. All the atoms in the structure occupy only the 4e Wyckoff positions, leaving all other sites, i.e., 2a, 2b, 2c, and 2d Wyckoff positions, vacant (see Fig. 1(e)). The arrangement of atoms is characterized by a slightly distorted hexagonal cubic packed (hcp) Cl− sublattice, where Li+ and Al3+ fill octahedral and tetrahedral interstices, respectively.1 In addition, each AlCl4 tetrahedron is linked to one Li–Cl dimer via two edges and to two other dimers through one corner each (Fig. 1(e)).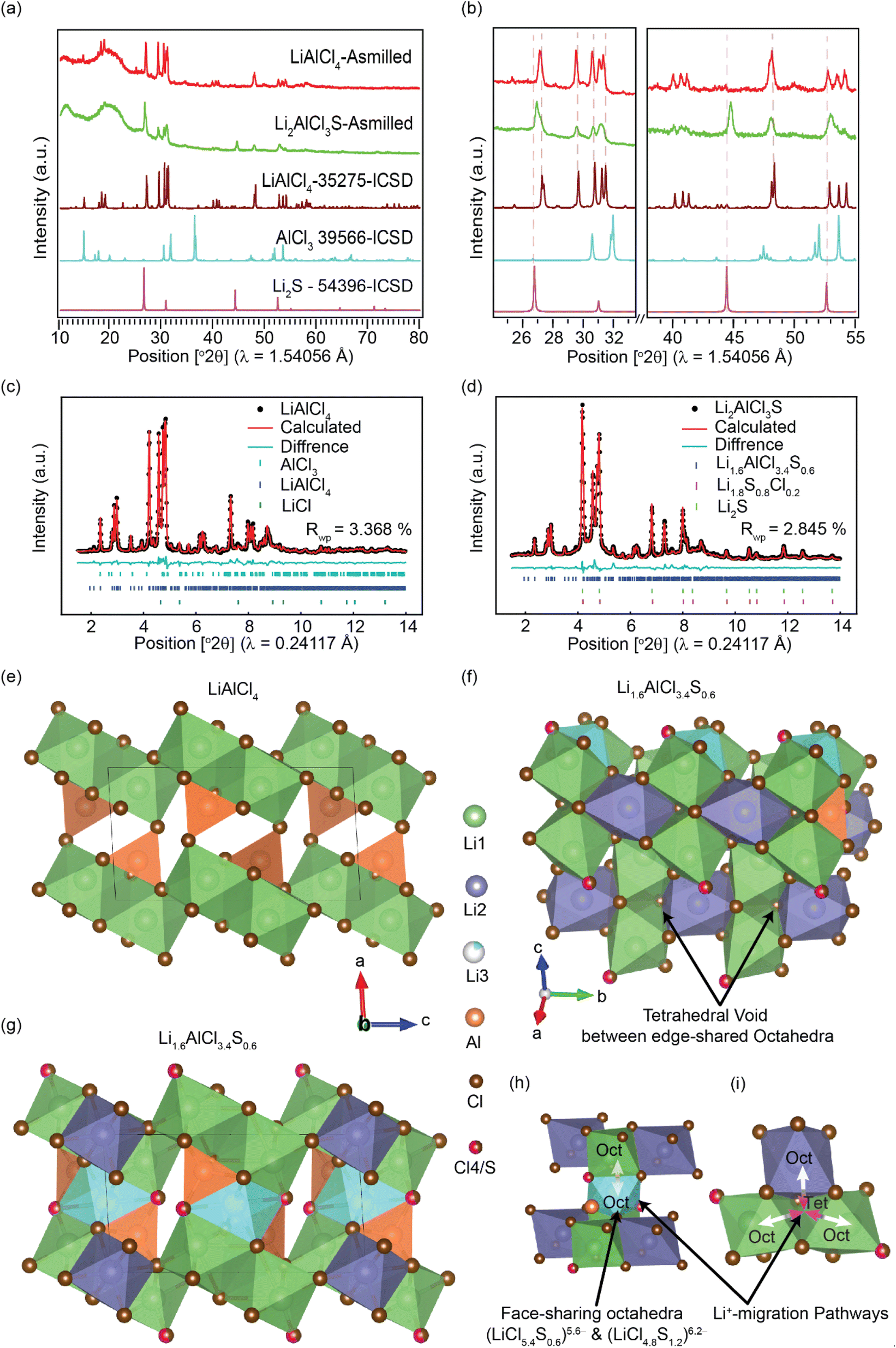 | ||
| Fig. 1 Long-range structures of LiAlCl4 and Li2AlCl3S, determined using lab-X-ray diffraction and high-resolution X-ray diffraction. (a) Lab X-ray diffraction patterns of the as-milled LiAlCl4 and Li2AlCl3S. The ICSD patterns of precursors and LiAlCl4 monoclinic phase (P21/c space group) are shown as references. (b) The magnified view of the selected 2θ ranges in (a). (c) and (d) High-resolution X-ray diffraction patterns and the corresponding Rietveld refinement of LiAlCl4 and Li2AlCl3S, respectively. (e) and (g) Monoclinic structures of LiAlCl4 and Li1.6AlCl3.4S0.6 with the P21/c space group, respectively, obtained from refined high-resolution XRD patterns. (f) The structure of Li1.6AlCl3.4S0.6 viewed along the a-axis showing tetrahedral voids face-shared with Li-octahedra. (h) The visualization of partially occupied octahedral lithium at Wyckoff 2b position face-shared with highly distorted octahedral sites at Wyckoff 4e positions. (i) Possible lithium migration pathways. | ||
Similarly, Li2AlCl3S crystallizes in the P21/c space group. However, different from LiAlCl4, the unit cell consists of three octahedrally coordinated cation sites – Li1 at 4e, Li2 at 2a, and Li3 at the 2a Wyckoff positions; and tetrahedrally coordinated Al at the 4e Wyckoff position. Sulfur and chlorine atoms co-occupy the 4e anionic site, yielding a disordered anion sublattice. Along the c-direction, the structure exhibits three distinct cation layers (Fig. 1(g)). In the first layer, Li2 octahedra (LiCl6)5− edge-share with Li1 (LiCl5.4S0.6)5.6− and corner-share with the tetrahedrally coordinated Al. In the second layer, Li3 octahedra (LiCl4.8S1.2)6.2− face-share with Li1 (Fig. 1(h)), and both Li1 and Li3 octahedra share corners with the tetrahedrally coordinated Al. In the third layer, the Al sites face-share with Li3 and edge-share with Li1 and Li2 octahedra. In addition, the edge-sharing Li1, Li2, and Li3 octahedra produce tetrahedral voids (Fig. 1(f)). The complete refinement parameters for LiAlCl4 and Li2AlCl3S are provided in Tables S1 to S3.† The refinement reveals the actual composition of the nominal Li2AlCl3S is Li1.6AlCl3.4S0.6, which exhibits mixed Cl−/S2− site occupancies in the structure. Consistent with the refined composition, a trace amount of Li2S is identified in the ball-milled sample. The refined composition, Li1.6AlCl3.4S0.6, is adopted hereafter.
Solid-state NMR
To understand the local structures of LiAlCl4 and Li1.6AlCl3.4S0.6, 6Li MAS NMR experiments were performed. As shown in Fig. 2, the 6Li MAS NMR spectrum of LiAlCl4 reveals a major resonance at −0.92 ppm assigned to octahedrally coordinated Li1. This is consistent with findings from a previous study.8 In addition, a minor resonance at −1.08 ppm is assigned to LiCl.26 After incorporating S2− in the parent material, the 6Li NMR spectra of Li1.6AlCl3.4S0.6 is characterized by five distinct resonances at −0.89, −1.24, −0.07, 1.48, and 2.21 ppm – indicating changes in the local Li environment. The minor peak observed at 2.21 ppm corresponds to the Li2S impurity.27 The phase quantification is given in Table S4.† The resonances at −0.89, −1.24, and −0.07 ppm are assigned to the octahedrally coordinated Li1, Li2, and Li3 sites, respectively – in agreement with the results from structural refinement. In addition, there is an extra peak at 1.48 ppm, likely from the Li1.66S0.66Cl0.34 phase identified with the Rietveld refinement of the high-resolution diffraction pattern (Fig. S1†). | ||
| Fig. 2 6Li MAS NMR spectra of LiAlCl4 and Li1.6AlCl3.4S0.6. | ||
7Li spin–lattice relaxation time (T1) is a useful indicator of ion dynamics.11,28 According to the Bloembergen, Purcell, and Pound (BPP) model, T1 relaxation time is a function of motional rate (τc−1),29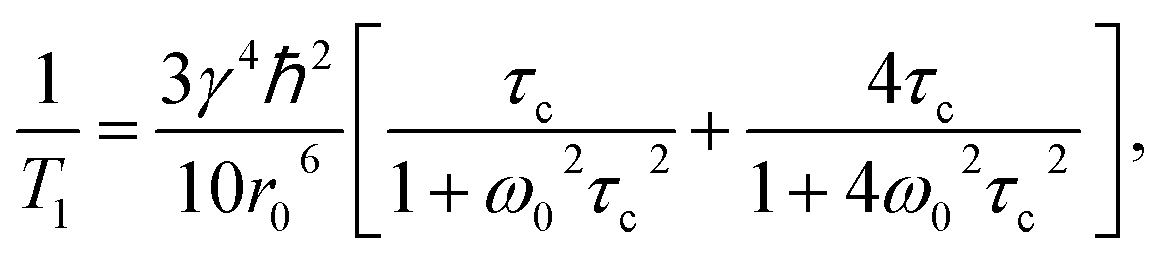 where γ is the gyromagnetic ratio, ħ is the reduced Planck's constant, r0 is the interatomic distance, ω0 = γB0 is the Larmor frequency, and B0 is the external magnetic field strength.
where γ is the gyromagnetic ratio, ħ is the reduced Planck's constant, r0 is the interatomic distance, ω0 = γB0 is the Larmor frequency, and B0 is the external magnetic field strength.
In the fast-motion regime (ω0τc ≪ 1), T1 increases with increasing motional rate, while in the slow-motion regime (ω0τc ≫ 1), T1 decreases with increasing motional rate. In addition, a resonance can lie in the intermediate region where ω0τc ≈ 1.29 Variable-temperature 7Li NMR T1 relaxation data of Li1.6AlCl3.4S0.6 in Fig. S2† reveals a decrease in T1 relaxation time with increasing temperature and, thus, suggesting Li+ dynamics in Li1.6AlCl3.4S0.6 lie in the slow-motion regime (ω0τc ≪ 1). Therefore, a shorter T1 value will correlate with faster ion mobility. As presented in Table 1, the 7Li T1 relaxation time significantly decreases from LiAlCl4 to Li1.6AlCl3.4S0.6, suggesting enhanced Li+ mobility30 with sulfur incorporation.
| Sample | 7Li T1 [s] |
|---|---|
| LiAlCl4 | 5.2 |
| Li1.6AlCl3.4S0.6 | 3.1 |
Ion transport pathways determined by tracer-exchange NMR
To directly probe the Li+ transport pathways in Li1.6AlCl3.4S0.6, tracer-exchange NMR is employed.31 By identifying and quantifying 6Li+ → 7Li+ exchange under an applied biased potential, the preferential pathway utilized by Li+ ions for migration is directly mapped out – facilitating the identification of active sites for Li+ transport.31–34 The experimental configuration involves sandwiching Li1.6AlCl3.4S0.6 pellet between two 6Li foils. An externally applied potential gradient establishes a driving force for 6Li+ ions in the 6Li foils to move toward and exchange with 7Li+ in the Li1.6AlCl3.4S0.6 pellet. Consequently, the preferential Li+ transport pathways are selectively enriched with 6Li+ ions. The 6Li NMR of the pristine and tracer-exchanged Li1.6AlCl3.4S0.6 pellet is displayed in Fig. S3a,† and the quantification of the Li sites before and after the tracer-exchange experiment is presented in Fig. S3b.† The 6Li NMR spectra reveal changes in the relative intensities of resonances assigned to distinct Li+ environments. Notably, a significant increase in the intensity of the Li1 and Li2 resonances is observed, suggesting the major involvement of Li1 and Li2 in ion conduction. In addition, the Li3 resonance shows a small intensity enhancement after cycling. The enhancement of these resonances suggests that Li1, Li2, and Li3 all participate in Li+-ion transport within the Li1.6AlCl3.4S0.6 solid electrolyte.AIMD simulations
To further understand the effect of Cl–S anion mixing on the Li+ density distribution and diffusion, AIMD simulations are employed for LiAlCl4 and Li1.6AlCl3.4S0.6 in a 2 × 2 × 1 cell. The mean square displacements (MSD) of Li+ (Fig. 3(a) and (b)) and distribution probability (Fig. 3(c) and (d)) for Li at 900 K were calculated. The MSD plots demonstrate that Li+ can diffuse in all three directions, with higher diffusion observed along the b direction in LiAlCl4. However, Li+ diffusion along the a and c directions increases significantly with Cl–S anion mixing, resulting in comparable MSDs in all three directions in Li1.6AlCl3.4S0.6. This transformation indicates that Li1.6AlCl3.4S0.6 is a 3D ion conductor with a uniform ion transport network in all three dimensions. Furthermore, the overall MSD of Li+ shows a 50% increase upon Cl–S anion diversification compared to LiAlCl4. The AIMD simulation of Li+ trajectories (i.e., Li+ probability density) in LiAlCl4 shows a localized “cage-like” Li+ diffusion pattern with few interstitial jumps, indicating limited long-range Li+ migration. In comparison, Li1.6AlCl3.4S0.6 exhibits a delocalized Li+ diffusion network, suggesting improved macroscopic Li+ migration and, consequently, enhanced Li+ conduction in Li1.6AlCl3.4S0.6. | ||
| Fig. 3 Mean square displacement (MSD) of Li+ in (a) LiAlCl4 and (b) Li1.6AlCl3.4S0.6 generated from AIMD simulations. Li+ (yellow) probability density map of LiAlCl4 (c) and Li1.6AlCl3.4S0.6 (d) in a 2 × 2 × 1 cell based on AIMD simulations at 900 K. | ||
Electrochemical properties
To examine ion transport properties of all prepared SEs, variable-temperature electrochemical impedance spectroscopy (EIS) was employed, and the corresponding Nyquist plots at 25 °C are presented in Fig. 4(a) with a corresponding exemplary equivalent circuit fitting shown in Fig. 4(b). The results from the EIS analysis are given in Table 2. Based on fitted resistances from the Nyquist plot using equivalent circuit modeling, the conductivities of LiAlCl4 and Li1.6AlCl3.4S0.6 are 0.008 mS cm−1 and 0.18 mS cm−1, respectively (Fig. 4(a)). In addition, an ionic conductivity of 0.12 mS cm−1 and 0.11 mS cm−1 were obtained at x = 0.5 and x = 0.8 in Li1+xAlCl4−xSx (Fig. S4†). The lower ionic conductivity observed at x = 0.5 is likely due to insufficient sulfur substitution to significantly perturb the lattice and create additional favorable Li+ migration pathways. Conversely, at x = 0.8, the solid-solution limit is reached yielding a high fraction of Li2S impurity, which likely contributes to the lower ionic conductivity. Therefore, the optimal composition for the highest ionic conductivity is Li1.6AlCl3.4S0.6. To confirm the conductivities are due to ionic transport, the DC polarization measurements25 were performed, and the plots for LiAlCl4 and Li1.6AlCl3.4S0.6 are shown in Fig. 4(c). The current value upon plateauing corresponds to electronic conductivities of 3.49 × 10−8 S cm−1 and 5.43 × 10−8 S cm−1 for LiAlCl4 and Li1.6AlCl3.4S0.6, respectively, thereby confirming negligible electronic contribution to the measured total conductivities of the samples. The ionic conductivity of Li1.6AlCl3.4S0.6 is approximately twenty-fold greater than LiAlCl4 at room temperature. The significant increase in ionic conductivity of Li1.6AlCl3.4S0.6 can be attributed to the local disorder and “Li-stuffing” of the Li3-octahedra that connect with neighboring Li1/Li2 octahedra and the substitution of Cl with more polarizable S anions. The energy barrier for Li-ion transport (Ea) was calculated from the variable-temperature EIS measurements between 0 °C to 70 °C, and representative Nyquist plots are shown in Fig. S5.† The Arrhenius-type plot of LiAlCl4 and Li1.6AlCl3.4S0.6 are shown in Fig. 4(d). A decrease in Ea from 0.51 eV to 0.44 eV is observed from LiAlCl4 to Li1.6AlCl3.4S0.6. This decrease in activation energy correlates with the increase in ionic conductivity.35 This trend agrees with the energy barrier obtained from the bond valence site energy (BVSE) calculation (Fig. S6†).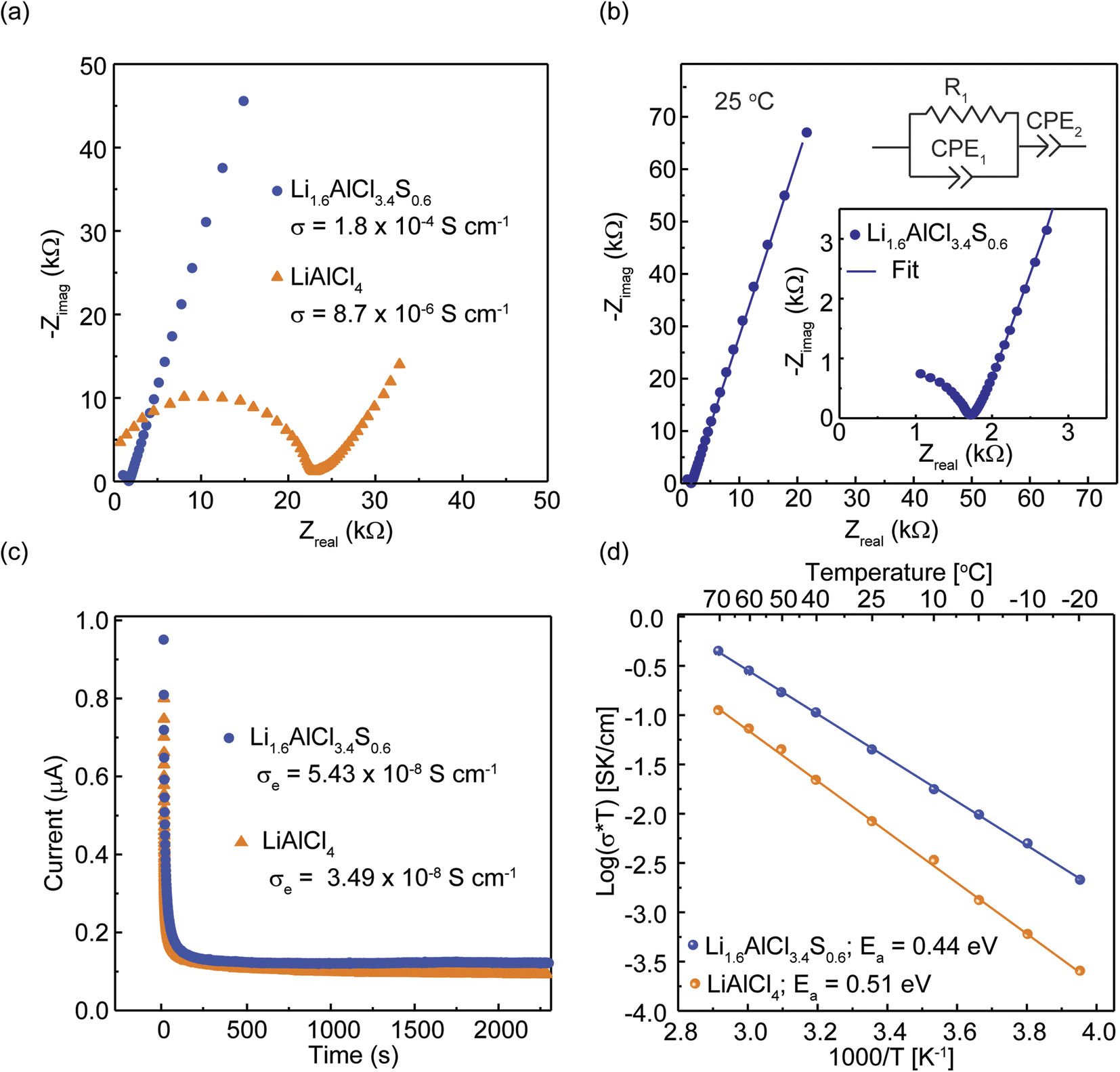 | ||
| Fig. 4 Charge transport properties of LiAlCl4 and Li1.6AlCl3.4S0.6. (a) Nyquist plots of LiAlCl4 and Li1.6AlCl3.4S0.6. (b) Exemplary equivalent circuit fitting of the Nyquist plot for Li1.6AlCl3.4S0.6 measured at 25 °C. (c) Electronic conductivities were determined using DC polarization, and (d) Arrhenius plots and activation energies for ion transport in LiAlCl4 and Li1.6AlCl3.4S0.6. | ||
| Composition | σ DC, 25°C [S cm−1] | σ e, 25°C [S cm−1] | E a [eV] | Log(σ0) [S cm−1 K] |
|---|---|---|---|---|
| LiAlCl4 | 8.65 × 10−6 | 3.49 × 10−8 | 0.51 | 6.55 |
| Li1.6AlCl3.4S0.6 | 1.80 × 10−4 | 5.43 × 10−8 | 0.44 | 6.07 |
Cyclic voltammetry and galvanostatic cycling of ASSB half-cells
Conventionally, CV measurements were done using stainless steel as the blocking electrode, which fails to accurately measure the oxidation and reduction current of SEs due to the limited electrical contact area of the SE and the planar ion-blocking electrode.11,36 To overcome this and estimate the oxidation–reduction reactions of the SE, we performed CV utilizing a 3SE:C (mass ratio) composite cathode in the Li–In|SE|3SE:C half-cell setup.37–39 In this setup, carbon serves as an electronic conductive medium in the composite cathode, enabling increased SE surface area in electrical contact with the electrode and consequential detection of degradation current.11,17,36–42 Fig. S7† shows the voltammograms of Li1.6AlCl3.4S0.6 with a scanning window of 0–4 V vs. Li–In. The first cathodic peak starts at the voltage of 1.03 V vs. Li–In. This voltage is assigned to the electrolyte reduction to form Li2S and AlCl3.11,17,43 The CV for Li1.6AlCl3.4S0.6 indicates the onset oxidation voltage of 2.4 V vs. Li–In. This is reasonable as most of the reported sulfide solid electrolytes have a lower stability window than halides (LiAlCl4).44,45 So, the electrochemical stability window of the prepared Li1.6AlCl3.4S0.6 is in the range of 1.03–2.40 V vs. Li–In, corresponding to the 1.63–3.00 V vs. Li/Li+. We have also performed LSV measurement (Fig. S8†), which shows the same electrochemical stability window of 1.63–3.00 V vs. Li/Li+
Titanium disulfide (TiS2) was employed as the cathode active material (CAM) in the half-cell configurations for electrochemical evaluation. Li–In|Li6PS5Cl|2(LiAlCl4):TiS2 and Li–In|Li6PS5Cl|2(Li1.6AlCl3.4S0.6):TiS2 cells were fabricated according to previous studies.11,17 A Li–In anode was employed for enhanced stability against SEs and limited dendrite formation through micropores – reducing the risk of short circuits.46 Li6PS5Cl was utilized as the separator due to its high ionic conductivity and stability against Li metal. The electrochemical performance of these half-cells was assessed through a series of rate capability tests, spanning charging/discharging currents from 0.1C to 2C conducted under galvanostatic conditions at ∼22 °C. Each rate was applied over five cycles (0.1C ∼0.14 mA cm−2, 0.2C ∼0.28 mA cm−2, 0.5C ∼0.70 mA cm−2, 1C ∼1.40 mA cm−2, and 2C ∼2.80 mA cm−2) followed by 125 cycles at 0.2C. A theoretical capacity of 239 mA h g−1 for TiS2 was used to calculate the charge–discharge rates.
Fig. 5(a) illustrates the variation in specific capacity with cycle number for the half-cell configurations, while Fig. 5(b) and (c) display the voltage profiles for select cycles. Initially, the cell incorporating LiAlCl4 demonstrated a discharge capacity of approximately 273 mA h g−1 and a charge capacity of 213 mA h g−1, whereas the Li1.6AlCl3.4S0.6-containing cell exhibited a comparable initial discharge capacity of 272 mA h g−1 and a higher charge capacity of 248 mA h g−1 (Fig. 5(a) and (b)). Upon the second discharge, the capacity for the LiAlCl4-based cell declined to 208 mA h g−1, whereas the Li1.6AlCl3.4S0.6-based cell showed a reduced yet more stable capacity of 240 mA h g−1, eventually stabilizing around 239 mA h g−1 (Fig. 5(a) and (b)). This is likely due to the formation of a stable solid–electrolyte interface (SEI) during the first cycle. The lower capacity observed in the LiAlCl4-based cell is primarily due to its low ionic conductivity of the solid electrolyte (SE), which restricts the efficient utilization of the active material – leading to a lower capacity.47 Interestingly, initial capacities recorded for both cells surpassed the theoretical capacity of TiS2, (239 mA h g−1). This is attributable to the unidentified reversible redox behavior of the SE in addition to the Ti3+/4+ redox (Fig. S9†).48,49
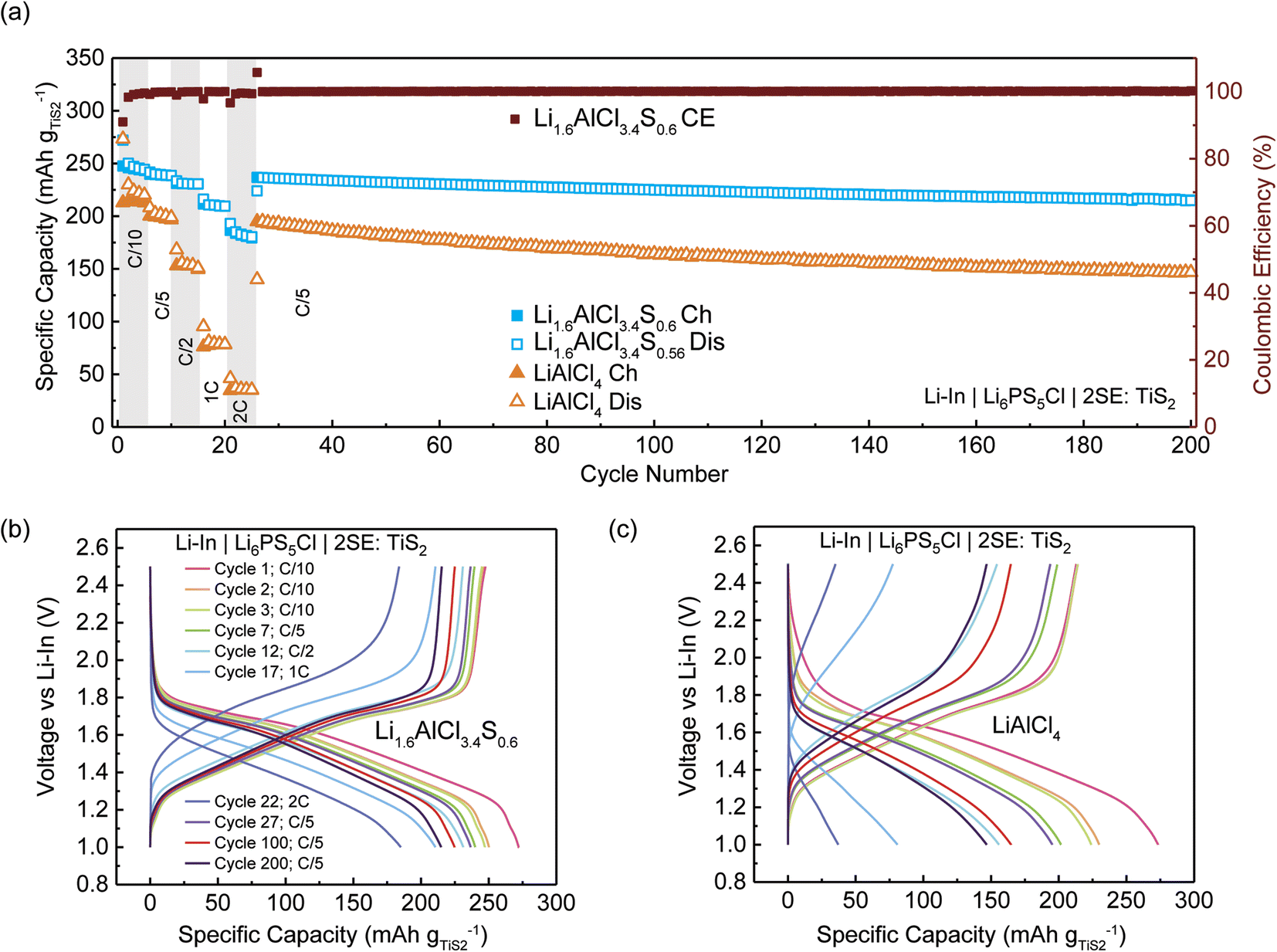 | ||
| Fig. 5 Galvanostatic cycling of 2SE:TiS2 catholyte with argyrodite Li6PS5Cl as the separator. (a) Rate performance of Li–In|Li6PS5Cl|2SE:TiS2 cell (SE = LiAlCl4 or Li1.6AlCl3.4S0.6) followed by long-term cycling at C/5, where C = 239 mA h g−1. Voltage profile of cycle 1 along with the 2nd cycles of each C-rate for a (b) Li–In|Li6PS5Cl|2(Li1.6AlCl3.4S0.6):TiS2 cell and (c) Li–In| Li6PS5Cl|2(LiAlCl4):TiS2 cell. | ||
The Li1.6AlCl3.4S0.6-containing cell exhibited a capacity of 180 mA h g−1 even at a high discharge rate of 2C, indicative of exceptional rate performance in contrast to the significantly reduced capacity of 37 mA h g−1 observed at a similar rate for the LiAlCl4-based cells. Upon returning to 0.2C after 26 cycles, both cell configurations demonstrated remarkable stability over 200 cycles. Furthermore, these cells maintained a high coulombic efficiency exceeding 99% throughout this extended cycle period (Fig. 5(a)). Between the 27th and 200th cycles, the Li1.6AlCl3.4S0.6-based cell demonstrated a capacity retention of approximately 90%, whereas the LiAlCl4-based cell exhibited a faster degradation rate. The superior performance of the Li–In|Li6PS5Cl|2(Li1.6AlCl3.4S0.6):TiS2 cell is attributed to the synergistic benefits of enhanced ionic conductivity, electrochemical stability, and improved utilization of cathode active materials.47
Conclusion
Developing inexpensive SEs using earth-abundant elements is imperative to reduce the cost of ASSBs for widespread applications in electric vehicles and consumer electronics. In this work, we synthesized a new lithium chalcohalide solid electrolyte, Li1.6AlCl3.4S0.6, with a room-temperature ionic conductivity of 0.18 mS cm−1. Structural characterization reveals that the enhanced ionic conductivity of Li1.6AlCl3.4S0.6 strongly correlates with the formation of face- and edge-sharing octahedrally coordinated lithium sites, which often create low-energy conduction pathways. AIMD simulations using the refined structures reveal incorporating the optimal amount of S into LiAlCl4 transitions it from a 1D to a 3D conductor. 6,7Li MAS NMR combined with tracer exchange and relaxometry reveals increased ion mobility and participation of all Li+ sites in ion conduction. Li1.6AlCl3.4S0.6 demonstrates good long-term cycling stability and rate performance in ASSBs, achieving a specific capacity of 180 mA h g−1 at a fast charging rate of 2C in a Li–In|Li6PS5Cl|2(Li1.6AlCl3.4S0.6):TiS2 battery cell, compared to 37 mA h g−1 in the LiAlCl4-containing cell. The cost-effectiveness, combined with the demonstrated high performance, makes Li1.6AlCl3.4S0.6 an excellent candidate as electrolytes for ASSBs.Data availability
The data that supports this manuscript will be available upon request.Author contributions
Tej Prasad Poudel: data curation: lead; formal analysis: lead; investigation: lead; methodology: lead; software: lead; validation: lead; visualization: lead; writing – original draft: lead. Ifeoluwa P. Oyekunle: data curation: equal; formal analysis: equal; investigation: equal; methodology: equal; software: equal; validation: equal; visualization: equal; writing – original draft: equal. Michael J. Deck: formal analysis: supporting; investigation: supporting; methodology: supporting; validation: supporting; visualization: supporting; writing – original draft: supporting. Yudan Chen: data curation: supporting; formal analysis: supporting; investigation: supporting; methodology: supporting; software: supporting; writing – original draft: supporting. Dewen Hou: data curation: supporting. Pawan K. Ojha: data curation: supporting; formal analysis: supporting; visualization: supporting; writing – review & editing: supporting. Bright O. Ogbolu: validation: supporting; writing – review & editing: supporting. Chen Huang: software: supporting. Hui Xiong: resources: supporting; supervision: supporting; validation: supporting. Yan-Yan Hu, PhD: conceptualization: lead; formal analysis: lead; funding acquisition: lead; methodology: lead; project administration: lead; resources: lead; supervision: lead; validation: lead; visualization: equal; writing – original draft: equal; writing – review & editing: lead.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the support from the National Science Foundation under grant no. DMR-1847038 and DMR-2319151. Hou and Xiong thank the support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences program under Award Number DE-SC0019121. All solid-state NMR experiments were performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement No. DMR-1644779 and DMR-2128556*. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.References
- X. Cao, S. Xu, Y. Zhang, X. Hu, Y. Yan, Y. Wang, H. Qian, J. Wang, H. Chang, F. Cheng and Y. Yang, Fluorine-Doped High-Performance Li6PS5Cl Electrolyte by Lithium Fluoride Nanoparticles for All-Solid-State Lithium-Metal Batteries, Trans. Tianjin Univ., 2024, 30, 250–261, DOI:10.1007/s12209-024-00394-1.
- K. Tuo, C. Sun and S. Liu, Recent Progress in and Perspectives on Emerging Halide Superionic Conductors for All-Solid-State Batteries, Electrochem. Energy Rev., 2023, 6(17), 1–46, DOI:10.1007/s41918-023-00179-5.
- N. Flores-González, N. Minafra, G. Dewald, H. Reardon, R. I. Smith, S. Adams, W. G. Zeier and D. H. Gregory, Mechanochemical Synthesis and Structure of Lithium Tetrahaloaluminates, LiAlX4 (X = Cl, Br, I): A Family of Li-Ion Conducting Ternary Halides, ACS Mater. Lett., 2021, 3(5), 652–657, DOI:10.1021/acsmaterialslett.1c00055.
- F. Scholz, W. Unkrig, P. Eiden, M. A. Schmidt, A. Garsuch and I. Krossing, Synthesis, Spectroscopic Characterization, Crystal Structures, Energetics, and Thermal Stabilities of Li[AlX4] (X = Cl, Br): Investigation and Performance of Their Electrolyte Solutions, Eur. J. Inorg. Chem., 2015,(19), 3128–3138, DOI:10.1002/ejic.201500254.
- T. Gao, B. Wang, L. Wang, G. Liu, F. Wang, H. Luo and D. Wang, LiAlCl4·3SO2 as a High Conductive, Non-Flammable and Inorganic Non-Aqueous Liquid Electrolyte for Lithium Ion Batteries, Electrochim. Acta, 2018, 286, 77–85, DOI:10.1016/j.electacta.2018.08.033.
- J. M. Weller, M. M. Li, E. Polikarpov, K. S. Han, N. Kidner, A. Patel, M. Nguyen, M. Stout, M. Gossett, K. Jung, D. M. Reed, V. L. Sprenkle and G. Li, Unlocking the NaCl-AlCl3 Phase Diagram for Low-Cost, Long-Duration Na-Al Batteries, Energy Storage Mater., 2023, 56, 108–120, DOI:10.1016/j.ensm.2023.01.009.
- J. M. S. D. Campos and L. F. Viccini, Cytotoxicity of Aluminum on Meristematic Cells of Zea Mays and Allium Cepa, Caryologia, 2003, 56(1), 65–73, DOI:10.1080/00087114.2003.10589309.
- N. Tanibata, S. Takimoto, K. Nakano, H. Takeda, M. Nakayama and H. Sumi, Metastable Chloride Solid Electrolyte with High Formability for Rechargeable All-Solid-State Lithium Metal Batteries, ACS Mater. Lett., 2020, 2(8), 880–886, DOI:10.1021/acsmaterialslett.0c00127.
- W. Weppner and R. A. Huggins, Ionic Conductivity of Solid and Liquid LiAlCl4, J. Electrochem. Soc., 1977, 124(1), 35–38, DOI:10.1149/1.2133238.
- B. C. Wood, J. B. Varley, K. E. Kweon, P. Shea, A. T. Hall, A. Grieder, M. Ward, V. P. Aguirre, D. Rigling, E. Lopez Ventura, C. Stancill and N. Adelstein, Paradigms of Frustration in Superionic Solid Electrolytes, Philos. Trans. R. Soc., A, 2021, 379(2211), 20190467, DOI:10.1098/rsta.2019.0467.
- T. P. Poudel, M. J. Deck, P. Wang and Y.-Y. Hu, Transforming Li3PS4 Via Halide Incorporation: A Path to Improved Ionic Conductivity and Stability in All-Solid-State Batteries, Adv. Funct. Mater., 2024, 34(4), 2309656, DOI:10.1002/adfm.202309656.
- D. Karabelli, K. P. Birke and M. Weeber, A Performance and Cost Overview of Selected Solid-State Electrolytes: Race between Polymer Electrolytes and Inorganic Sulfide Electrolytes, Batteries, 2021, 7(18), 1–13, DOI:10.3390/batteries7010018.
- S. V. Patel, S. Banerjee, H. Liu, P. Wang, P.-H. Chien, X. Feng, J. Liu, S. P. Ong and Y.-Y. Hu, Tunable Lithium-Ion Transport in Mixed-Halide Argyrodites Li6−xPS5−xClBrx: An Unusual Compositional Space, Chem. Mater., 2021, 33(4), 1435–1443, DOI:10.1021/acs.chemmater.0c04650.
- X. Feng, P.-H. Chien, Z. Zhu, I.-H. Chu, P. Wang, M. Immediato-Scuotto, H. Arabzadeh, S. P. Ong and Y.-Y. Hu, Studies of Functional Defects for Fast Na-Ion Conduction in Na3−yPS4−xClx with a Combined Experimental and Computational Approach, Adv. Funct. Mater., 2019, 29(9), 1807951, DOI:10.1002/adfm.201807951.
- A. Shastri, N. Rons, Q.-P. Ding, S. J. Kmiec, M. Olson, Y. Furukawa and S. W. Martin, The Novel Oxy-Sulfide Glassy Ionic Conductors Na4P2S7-xOx 0 ≤ x ≤ 7: Understanding the Features of Static and Dynamic Cations, Solid State Ionics, 2023, 402, 116363, DOI:10.1016/j.ssi.2023.116363.
- M. Olson, S. Kmiec and S. W. Martin, NaPON Doping of Na4P2S7 Glass and Its Effects on the Structure and Properties of Mixed Oxy-Sulfide-Nitride Phosphate Glass, Inorg. Chem., 2022, 61(44), 17469–17484, DOI:10.1021/acs.inorgchem.2c02300.
- M. J. Deck, P.-H. Chien, T. P. Poudel, Y. Jin, H. Liu and Y.-Y. Hu, Oxygen-Induced Structural Disruption for Improved Li+ Transport and Electrochemical Stability of Li3PS4, Adv. Energy Mater., 2024, 14(4), 2302785, DOI:10.1002/aenm.202302785.
- Q. Zhang, W. Arnold, Z. D. Hood, Y. Li, R. DeWees, M. Chi, Z. Chen, Y. Chen and H. Wang, Li 0.625 Al 0.125 H 0.25 Cl 0.75 O 0.25 Superionic Conductor with Disordered Rock-Salt Structure, ACS Appl. Energy Mater., 2021, 4(8), 7674–7680, DOI:10.1021/acsaem.1c01011.
- X. Li, J. T. Kim, J. Luo, C. Zhao, Y. Xu, T. Mei, R. Li, J. Liang and X. Sun, Structural Regulation of Halide Superionic Conductors for All-Solid-State Lithium Batteries, Nat. Commun., 2024, 15(53), 1–8, DOI:10.1038/s41467-023-43886-9.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B, 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979, DOI:10.1103/PhysRevB.50.17953.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (Pymatgen): A Robust, Open-Source Python Library for Materials Analysis, Comput. Mater. Sci., 2013, 68, 314–319, DOI:10.1016/j.commatsci.2012.10.028.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Design Principles for Solid-State Lithium Superionic Conductors, Nature Mater., 2015, 14(10), 1026–1031, DOI:10.1038/nmat4369.
- S. Ohno, C. Rosenbach, G. F. Dewald, J. Janek and W. G. Zeier, Linking Solid Electrolyte Degradation to Charge Carrier Transport in the Thiophosphate-Based Composite Cathode toward Solid-State Lithium-Sulfur Batteries, Adv. Funct. Mater., 2021, 31(18), 2010620, DOI:10.1002/adfm.202010620.
- J. Gamon, M. S. Dyer, B. B. Duff, A. Vasylenko, L. M. Daniels, M. Zanella, M. W. Gaultois, F. Blanc, J. B. Claridge and M. J. Rosseinsky, Li4.3AlS3.3Cl0.7 : A Sulfide–Chloride Lithium Ion Conductor with Highly Disordered Structure and Increased Conductivity, Chem. Mater., 2021, 33(22), 8733–8744, DOI:10.1021/acs.chemmater.1c02751.
- L. A. Huff, J. L. Rapp, J. A. Baughman, P. L. Rinaldi and A. A. Gewirth, Identification of Lithium–Sulfur Battery Discharge Products through 6Li and 33S Solid-State MAS and 7Li Solution NMR Spectroscopy, Surf. Sci., 2015, 631, 295–300, DOI:10.1016/j.susc.2014.07.027.
- M. J. Deck and Y.-Y. Hu, Leveraging Local Structural Disorder for Enhanced Ion Transport, J. Mater. Res., 2023, 38(10), 2631–2644, DOI:10.1557/s43578-023-01023-8.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Relaxation Effects in Nuclear Magnetic Resonance Absorption, Phys. Rev., 1948, 73(7), 679–712, DOI:10.1103/PhysRev.73.679.
- N. Wu, P. Chien, Y. Qian, Y. Li, H. Xu, N. S. Grundish, B. Xu, H. Jin, Y. Hu, G. Yu and J. B. Goodenough, Enhanced Surface Interactions Enable Fast Li + Conduction in Oxide/Polymer Composite Electrolyte, Angew. Chem., Int. Ed., 2020, 59(10), 4131–4137, DOI:10.1002/anie.201914478.
- J. Zheng and Y.-Y. Hu, New Insights into the Compositional Dependence of Li-Ion Transport in Polymer–Ceramic Composite Electrolytes, ACS Appl. Mater. Interfaces, 2018, 10(4), 4113–4120, DOI:10.1021/acsami.7b17301.
- P. Wang, H. Liu, S. Patel, X. Feng, P.-H. Chien, Y. Wang and Y.-Y. Hu, Fast Ion Conduction and Its Origin in Li6 − xPS5 − xBr1 + x, Chem. Mater., 2020, 32(9), 3833–3840, DOI:10.1021/acs.chemmater.9b05331.
- J. Zheng, H. Dang, X. Feng, P.-H. Chien and Y.-Y. Hu, Li-Ion Transport in a Representative Ceramic–Polymer–Plasticizer Composite Electrolyte: Li7La3Zr2O12 –Polyethylene Oxide–Tetraethylene Glycol Dimethyl Ether, J. Mater. Chem. A, 2017, 5(35), 18457–18463, 10.1039/C7TA05832B.
- J. Zheng, M. Tang and Y. Hu, Lithium Ion Pathway within Li7La3Zr2O12 -Polyethylene Oxide Composite Electrolytes, Angew., Chem. Int. Ed., 2016, 55(40), 12538–12542, DOI:10.1002/anie.201607539.
- Y. Gao, A. M. Nolan, P. Du, Y. Wu, C. Yang, Q. Chen, Y. Mo and S.-H. Bo, Classical and Emerging Characterization Techniques for Investigation of Ion Transport Mechanisms in Crystalline Fast Ionic Conductors, Chem. Rev., 2020, 120(13), 5954–6008, DOI:10.1021/acs.chemrev.9b00747.
- G. F. Dewald, S. Ohno, M. A. Kraft, R. Koerver, P. Till, N. M. Vargas-Barbosa, J. Janek and W. G. Zeier, Experimental Assessment of the Practical Oxidative Stability of Lithium Thiophosphate Solid Electrolytes, Chem. Mater., 2019, 31(20), 8328–8337, DOI:10.1021/acs.chemmater.9b01550.
- D. H. S. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. T. Marple, J.-M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, Elucidating Reversible Electrochemical Redox of Li6PS5Cl Solid Electrolyte, ACS Energy Lett., 2019, 4(10), 2418–2427, DOI:10.1021/acsenergylett.9b01693.
- Y. Tian, T. Shi, W. D. Richards, J. Li, J. Chul Kim, S.-H. Bo and G. Ceder, Compatibility Issues between Electrodes and Electrolytes in Solid-State Batteries, Energy Environ. Sci., 2017, 10(5), 1150–1166, 10.1039/C7EE00534B.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Electrochemical Stability of Li10GeP2 S12 and Li7La3Zr2O12 Solid Electrolytes, Adv. Energy Mater., 2016, 6, 1501590, DOI:10.1002/aenm.201501590.
- Y. Chen, P. Wang, E. Truong, B. Ogbolu, Y. Jin, I. Oyekunle, H. Liu, M. M. Islam, T. Poudel, C. Huang, I. Hung, Z. Gan and Y.-Y. Hu, Superionic Conduction in K3SbS4 Enabled by Cl-Modified Anion Lattice, Angew. Chem., Int. Ed., 2024, 63(35), e202408574, DOI:10.1002/anie.202408574.
- S. Wang, R. Fang, Y. Li, Y. Liu, C. Xin, F. H. Richter and C.-W. Nan, Interfacial Challenges for All-Solid-State Batteries Based on Sulfide Solid Electrolytes, J. Materiomics, 2021, 7(2), 209–218, DOI:10.1016/j.jmat.2020.09.003.
- A. D. Bui, S.-H. Choi, H. Choi, Y.-J. Lee, C.-H. Doh, J.-W. Park, B. G. Kim, W.-J. Lee, S.-M. Lee and Y.-C. Ha, Origin of the Outstanding Performance of Dual Halide Doped Li7P2S8X (X = I, Br) Solid Electrolytes for All-Solid-State Lithium Batteries, ACS Appl. Energy Mater., 2021, 4(1), 1–8, DOI:10.1021/acsaem.0c02321.
- S. Banerjee, M. L. Holekevi Chandrappa and S. P. Ong, Role of Critical Oxygen Concentration in the β-Li3PS4–xOx Solid Electrolyte, ACS Appl. Energy Mater., 2022, 5(1), 35–41, DOI:10.1021/acsaem.1c03795.
- S. Wang, Q. Bai, A. M. Nolan, Y. Liu, S. Gong, Q. Sun and Y. Mo, Lithium Chlorides and Bromides as Promising Solid-State Chemistries for Fast Ion Conductors with Good Electrochemical Stability, Angew. Chem., Int. Ed., 2019, 58(24), 8039–8043, DOI:10.1002/anie.201901938.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Interface Stability in Solid-State Batteries, Chem. Mater., 2016, 28(1), 266–273, DOI:10.1021/acs.chemmater.5b04082.
- J. Qu, J. Xiao, T. Wang, D. Legut and Q. Zhang, High Rate Transfer Mechanism of Lithium Ions in Lithium–Tin and Lithium–Indium Alloys for Lithium Batteries, J. Phys. Chem. C, 2020, 124(45), 24644–24652, DOI:10.1021/acs.jpcc.0c07880.
- P. Minnmann, L. Quillman, S. Burkhardt, F. H. Richter and J. Janek, Editors' Choice—Quantifying the Impact of Charge Transport Bottlenecks in Composite Cathodes of All-Solid-State Batteries, J. Electrochem. Soc., 2021, 168(4), 040537, DOI:10.1149/1945-7111/abf8d7.
- D. Y. Oh, Y. E. Choi, D. H. Kim, Y.-G. Lee, B.-S. Kim, J. Park, H. Sohn and Y. S. Jung, All-Solid-State Lithium-Ion Batteries with TiS2 Nanosheets and Sulphide Solid Electrolytes, J. Mater. Chem. A, 2016, 4(26), 10329–10335, 10.1039/C6TA01628F.
- B. R. Shin, Y. J. Nam, J. W. Kim, Y.-G. Lee and Y. S. Jung, Interfacial Architecture for Extra Li+ Storage in All-Solid-State Lithium Batteries, Sci. Rep., 2014, 4(1), 5572, DOI:10.1038/srep05572.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07151d |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |