
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTungsten-catalyzed stereodivergent isomerization of terminal olefins†
Tanner C.
Jankins‡
 ,
Camille Z.
Rubel‡
,
Hang Chi
Ho
,
Raul
Martin-Montero
and
Keary M.
Engle
*
,
Camille Z.
Rubel‡
,
Hang Chi
Ho
,
Raul
Martin-Montero
and
Keary M.
Engle
*
Department of Chemistry, The Scripps Research Institute, 10550N. Torrey Pines Road, La Jolla, CA 92037, USA. E-mail: keary@scripps.edu
First published on 4th December 2024
Abstract
Catalytic alkene isomerization is a powerful synthetic strategy for preparing valuable internal alkenes from simple feedstocks. The utility of olefin isomerization hinges on the ability to control both positional and stereoisomerism to access a single product among numerous potential isomers. Within base-metal catalysis, relatively little is known about how to modulate reactivity and selectivity with group 6 metal-catalyzed isomerization. Here, we describe a tungsten-catalyzed, positionally selective alkene isomerization reaction in which tuning the ligand environment grants access to either the E- or Z-stereoisomer. The reactions employ simple, commercially available precatalysts and ligands. Preliminary mechanistic studies suggest that the ligand environment around 7-coordinate tungsten is crucial for stereoselectivity, and that substrate directivity prevents over-isomerization to the conjugated alkene. These features allow for exclusive formation of β,γ-unsaturated carbonyl compounds that are otherwise difficult to prepare.
Alkenes are common functional groups in natural products,1 fragrances,2 and materials, and they are versatile intermediates in organic synthesis.3 The ability to access alkenes as a single positional and E/Z-stereoisomer is key for many applications; isomers have different physical and chemical properties. Further, internal alkenes are pro-chiral intermediates in chemical synthesis, so stereoselective preparation is needed to maximize enantiomeric excess in downstream functionalizations.4
While catalytic alkene transposition of terminal olefins is an atom-economical way to access internal olefins,3a–c,5 control of stereoselectivity and regioselectivity remains challenging,6 especially when performing isomerization to unactivated positions. Due to the importance of these motifs, chemical methods that can control the formation of either E- or Z-alkenes from a single starting material are highly desirable.
Divergent synthetic methods are an efficient way to access multiple products from a single intermediate (Fig. 1A).7 Stereodivergent synthesis is particularly important for the derivatization of late-stage intermediates.8 Particular attention has been paid to the stereodivergent preparation of internal olefins. Stereodivergent Markovnikov hydrofunctionalizations of terminal alkynes and hydrogenations of internal alkynes have been reported to access E- or Z-olefin products (Fig. 1(B1)).8a
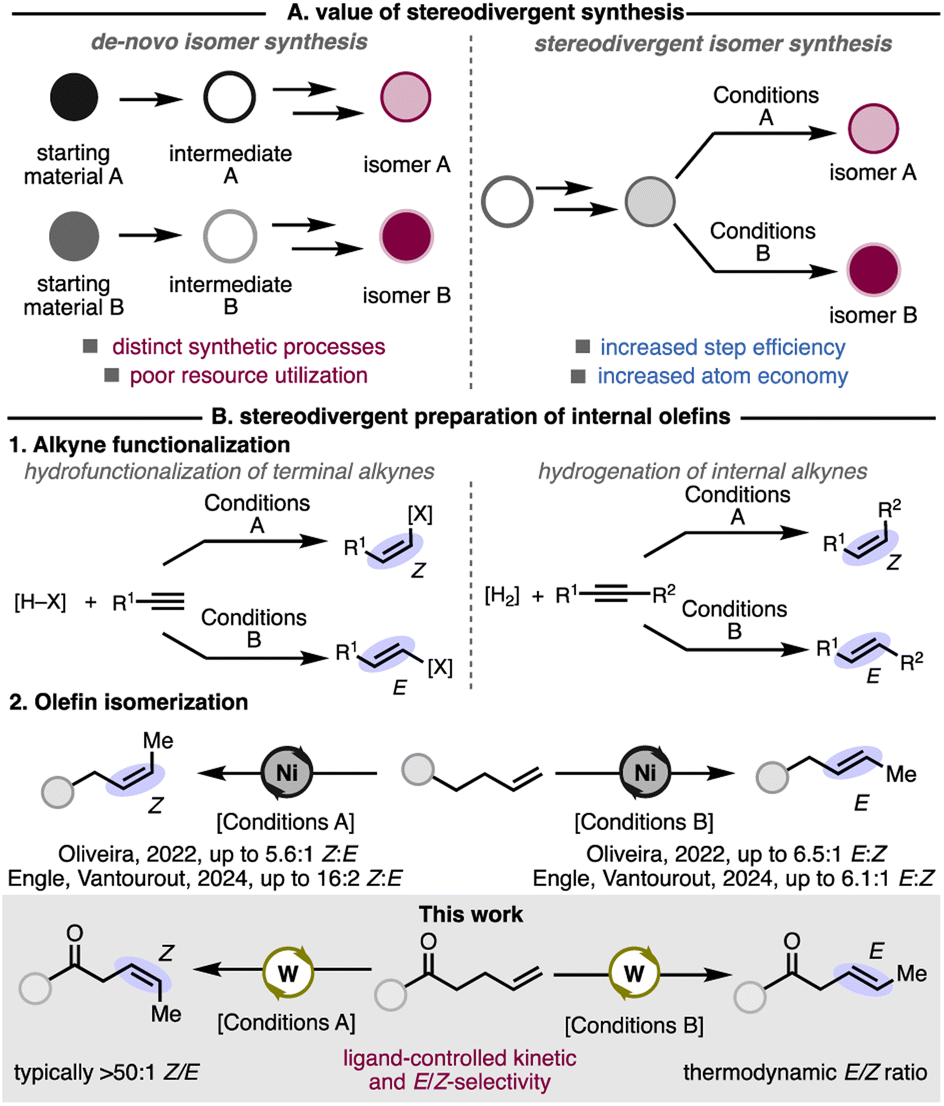 | ||
| Fig. 1 Stereodivergent synthesis and its application to the preparation of internal olefins. | ||
Compared to transformations of alkynes, stereodivergent kinetic isomerization to access E- or Z-alkenes from unsubstituted, 1-alkenes are rare. Nevertheless, impressive examples of alkene isomerization that achieve either E- or Z-selectivity have been disclosed. Several examples of E-selective kinetic alkene isomerization are reported with precious metals Ru,1a,9 Ir,10 and Pd.6s,11 Kinetic isomerization methods toward E-olefins with base-metal Fe6b,12 and Mn13 catalysts have also been demonstrated. Ligand and reagent control has enabled access to Z-selective and E-selective kinetic isomerization processes with Co6g,14 and Ni.6m,11a,15 Our lab and the Oliveira lab have demonstrated stereodivergent alkene isomerization with Ni, which rely on changes to the ligand, solvent, and additives to achieve E- or Z-selectivity from a common Ni(0) precatalyst (Fig. 1(B2)).16
Group 6 metals are considered base metals due to their abundance in Earth's crust and are therefore attractive candidates as homogeneous catalysts. However, apart from olefin metathesis, applications of these metals as catalysts in olefin functionalization and kinetic olefin isomerization remains underexplored (Scheme 1).17 Zeng and Dobereiner have independently reported kinetic olefin isomerizations with chromium18 and molybdenum,19 respectively. A seminal study by Gray in the 1970s demonstrated olefin isomerization with W(CO)6.20 Despite these pioneering reports, a significant knowledge gap remains about how to design and employ ligands to control selectivity with these metals.19a The high affinity for π-accepting ligands (e.g. CO) of group 6 metals compared to later transition metals, and their propensity to adopt geometries up to seven-coordinate, suggest that these systems may require a new conceptual framework to achieve regio- and stereoselectivity.
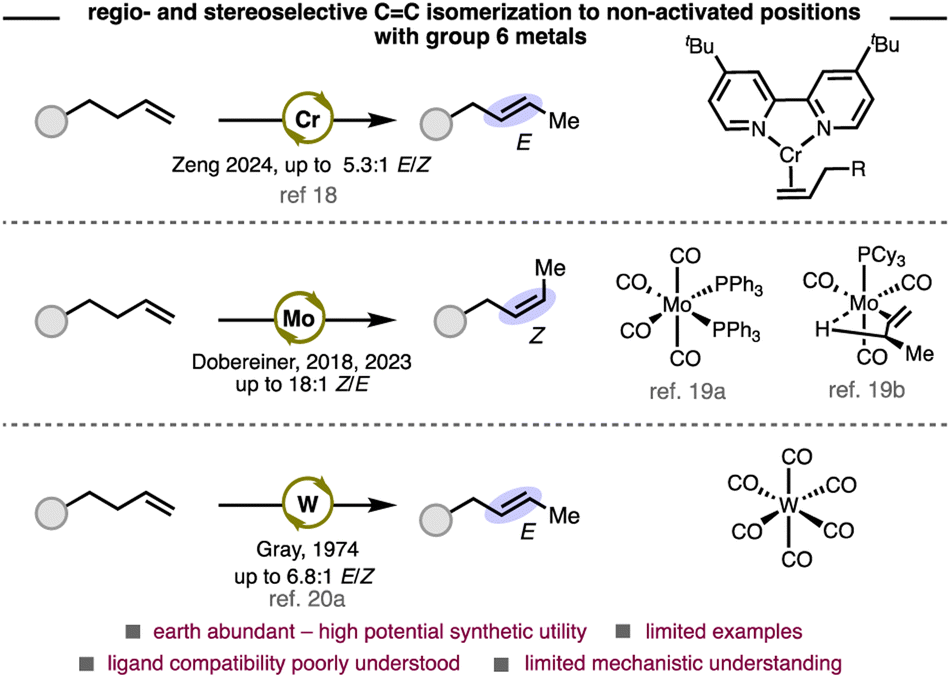 | ||
| Scheme 1 Reported methods that use group 6 metals for the preparation of internal olefins. | ||
Based on our studies of the W(0)/W(II)-catalyzed tandem alkene isomerization/functionalization,21 we envisioned that the different molecular geometries and ligand configurations of 7-coordinate W(II) could be exploited to control the stereochemical outcome of olefin isomerization. Specifically, we sought to develop an alkene isomerization whereby the ligands control formation of either the E- or Z-product. Previous work in our lab has established the ability of amide carbonyls to coordinate to a W(0) center.21b Accordingly, a γ,δ-unsaturated amide was selected as model substrate. γ,δ-Unsaturated carbonyls are simple to access via the Johnson–Claisen rearrangement, whereas the β,γ-unsaturated isomers are much harder to make due to competitive base- or transition-metal-promoted isomerization into conjugation. In addition to isomerization from terminal olefins, β,γ-unsaturated ketones, esters, imines, and analogous species can be accessed through deconjugative isomerization in the presence of light or transition-metal catalysts, but reports rely on thermodynamics to form trisubstituted or conjugated alkenes from α,β-unsaturated starting materials, making such methods complimentary to this work.22
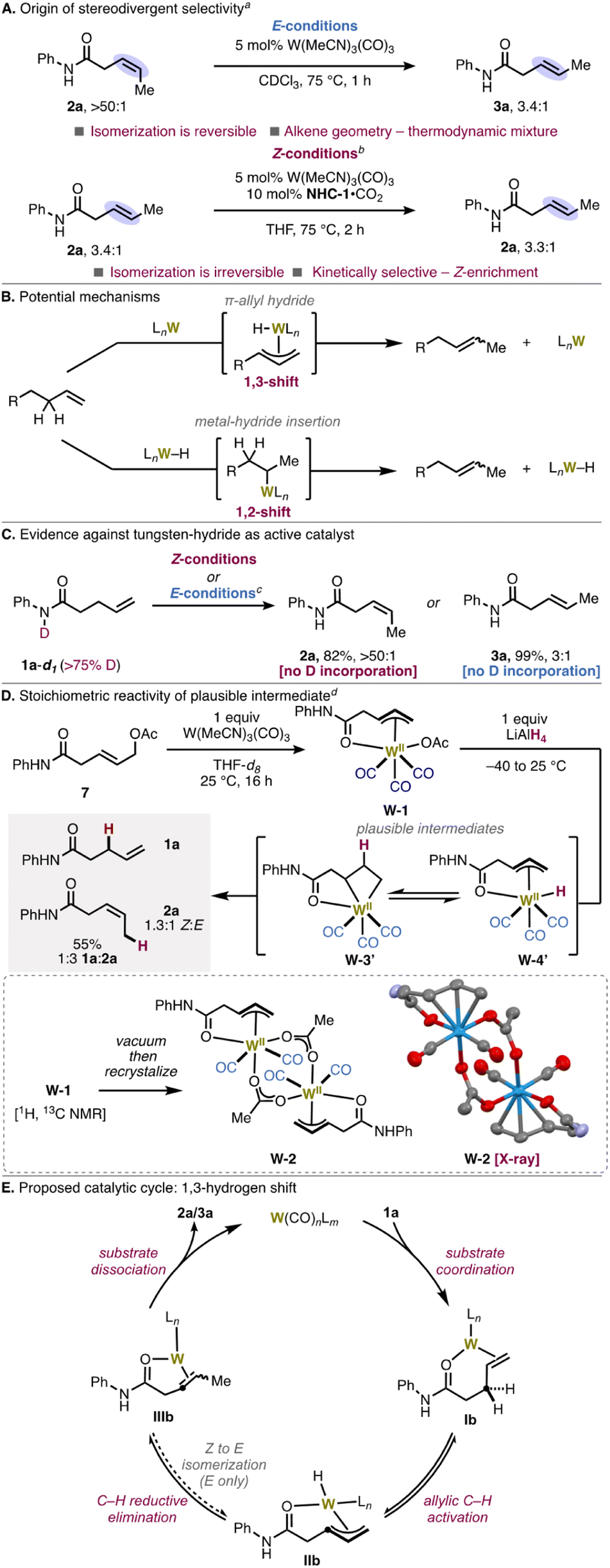
From amide substrate 1a, isomerization to the Z-isomer occurred with 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity in THF (tetrahydrofuran) as solvent (Scheme 2, entry 1). Use of the less-coordinating 2-MeTHF gave the E-isomer as the major product, which suggests that THF acts as a ligand and affects the selectivity (entry 2). Use of N-heterocyclic carbene (NHC) ligands, which, like THF, have a 5-membered ring structure and engage in L-type coordination to metals, further enhanced Z-selectivity. We identified NHC-1 as the optimal ligand, which provided 18:1 Z-selectivity (entry 3). Excess ligand (entry 4) was used to ensure that no background isomerization occurred from a non-ligated tungsten species, resulting in >20:1 Z-selectivity. Increasing the steric bulk of the nitrogen substituents on the NHC decreased the Z-selectivity and yield (entries 5–6). Starting from the carboxylate “pre-NHC” allows for base-free activation to form the free carbene and thus circumvents base-mediated over-isomerization observed with KOt-Bu and the imidazolium salt.23,24 Notably, the corresponding conjugated isomer (2ab) was not observed, which indicates high kinetic selectivity.
:1 selectivity in THF (tetrahydrofuran) as solvent (Scheme 2, entry 1). Use of the less-coordinating 2-MeTHF gave the E-isomer as the major product, which suggests that THF acts as a ligand and affects the selectivity (entry 2). Use of N-heterocyclic carbene (NHC) ligands, which, like THF, have a 5-membered ring structure and engage in L-type coordination to metals, further enhanced Z-selectivity. We identified NHC-1 as the optimal ligand, which provided 18:1 Z-selectivity (entry 3). Excess ligand (entry 4) was used to ensure that no background isomerization occurred from a non-ligated tungsten species, resulting in >20:1 Z-selectivity. Increasing the steric bulk of the nitrogen substituents on the NHC decreased the Z-selectivity and yield (entries 5–6). Starting from the carboxylate “pre-NHC” allows for base-free activation to form the free carbene and thus circumvents base-mediated over-isomerization observed with KOt-Bu and the imidazolium salt.23,24 Notably, the corresponding conjugated isomer (2ab) was not observed, which indicates high kinetic selectivity.
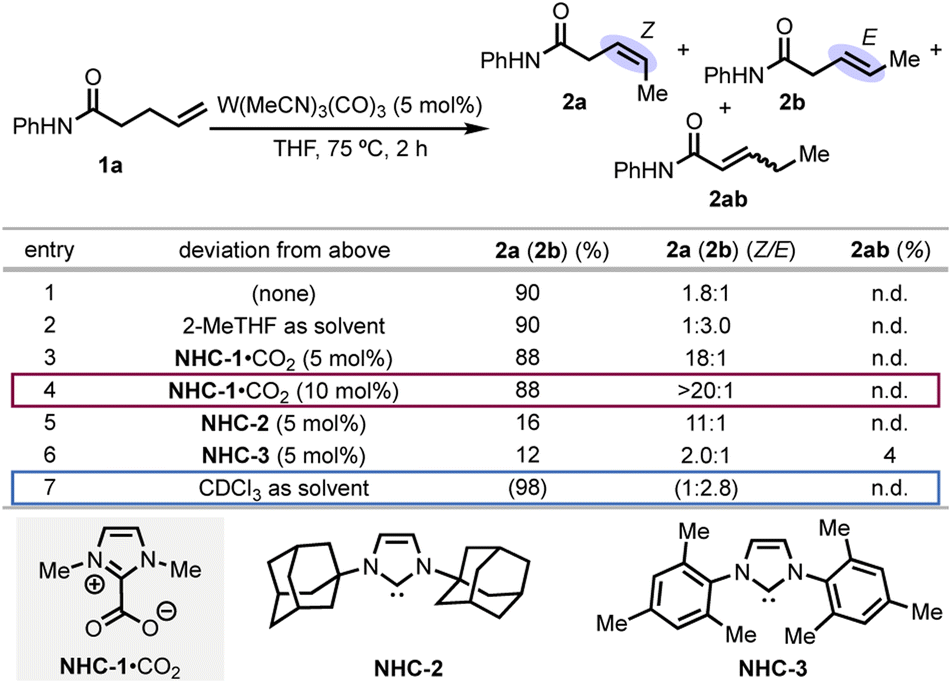 | ||
| Scheme 2 Optimization of the reaction conditions. Reactions were performed on 0.100 mmol scale. Selectivity and conversions were determined by 1H NMR (n.d. = not detected). Remaining mass balance was unreacted 1a. | ||
Ligand-free conditions and noncoordinating solvent led to E-selectivity in a 2.8:1 E/Z ratio (entry 3). The E/Z ratios obtained under these optimal conditions are in line with expected thermodynamic ratios based on an energy difference between the E- and Z-isomers of non-conjugated alkenes of ∼0.4 kcal mol−1 at 75 °C.25 Thus, while the E-selective reaction remains under kinetic regiocontrol, the stereochemistry of the products is under thermodynamic control (vide infra). Further optimization attempts revealed that although substoichiometric coordinating additives could enhance the E-selectivity above the thermodynamic ratio, regioselectivity suffered.23
The generality of the stereodivergent isomerization was investigated with various γ,δ-unsaturated carboxamides, esters, and ketones (Fig. 2). Under the Z-selective conditions, a range of secondary N-aryl amides with functional groups at the para position proceeded in high yield with >20:1 Z-selectivity (2a–d). Potentially reactive functional groups were tolerated, including an iodo substituent (2f), which provides a handle for further derivatization via cross-coupling (vide infra). An unhindered ketone, which could be reduced by a propagating M–H, was tolerated here to form product 2g in high yield and selectivity. Additionally, secondary N-alkyl alkenyl amides were effective substrates, offering very good Z-selectivity (10:1 to >20:1, 2h–j), even for a substrate that contained an N-t-butyl group (2i). We envisioned that the substrate-directed nature of the reaction systems could be leveraged to achieve selective isomerization in a substrate with multiple terminal olefins. To this end, exposing diene 1k to the reaction conditions brought about exclusive isomerization at the carbonyl side of the amide, which shows that the catalyst can differentiate solely by the presence of an intervening NH group. Tertiary amides with pharmaceutically-relevant heterocycles provided high yields but slightly diminished selectivities (8:1–19:1, 2l and 2m). In general, alkyl substituents on the amide decreased Z-selectivity, which could be due to stronger coordination of the more Lewis basic amide carbonyl to tungsten, which enables the catalyst to reengage the product in a secondary Z-to-E isomerization. α-Branched products 2n and 2o formed in >20:1 Z-selectivity despite the increased allylic strain. In addition to amides, esters (2p and 2q) and ketones (2r and 2s) also gave excellent selectivity and high yields. Notably, 2q and the corresponding E-product 3q are derivatives of a natural product derived from the dark-spotted cherry tree (Prunus phaeosticta), which is relevant to fragrance production.26
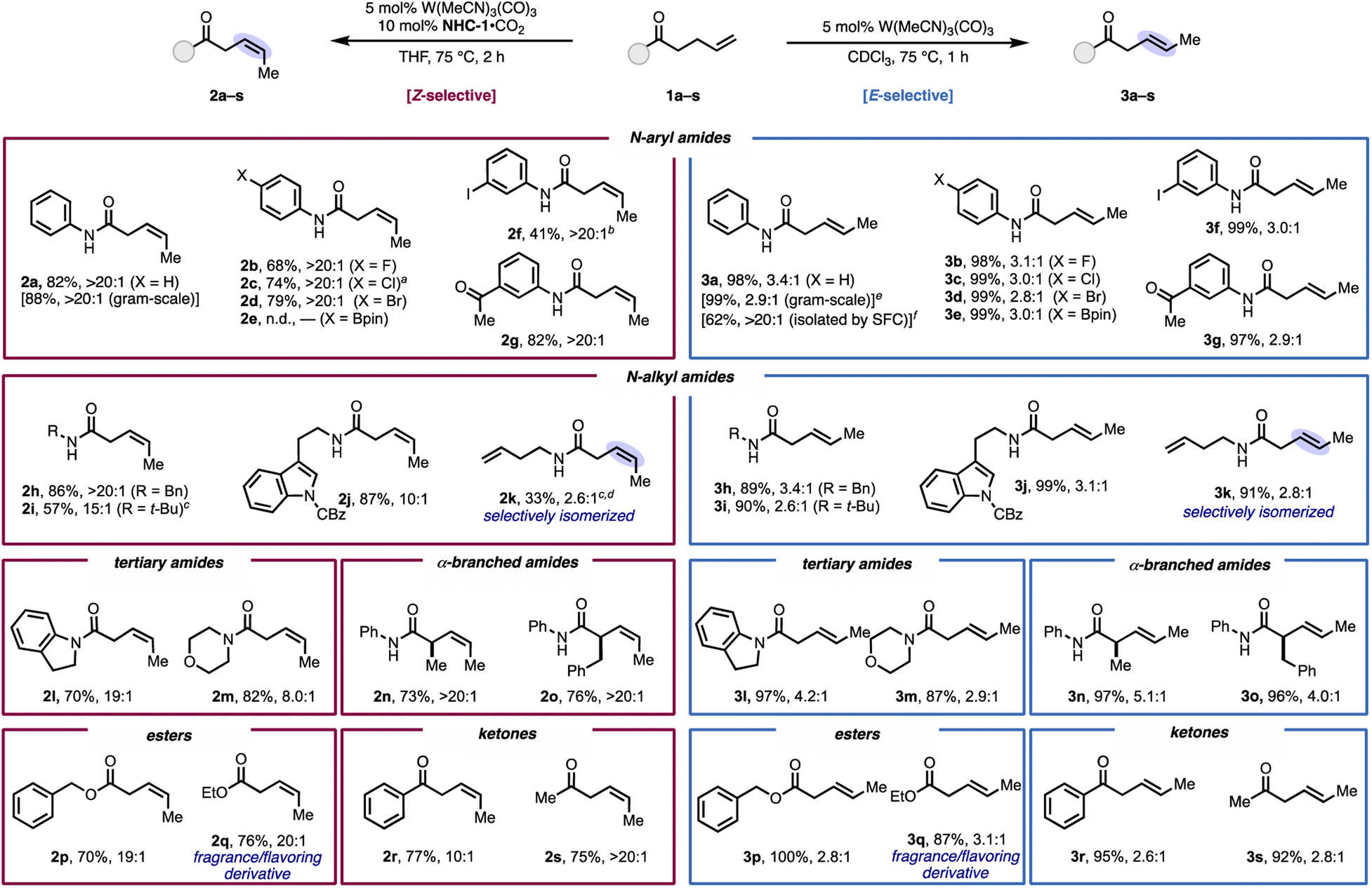 | ||
| Fig. 2 Substrate scope. Reactions performed on 0.100 mmol scale unless otherwise noted. Percentages represent yields of isolated products (obtained as mixtures of E and Z isomers where indicated) unless otherwise noted and are adjusted for remaining starting material. Selectivities were determined by 1H NMR. a Reaction performed using 15 mol% NHC-1·CO2 and 10 mol% W(MeCN)3(CO)3. b Reaction performed using 5 mol% NHC-1·CO2. c Reaction performed at room temperature. d Yield was determined by 1H NMR. e CHCl3 used instead of CDCl3. f Reaction performed on 0.200 mmol scale. | ||
We then assessed the substrate scope with the E-selective conditions and found the protocol to give high to quantitative yields with the same alkenyl amides, esters, and ketones assessed with the Z-selective conditions (Fig. 2, right).27 Notably, substrate 3e, which possesses an aryl boronate, performed well under these conditions, despite not reacting under the Z-selective conditions. For all substrates subjected to the E-selective conditions, the stereoselectivity reflects the thermodynamic ratio for internal 1,2-dialkyl-substituted alkenes.25 The stereoselectivity could be further improved by isolating the product using preparative SFC, which was demonstrated on 3a to provide >20:1 E/Z ratio of the product in 62% isolated yield. The stereodivergent isomerization was scalable; on 1 gram scale, high yield and only modest erosion of E- or Z-selectivity occurred.
To access internal β,γ-unsaturated amides that are otherwise difficult to prepare in a stereoselective fashion, we parlayed the stereoselective isomerization into cross-metathesis (Scheme 3A). Evaluation of several stereoretentive ruthenium metathesis catalysts revealed that dithiolate catalyst28HG-Z could convert the Z-alkenyl amide into the desired cross-metathesis product with >20:1 selectivity, with no erosion of stereochemistry from the starting Z-olefin. Moreover, the Grubbs third-generation bis-pyridine catalyst29 could be used to furnish the E-enriched internal alkene product in the thermodynamic 2.7:1 product ratio. The utility of this approach stems from the widespread availability of terminal alkenes and γ,δ-unsaturated carboxylic acid derivatives, the latter of which can be conveniently prepared via the Johnson–Claisen rearrangement, among other methods.
 | ||
| Scheme 3 Product derivatization. Reactions were performed on 0.100 mmol scale. Yields are isolated and adjusted for remaining starting material. Selectivities were determined by crude 1H NMR. See ESI Schemes S7–S10† for detailed reaction conditions. | ||
Taking advantage of the isomerization reactions' tolerance of arenes that contain potentially reactive functional groups, further product derivatization was performed. Namely, aryl iodides 2f and 3f, generated in situ from 1f, smoothly underwent a one-pot Pd/Cu-catalyzed Sonogashira coupling, maintaining the alkene stereochemistry established during isomerization (Scheme 3B).
To determine the origins of stereoselectivity, we sought to understand how the NHC ligand controls the olefin geometry of the product. In initial control experiments, subjecting pure Z-alkene 2a to the E-selective conditions resulted in a 3.4:1 E/Z ratio, identical to that obtained in the E-selective isomerization of 1a (3a; E/Z 3.4:1),25 which establishes that 2a is a competent intermediate en route to 3a (Fig. 3A, top). As expected, when E-alkene 3a was subjected to the Z-selective reaction conditions, the E/Z ratio remained unchanged, which demonstrates that 3a is not an intermediate to 2a under the Z-selective conditions (Fig. 3A, bottom). These findings suggest that the presence of coordinating ligands like NHCs disfavors reassociation of the product to the metal center, leading to kinetic selectivity under the Z-selective conditions. On the other hand, under the E-selective conditions, the product can reengage the metal center, leading to secondary isomerization to the thermodynamic distribution of E/Z isomers. In this system, the product is prevented from isomerizing into conjugation due to the coordination of the amide. As such, the Z-selective reaction is doubly kinetically selective, in positional and geometric isomerism, whereas the E-selective reaction is only positionally kinetically selective.
 | ||
| Fig. 3 Mechanistic investigations. Reactions were performed on 0.100 mmol scale unless otherwise noted. Yields are isolated and are adjusted for remaining starting material unless otherwise noted. Selectivities were determined by crude 1H NMR. a Reactions were not isolated and were analyzed by crude 1H NMR only. b Reaction performed on 0.06 mmol scale. c CHCl3 used as solvent. d Reaction performed on 0.05 mmol scale. | ||
A priori, we envisioned two potential mechanisms for alkene transposition; (1) a metal–hydride insertion–elimination pathway, and (2) a W(π-allyl)(H) pathway (Fig. 3B).3a,b,5a–e,30 Radical based pathways are implausible; it is energetically infeasible for low-valent tungsten to have unpaired electrons in the presence of strong-field CO ligands.6d,20,31 To differentiate between the two closed-shell mechanisms above, an initial experiment with N–D labeled secondary amide 1a–d1 was performed. Reaction with 1a–d1 did not lead to deuterium incorporation under E- or Z-selective conditions, suggesting that an N–H/D oxidative addition for generation of a W(II)(H/D) species is unlikely (Fig. 3C). This result contrasts with previous findings from our lab in which a similar experiment positively implicated N–H/D oxidative addition in W-catalyzed hydrocarboxylation.21a The compatibility of ketone and ester substrates also evince a mechanism that does not require X–H oxidative addition to form the active catalyst.
Efforts to detect a resting state or catalytically relevant organotungsten species by in situ NMR were inconclusive.20b When performing NMR monitoring over a range of temperatures (4–75 °C), no alkene-bound metal species could be observed.32In situ13C NMR revealed multiple peaks corresponding to W(NHC-1)n species that may or may not be catalytically relevant and could not be characterized by NMR alone. Instead, we turned to independent synthesis to shed light on the underlying coordination chemistry and reactive intermediates. A stable progenitor complex that could be converted into a reactive W(II)(π-allyl)(H) was devised. To this end, treatment of amide-containing allyl acetate 7 with stoichiometric W(MeCN)3(CO)3 in THF at room temperature yielded chelated W(II)(π-allyl)(acetate) oxidative addition adduct W-1 (Fig. 3D). Concentration of the solution and recrystallization afforded the dimeric species W-2. This structure supports coordination of tungsten through the lone pair of the amide carbonyl, suggesting that this directivity effect prevents over-isomerization into conjugation.33 Tungsten coordination to the carbonyl in this structure also clarifies why sterically bulky groups on nitrogen have minimal effect on the reaction. In each of the monomeric units of W-2, the metal center has approximately capped octahedral geometry, with the CO ligands oriented in the most thermodynamically favored configuration toward the “open face” of the π-allyl fragment.34 While it is difficult to determine the exact geometries and 3-D ligand configurations responsible for E versus Z-selectivity at present, it appears that the CO ligands and the amide carbonyl play a critical role in controlling the geometry of the W(II) intermediate.
Reaction between W-1 and LiAlH4 at −40 °C furnished a mixture of E/Z-β,γ-unsaturated amides 2a and 3a and γ,δ-unsaturated amide 1a. Mechanistically, this is consistent with a sequence involving transmetalation to furnish reactive W(II)(π-allyl)(H) intermediate W-3′ (which could further react via reversible hydride insertion to form metallocyclobutane W-4′).35 C(allyl)–H reductive elimination from W-4′ forms 1a, 2a, or 3a. Interestingly, the Z/E ratio (2a:3a = 1.3:1) is approximately equal to the ratio obtained when only THF is used as solvent without additional ligand (1.8:1, see Scheme 2, entry 1), which corroborates the validity of this model system. Formation of starting material 1a further supports that the C(allyl)–H activation step may be reversible, leading to erosion of geometric selectivity in the absence of NHC ligand. Attempts to exchange a CO ligand for NHC-1 or to exchange the acetate for another X-type ligand were unsuccessful, resulting in decomposition and formation of intractable product mixtures. A catalytic cycle consistent with the involvement of these key organometallic intermediates is depicted in Fig. 3E.
Conclusions
In summary, this work demonstrates how the stereoselectivity of kinetic alkene isomerization can be affected by different ligand environments, highlighted by the ability to achieve E/Z-stereodivergence by changing the coordination environment in these tungsten-catalyzed reactions. Mechanistic studies, including stoichiometric mechanistic probes, support a 1,3-hydride shift via a W(allyl)–H intermediate. Overall, this work contributes to the growing field of group 6 metal catalysis and alkene isomerization. We anticipate this method will be useful, particularly when paired with stereoretentive cross-metathesis, to form a variety of internal alkenes in a highly selective manner.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for W-S1 and W-2 has been deposited at the CCDC under 2144251 and 2224919, respectively, and can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures/.Author contributions
Tanner C. Jankins: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft. Camille Z. Rubel: data curation, investigation, validation, visualization, writing – original draft. Hang Chi Ho: data curation, investigation, validation, writing – review & editing. Raul Martin-Montero: data curation, investigation. Keary M. Engle: funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Science Foundation (CHE-2046286). We thank the Schimmel Family Endowed Scholarship Fund for a graduate fellowship (C. Z. R.), The Lee Hysan Foundation for a summer research fellowship (H. C. H.), and La Caixa Foundation for a predoctoral fellowship (R. M.-M.). We thank Materia, Inc. for the donation of metathesis catalysts. Dr Milan Gembicky and Jake Bailey (UCSD) are acknowledged for the collection and analysis of X-ray structures. Dr Jason Chen (Scripps Research) assisted in the separation of E and Z-isomers for some examples. Dr Skyler D. Mendoza is acknowledged for helpful proofreading of this manuscript.Notes and references
- (a) T. J. Donohoe, T. J. C. O'Riordan and C. P. Rosa, Ruthenium-Catalyzed Isomerization of Terminal Olefins: Applications to Synthesis, Angew. Chem., Int. Ed., 2009, 48, 1014–1017 CrossRef CAS; (b) C. D. Vanderwal and B. R. Atwood, Recent Advances in Alkene Metathesis for Natural Product Synthesis—Striking Achievements Resulting from Increased Sophistication in Catalyst Design and Synthesis Strategy, Aldrichimica Acta, 2017, 50, 17–27 CAS; (c) I. Cheng-Sánchez and F. Sarabia, Recent Advances in Total Synthesis via Metathesis Reactions, Synthesis, 2018, 50, 3749–3786 CrossRef; (d) M. M. Heravi, M. Ghanbarian, V. Zadsirjan and B. Alimadadi Jani, Recent Advances in the Applications of Wittig Reaction in the Total Synthesis of Natural Products Containing Lactone, Pyrone, and Lactam as a Scaffold, Monatsh. Chem., 2019, 150, 1365–1407 CrossRef CAS; (e) M. Tomanik, I. T. Hsu and S. B. Herzon, Fragment Coupling Reactions in Total Synthesis That Form Carbon–Carbon Bonds via Carbanionic or Free Radical Intermediates, Angew. Chem., Int. Ed., 2021, 60, 1116–1150 CrossRef CAS.
- N. Armanino, J. Charpentier, F. Flachsmann, A. Goeke, M. Liniger and P. Kraft, What's Hot, What's Not: The Trends of the Past 20 Years in the Chemistry of Odorants, Angew. Chem., Int. Ed., 2020, 59, 16310–16344 CrossRef CAS PubMed.
- (a) A. Vasseur, J. Bruffaerts and I. Marek, Remote Functionalization through Alkene Isomerization, Nat. Chem., 2016, 8, 209–219 CrossRef CAS; (b) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, Walking Metals for Remote Functionalization, ACS Cent. Sci., 2018, 4, 153–165 CrossRef CAS; (c) D. Fiorito, S. Scaringi and C. Mazet, Transition Metal-Catalyzed Alkene Isomerization as an Enabling Technology in Tandem, Sequential and Domino Processes, Chem. Soc. Rev., 2021, 50, 1391–1406 RSC; (d) S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed; (e) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Cascade Reactions in Total Synthesis, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS.
- I. Massad and I. Marek, Alkene Isomerization through Allylmetals as a Strategic Tool in Stereoselective Synthesis, ACS Catal., 2020, 10, 5793–5804 CrossRef CAS.
- (a) C. R. Larsen and D. B. Grotjahn, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. H. Cornils, Wolfgang A., Beller, M. and Paciello, R., Wiley-VCH, Weinheim, 3 edn, 2017, ch. 25, vol. 4, pp. 1365–1378 Search PubMed; (b) E. Larionov, H. Li and C. Mazet, Well-Defined Transition Metal Hydrides in Catalytic Isomerizations, Chem. Commun., 2014, 50, 9816–9826 RSC; (c) X. Liu and Q. Liu, Catalytic Asymmetric Olefin Isomerization: Facile Access to Chiral Carbon-Stereogenic Olefinic Compounds, Chem Catal., 2022, 2, 2852–2864 CrossRef CAS; (d) G. Hilt, Double Bond Isomerisation and Migration—New Playgrounds for Transition Metal-Catalysis, ChemCatChem, 2014, 6, 2484–2485 CrossRef CAS; (e) J. J. Molloy, T. Morack and R. Gilmour, Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef CAS; (f) A. S.-m. Chang, M. A. Kascoutas, Q. P. Valentine, K. I. How, R. M. Thomas and A. K. Cook, Alkene Isomerization Using a Heterogeneous Nickel-Hydride Catalyst, J. Am. Chem. Soc., 2024, 146, 15596–15608 CrossRef CAS; (g) S. Otsuka and K. Tani, in Transition Metals for Organic Synthesis, ed. M. B. Beller, Wiley-VCH, Weinheim, 1998, ch. 2.9, vol. 1, pp. 147–157 Search PubMed.
- (a) S. Zhang, D. Bedi, L. Cheng, D. K. Unruh, G. Li and M. Findlater, Cobalt(II)-Catalyzed Stereoselective Olefin Isomerization: Facile Access to Acyclic Trisubstituted Alkenes, J. Am. Chem. Soc., 2020, 142, 8910–8917 CrossRef CAS; (b) X. Yu, H. Zhao, P. Li and M. J. Koh, Iron-Catalyzed Tunable and Site-Selective Olefin Transposition, J. Am. Chem. Soc., 2020, 142, 18223–18230 CrossRef CAS PubMed; (c) J. Zhao, B. Cheng, C. Chen and Z. Lu, Cobalt-Catalyzed Migrational Isomerization of Styrenes, Org. Lett., 2020, 22, 837–841 CrossRef CAS PubMed; (d) A. Kapat, T. Sperger, S. Guven and F. Schoenebeck, E-Olefins Through Intramolecular Radical Relocation, Science, 2019, 363, 391–396 CrossRef CAS PubMed; (e) M. Murai, K. Nishimura and K. Takai, Palladium-Catalyzed Double-Bond Migration of Unsaturated Hydrocarbons Accelerated by Tantalum Chloride, Chem. Commun., 2019, 55, 2769–2772 RSC; (f) B. M. Trost, J. J. Cregg and N. Quach, Isomerization of N-Allyl Amides To Form Geometrically Defined Di-, Tri-, and Tetrasubstituted Enamides, J. Am. Chem. Soc., 2017, 139, 5133–5139 CrossRef CAS PubMed; (g) S. W. M. Crossley, F. Barabé and R. A. Shenvi, Simple, Chemoselective, Catalytic Olefin Isomerization, J. Am. Chem. Soc., 2014, 136, 16788–16791 CrossRef CAS; (h) L.-G. Zhuo, Z.-K. Yao and Z.-X. Yu, Synthesis of Z-Alkenes from Rh(I)-Catalyzed Olefin Isomerization of β,γ-Unsaturated Ketones, Org. Lett., 2013, 15, 4634–4637 CrossRef CAS PubMed; (i) P. Mamone, M. F. Grünberg, A. Fromm, B. A. Khan and L. J. Gooßen, [Pd(μ-Br)(PtBu3)]2 as a Highly Active Isomerization Catalyst: Synthesis of Enol Esters from Allylic Esters, Org. Lett., 2012, 14, 3716–3719 CrossRef CAS; (j) M. Mayer, A. Welther and A. Jacobi von Wangelin, Iron-Catalyzed Isomerizations of Olefins, ChemCatChem, 2011, 3, 1567–1571 CrossRef CAS; (k) D. B. Grotjahn, C. R. Larsen, J. L. Gustafson, R. Nair and A. Sharma, Extensive Isomerization of Alkenes Using a Bifunctional Catalyst: An Alkene Zipper, J. Am. Chem. Soc., 2007, 129, 9592–9593 CrossRef CAS PubMed; (l) G. Li, J. L. Kuo, A. Han, J. M. Abuyuan, L. C. Young, J. R. Norton and J. H. Palmer, Radical Isomerization and Cycloisomerization Initiated by H· Transfer, J. Am. Chem. Soc., 2016, 138, 7698–7704 CrossRef CAS PubMed; (m) K. E. Kawamura, A. S.-m. Chang, D. J. Martin, H. M. Smith, P. T. Morris and A. K. Cook, Modular Ni(0)/Silane Catalytic System for the Isomerization of Alkenes, Organometallics, 2022, 41, 486–496 CrossRef CAS; (n) Q. Wu, L. Wang, R. Jin, C. Kang, Z. Bian, Z. Du, X. Ma, H. Guo and L. Gao, Nickel-Catalyzed Allylic C(sp2)–H Activation: Stereoselective Allyl Isomerization and Regiospecific Allyl Arylation of Allylarenes, Eur. J. Org Chem., 2016, 2016, 5415–5422 CrossRef CAS; (o) H. Iwamoto, T. Tsuruta and S. Ogoshi, Development and Mechanistic Studies of (E)-Selective Isomerization/Tandem Hydroarylation Reactions of Alkenes with a Nickel(0)/Phosphine Catalyst, ACS Catal., 2021, 11, 6741–6749 CrossRef CAS; (p) S. Krompiec, M. Pigulla, M. Krompiec, B. Marciniec and D. Chadyniak, Highly Selective Isomerization of N-Allylamines Catalyzed by Ruthenium and Rhodium Complexes, J. Mol. Catal. A: Chem., 2005, 237, 17–25 CrossRef CAS; (q) K. Tani, T. Yamagata, S. Akutagawa, H. Kumobayashi, T. Taketomi, H. Takaya, A. Miyashita, R. Noyori and S. Otsuka, Metal-Assisted Terpenoid Synthesis. 7. Highly Enantioselective Isomerization of Prochiral Allylamines Catalyzed by Chiral Diphosphine Rhodium(I) Complexes. Preparation of Optically Active Enamines, J. Am. Chem. Soc., 1984, 106, 5208–5217 CrossRef CAS; (r) T. Ohmura, Y. Yamamoto and N. Miyaura, A Stereoselective Isomerization of Allyl Silyl Ethers to (E)- or (Z)-Silyl Enol Ethers using Cationic Iridium Complexes, Chem. Commun., 1998, 1337–1338 RSC; (s) D. Gauthier, A. T. Lindhardt, E. P. K. Olsen, J. Overgaard and T. Skrydstrup, In Situ Generated Bulky Palladium Hydride Complexes as Catalysts for the Efficient Isomerization of Olefins. Selective Transformation of Terminal Alkenes to 2-Alkenes, J. Am. Chem. Soc., 2010, 132, 7998–8009 CrossRef CAS; (t) S. Krompiec, N. Kuźnik, R. Penczek, J. Rzepa and J. Mrowiec-Białoń, Isomerization of Allyl Aryl Ethers to Their 1-Propenyl Derivatives Catalysed by Ruthenium Complexes, J. Mol. Catal. A: Chem., 2004, 219, 29–40 CrossRef CAS; (u) A. Wille, S. Tomm and H. Frauenrath, A Highly Z-Selective Isomerization (Double-Bond Migration) Procedure for Allyl Acetals and Allyl Ethers Mediated by Nickel Complexes, Synthesis, 1998, 1998, 305–308 CrossRef; (v) A. Scarso, M. Colladon, P. Sgarbossa, C. Santo, R. A. Michelin and G. Strukul, Highly Active and Selective Platinum(II)-Catalyzed Isomerization of Allylbenzenes: Efficient Access to (E)-Anethole and Other Fragrances via Unusual Agostic Intermediates, Organometallics, 2010, 29, 1487–1497 CrossRef CAS; (w) D. Baudry, M. Ephritikhine and H. Felkin, Isomerisation of Allyl Ethers Catalysed by the Cationic Iridium Complex [Ir(cyclo-octa-1,5-diene)(PMePh2)2]PF6. A Highly Stereoselective Route to trans-Propenyl Ethers, J. Chem. Soc., Chem. Commun., 1978, 694–695 RSC; (x) A. Sivaramakrishna, P. Mushonga, I. L. Rogers, F. Zheng, R. J. Haines, E. Nordlander and J. R. Moss, Selective Isomerization of 1-Alkenes by Binary Metal Carbonyl Compounds, Polyhedron, 2008, 27, 1911–1916 CrossRef CAS.
- (a) C. Nájera, I. P. Beletskaya and M. Yus, Metal-Catalyzed Regiodivergent Organic Reactions, Chem. Soc. Rev., 2019, 48, 4515–4618 RSC; (b) N. Funken, Y.-Q. Zhang and A. Gansäuer, Regiodivergent Catalysis: A Powerful Tool for Selective Catalysis, Chem.–Eur. J., 2017, 23, 19–32 CrossRef CAS PubMed; (c) L. Ping, D. S. Chung, J. Bouffard and S.-g. Lee, Transition Metal-Catalyzed Site- and Regio-Divergent C–H Bond Functionalization, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC; (d) G. Zhan, W. Du and Y.-C. Chen, Switchable Divergent Asymmetric Synthesis via Organocatalysis, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC; (e) M. Viji, S. Lanka, J. Sim, C. Jung, H. Lee, M. Vishwanath and J.-K. Jung, Regiodivergent Organocatalytic Reactions, Catalysts, 2021, 11, 1013 CrossRef CAS; (f) L. Segura, I. Massad, M. Ogasawara and I. Marek, Stereodivergent Access to Trisubstituted Alkenylboronate Esters through Alkene Isomerization, Org. Lett., 2021, 23, 9194–9198 CrossRef CAS PubMed.
- (a) I. P. Beletskaya, C. Nájera and M. Yus, Stereodivergent Catalysis, Chem. Rev., 2018, 118, 5080–5200 CrossRef CAS PubMed; (b) X. Huo, G. Li, X. Wang and W. Zhang, Bimetallic Catalysis in Stereodivergent Synthesis, Angew. Chem., Int. Ed., 2022, 61, e202210086 CrossRef CAS PubMed; (c) A. Kumari, A. Jain and N. K. Rana, A Review on Solvent-Controlled Stereodivergent Catalysis, Tetrahedron, 2024, 150, 133754 CrossRef CAS; (d) M. Bihani and J. C.-G. Zhao, Advances in Asymmetric Diastereodivergent Catalysis, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS; (e) L. Lin and X. Feng, Catalytic Strategies for Diastereodivergent Synthesis, Chem.–Eur. J., 2017, 23, 6464–6482 CrossRef CAS.
- (a) C. R. Larsen, G. Erdogan and D. B. Grotjahn, General Catalyst Control of the Monoisomerization of 1-Alkenes to trans-2-Alkenes, J. Am. Chem. Soc., 2014, 136, 1226–1229 CrossRef CAS; (b) C. R. Larsen and D. B. Grotjahn, Stereoselective Alkene Isomerization over One Position, J. Am. Chem. Soc., 2012, 134, 10357–10360 CrossRef CAS PubMed; (c) M. Tuner, J. v. Jouanne, H.-D. Brauer and H. Kelm, The Isomerization of n-Butenes Catalyzed by RhCl(PPh3)3. II. The Isomerization of 1-Butene, J. Mol. Catal., 1979, 5, 433–445 CrossRef CAS; (d) E. R. Paulson, C. E. Moore, A. L. Rheingold, D. P. Pullman, R. W. Sindewald, A. L. Cooksy and D. B. Grotjahn, Dynamic π-Bonding of Imidazolyl Substituent in a Formally 16-Electron Cp*Ru(κ2-P,N)+ Catalyst Allows Dramatic Rate Increases in (E)-Selective Monoisomerization of Alkenes, ACS Catal., 2019, 9, 7217–7231 CrossRef CAS; (e) C. Pandya, R. R. Panicker, P. Senjaliya, M. K. H. Hareendran, P. V. Anju, S. Sarkar, H. Bhat, P. C. Jha, K. P. Rao, G. S. Smith and A. Sivaramakrishna, Designing and Synthesis of Phosphine Derivatives of Ru3(CO)12 – Studies on Catalytic Isomerization of 1-Alkenes, Inorg. Chim. Acta, 2021, 518, 120211 CrossRef CAS; (f) S. Hanessian, S. Giroux and A. Larsson, Efficient Allyl to Propenyl Isomerization in Functionally Diverse Compounds with a Thermally Modified Grubbs Second-Generation Catalyst, Org. Lett., 2006, 8, 5481–5484 CrossRef CAS; (g) M. Arisawa, Y. Terada, M. Nakagawa and A. Nishida, Selective Isomerization of a Terminal Olefin Catalyzed by a Ruthenium Complex: The Synthesis of Indoles through Ring-Closing Metathesis, Angew. Chem., Int. Ed., 2002, 41, 4732–4734 CrossRef CAS PubMed.
- (a) Y. Wang, C. Qin, X. Jia, X. Leng and Z. Huang, An Agostic Iridium Pincer Complex as a Highly Efficient and Selective Catalyst for Monoisomerization of 1-Alkenes to trans-2-Alkenes, Angew. Chem., Int. Ed., 2017, 56, 1614–1618 CrossRef CAS; (b) A. M. Camp, M. R. Kita, P. T. Blackburn, H. M. Dodge, C.-H. Chen and A. J. M. Miller, Selecting Double Bond Positions with a Single Cation-Responsive Iridium Olefin Isomerization Catalyst, J. Am. Chem. Soc., 2021, 143, 2792–2800 CrossRef CAS; (c) S. De-Botton, O. A. Filippov, E. S. Shubina, N. V. Belkova and D. Gelman, Regioselective Isomerization of Terminal Alkenes Catalyzed by a PC(sp3)Pincer Complex with a Hemilabile Pendant Arm, ChemCatChem, 2020, 12, 5959–5965 CrossRef CAS.
- (a) H. J. Lim, C. R. Smith and T. V. RajanBabu, Facile Pd(II)- and Ni(II)-Catalyzed Isomerization of Terminal Alkenes into 2-Alkenes, J. Org. Chem., 2009, 74, 4565–4572 CrossRef CAS; (b) W. Ren, F. Sun, J. Chu and Y. Shi, A Pd-Catalyzed Site-Controlled Isomerization of Terminal Olefins, Org. Lett., 2020, 22, 1868–1873 CrossRef CAS; (c) T. Ohmura, K. Oshima and M. Suginome, (E)- and (Z)-β-Borylallylsilanes by Alkyne Silaboration Followed by Regio- and Stereoselective Double-Bond Migration, Angew. Chem., Int. Ed., 2011, 50, 12501–12504 CrossRef CAS.
- (a) S. Garhwal, A. Kaushansky, N. Fridman and G. de Ruiter, Part Per Million Levels of an Anionic Iron Hydride Complex Catalyzes Selective Alkene Isomerization via Two-State Reactivity, Chem Catal., 2021, 1, 631–647 CrossRef CAS; (b) C. R. Woof, D. J. Durand, N. Fey, E. Richards and R. L. Webster, Iron Catalyzed Double Bond Isomerization: Evidence for an FeI/FeIII Catalytic Cycle, Chem.–Eur. J., 2021, 27, 5972–5977 CrossRef CAS PubMed; (c) S. A. Lutz, A. K. Hickey, Y. Gao, C.-H. Chen and J. M. Smith, Two-State Reactivity in Iron-Catalyzed Alkene Isomerization Confers σ-Base Resistance, J. Am. Chem. Soc., 2020, 142, 15527–15535 CrossRef CAS; (d) R. Jennerjahn, R. Jackstell, I. Piras, R. Franke, H. Jiao, M. Bauer and M. Beller, Benign Catalysis with Iron: Unique Selectivity in Catalytic Isomerization Reactions of Olefins, ChemSusChem, 2012, 5, 734–739 CrossRef CAS PubMed.
- W. Yang, I. Y. Chernyshov, M. Weber, E. A. Pidko and G. A. Filonenko, Switching between Hydrogenation and Olefin Transposition Catalysis via Silencing NH Cooperativity in Mn(I) Pincer Complexes, ACS Catal., 2022, 12, 10818–10825 CrossRef CAS PubMed.
- (a) C. Chen, T. R. Dugan, W. W. Brennessel, D. J. Weix and P. L. Holland, Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes, J. Am. Chem. Soc., 2014, 136, 945–955 CrossRef CAS PubMed; (b) D. Kim, G. Pillon, D. J. DiPrimio and P. L. Holland, Highly Z-Selective Double Bond Transposition in Simple Alkenes and Allylarenes through a Spin-Accelerated Allyl Mechanism, J. Am. Chem. Soc., 2021, 143, 3070–3074 CrossRef CAS; (c) T. Kobayashi, H. Yorimitsu and K. Oshima, Cobalt-Catalyzed Isomerization of 1-Alkenes to (E)-2-Alkenes with Dimethylphenylsilylmethylmagnesium Chloride and Its Application to the Stereoselective Synthesis of (E)-Alkenylsilanes, Chem.–Asian J., 2009, 4, 1078–1083 CrossRef CAS; (d) X. Liu, W. Zhang, Y. Wang, Z.-X. Zhang, L. Jiao and Q. Liu, Cobalt-Catalyzed Regioselective Olefin Isomerization Under Kinetic Control, J. Am. Chem. Soc., 2018, 140, 6873–6882 CrossRef CAS PubMed; (e) S. Raje, T. Sheikh Mohammad and G. de Ruiter, A Neutral PCNHCP Co(I)–Me Pincer Complex as a Catalyst for N-Allylic Isomerization with a Broad Substrate Scope, J. Org. Chem., 2024, 89, 4319–4325 CrossRef CAS PubMed.
- (a) H. Kanai, Selective cis-Isomerisation of But-1-ene by Homogeneous Catalysis with Triphenylphosphinenickel Complexes, J. Chem. Soc., Chem. Commun., 1972, 203–204, 10.1039/C39720000203; (b) H. Kanai, K. Kushi, K. Sakanoue and N. Kishimoto, Selective cis-Isomerization of 1-Pentene Catalyzed by Ni(I)-Triphenylphosphine Complexes, Bull. Chem. Soc. Jpn., 1980, 53, 2711–2715 CrossRef CAS; (c) F. Weber, A. Schmidt, P. Röse, M. Fischer, O. Burghaus and G. Hilt, Double-Bond Isomerization: Highly Reactive Nickel Catalyst Applied in the Synthesis of the Pheromone (9Z,12Z)-Tetradeca-9,12-dienyl Acetate, Org. Lett., 2015, 17, 2952–2955 CrossRef CAS PubMed.
- (a) C. Z. Rubel, A. K. Ravn, H. C. Ho, S. Yang, Z.-Q. Li, K. M. Engle and J. C. Vantourout, Stereodivergent, Kinetically Controlled Isomerization of Terminal Alkenes via Nickel Catalysis, Angew. Chem., Int. Ed., 2024, 63, e202320081 CrossRef CAS PubMed; (b) E. J. de Carvalho Junior and C. Oliveira, Nickel-Catalyzed Double Bond Transposition under Kinetic Control, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-w9n02.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Using Nature's Blueprint to Expand Catalysis with Earth-Abundant Metals, Science, 2020, 369, eabc3183 CrossRef CAS.
- J. Zhong, X. Wang, M. Luo and X. Zeng, Chromium-Catalyzed Alkene Isomerization with Switchable Selectivity, Org. Lett., 2024, 26, 3124–3129 CrossRef CAS.
- (a) S. E. Jenny, J. M. Serviano, A. Nova and G. E. Dobereiner, A Hydride Migration Mechanism for the Mo-Catalyzed Z-2-Selective Isomerization of Terminal Alkenes, ChemCatChem, 2023, 15, e202301052 CrossRef CAS; (b) J. Becica, O. D. Glaze, D. I. Wozniak and G. E. Dobereiner, Selective Isomerization of Terminal Alkenes to (Z)-2-Alkenes Catalyzed by an Air-Stable Molybdenum(0) Complex, Organometallics, 2018, 37, 482–490 CrossRef CAS.
- (a) M. Wrighton, G. S. Hammond and H. B. Gray, Group VI Metal Carbonyl Photoassisted Isomerization of Olefins, J. Organomet. Chem., 1974, 70, 283–301 CrossRef CAS; (b) T. Szymańska-Buzar, M. Jaroszewski, M. Wilgocki and J. J. Ziółkowski, Reactivity of Bis(Alkene) Tetracarbonyl Complexes of Tungsten: Evidence for Alkene to π-allyl Hydride Rearrangement, J. Mol. Catal. A: Chem., 1996, 112, 203–210 CrossRef.
- (a) T. C. Jankins, W. C. Bell, Y. Zhang, Z.-Y. Qin, J. S. Chen, M. Gembicky, P. Liu and K. M. Engle, Low-Valent Tungsten Redox Catalysis Enables Controlled Isomerization and Carbonylative Functionalization of Alkenes, Nat. Chem., 2022, 14, 632–639 CrossRef CAS PubMed; (b) T. C. Jankins, R. Martin-Montero, P. Cooper, R. Martin and K. M. Engle, Low-Valent Tungsten Catalysis Enables Site-Selective Isomerization–Hydroboration of Unactivated Alkenes, J. Am. Chem. Soc., 2021, 143, 14981–14986 CrossRef CAS.
- (a) T. Morack, C. Onneken, H. Nakakohara, C. Mück-Lichtenfeld and R. Gilmour, Enantiodivergent Prenylation via Deconjugative Isomerization, ACS Catal., 2021, 11, 11929–11937 CrossRef CAS; (b) N. C. Yang and M. J. Jorgenson, Photochemical Isomerization of Simple α,β-Unsaturated Ketones, Tetrahedron Lett., 1964, 5, 1203–1207 CrossRef; (c) Y. Liu, L. Zhang, Y. Zhang, S. Cao, X. Ban, Y. Yin, X. Zhao and Z. Jiang, Asymmetric Olefin Isomerization via Photoredox Catalytic Hydrogen Atom Transfer and Enantioselective Protonation, J. Am. Chem. Soc., 2023, 145, 18307–18315 CrossRef CAS; (d) R. R. Rando and W. v. E. Doering, β,γ-Unsaturated Acids and Esters by Photochemical Isomerization of α,β-Congeners, J. Org. Chem., 1968, 33, 1671–1673 CrossRef CAS; (e) J.-P. Pete, F. Henin, R. Mortezaei, J. Muzart and O. Piva, Enantioselective Photodeconjugation of Conjugated Esters and Lactones, Pure Appl. Chem., 1986, 58, 1257–1262 CrossRef CAS; (f) O. Piva, R. Mortezaei, F. Henin, J. Muzart and J. P. Pete, Highly Enantioselective Photodeconjugation of α,β-Unsaturated Esters. Origin of the Chiral Discrimination, J. Am. Chem. Soc., 1990, 112, 9263–9272 CrossRef CAS; (g) O. Piva and J.-P. Pete, Diacetone D-Glucose: Efficient Chiral Building Block for Asymmetric Photodeconjugation, Tetrahedron: Asymmetry, 1992, 3, 759–768 CrossRef CAS.
- A. M. Voutchkova, M. Feliz, E. Clot, O. Eisenstein and R. H. Crabtree, Imidazolium Carboxylates as Versatile and Selective N-Heterocyclic Carbene Transfer Agents: Synthesis, Mechanism, and Applications, J. Am. Chem. Soc., 2007, 129, 12834–12846 CrossRef CAS PubMed.
- See ESI Table S10,† entry 8.
- J. B. Pedley, R. D. Naylor and S. P. Kirby, Thermochemical Data of Organic Compounds, Chapman and Hall, London, 2nd edn, 1986 Search PubMed.
- C.-L. Ho, E. I.-C. Wang and Y.-C. Su, Composition of the Leaf Oils of Prunus phaeosticta var. phaeosticta From Taiwan, J. Essent. Oil Res., 2009, 21, 345–347 CrossRef CAS.
- The percentages represent yields of isolated products (obtained as a mixture of E- and Z-isomers where indicated), as well as starting material in some cases, which could not be separated with silica gel chromatography. To demonstrate that separation to access isomerically pure E-product is possible, preparative 2D-SFC was performed, and 3a was isolated with >20 : 1 E/Z ratio.
- M. J. Koh, R. K. M. Khan, S. Torker, M. Yu, M. S. Mikus and A. H. Hoveyda, High-Value Alcohols and Higher-Oxidation-State Compounds by Catalytic Z-Selective Cross-Metathesis, Nature, 2015, 517, 181–186 CrossRef CAS.
- M. S. Sanford, J. A. Love and R. H. Grubbs, A Versatile Precursor for the Synthesis of New Ruthenium Olefin Metathesis Catalysts, Organometallics, 2001, 20, 5314–5318 CrossRef CAS.
- (a) S. Biswas, Mechanistic Understanding of Transition-Metal-Catalyzed Olefin Isomerization: Metal-Hydride Insertion–Elimination vs. π-Allyl Pathways, Comments Inorg. Chem., 2015, 35, 300–330 CrossRef CAS; (b) X. Liu, B. Li and Q. Liu, Base-Metal-Catalyzed Olefin Isomerization Reactions, Synthesis, 2019, 51, 1293–1310 CrossRef CAS.
- S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Catalytic Hydrogen Atom Transfer to Alkenes: A Roadmap for Metal Hydrides and Radicals, Chem. Sci., 2020, 11, 12401–12422 RSC.
- See ESI Fig. S1† for isolation, characterization, and reactivity of W-S1, which formed under the E-selective reaction conditions and was not catalytically active.
- R. Matsuura, M. K. Karunananda, M. Liu, N. Nguyen, D. G. Blackmond and K. M. Engle, Mechanistic Studies of Pd(II)-Catalyzed E/Z Isomerization of Unactivated Alkenes: Evidence for a Monometallic Nucleopalladation Pathway, ACS Catal., 2021, 11, 4239–4246 CrossRef CAS.
- (a) D. E. Ryan, D. J. Cardin and F. Hartl, η3-Allyl Carbonyl Complexes of Group 6 Metals: Structural Aspects, Isomerism, Dynamic Behaviour and Reactivity, Coord. Chem. Rev., 2017, 335, 103–149 CrossRef CAS; (b) M. J. Calhorda and P. J. Costa, Structure, Bonding and Reactivity of Seven-Coordinate Allylic Mo(II) and W(II) Complexes, Coord. Chem. Rev., 2017, 344, 83–100 CrossRef CAS.
- M. Ephritikhine, M. L. H. Green and R. E. MacKenzie, Some η1 and η3-Allylic and Metallocyclobutane Derivatives of Molybdenum and Tungsten, J. Chem. Soc., Chem. Commun., 1976, 619–621 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2144251 and 2224919. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07093c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |