
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConjugated core–shell bottlebrush polymers that exhibit crystallization-driven self-assembly†
Victor
Lotocki
a,
Alicia M.
Battaglia
a,
Nahye
Moon
a,
Hatem M.
Titi
 b and
Dwight S.
Seferos
*ac
b and
Dwight S.
Seferos
*ac
aDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: dwight.seferos@utoronto.ca
bDepartment of Chemistry, McGill University, 801 Sherbrooke St. W., Montreal, Quebec H3A 0B8, Canada
cDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, Toronto, Ontario M5S 3E5, Canada
First published on 9th December 2024
Abstract
Bottlebrush polymers are complex architectures with densely grafted polymer side chains along polymeric backbones. The dense and conformationally extended chains in bottlebrush polymers give rise to unique properties, including low chain entanglement, low critical aggregation concentrations, and elastomeric properties in the bulk phase. Conjugated polymers have garnered attention as lightweight, processible, and flexible semi-conducting materials. They are promising candidates in electronic devices and sensors, but their optoelectronic properties depend on adequate polymer ordering, π–π interactions, and crystallization. Crystallization-driven self-assembly of conjugated polymers has become a prominent method to optimize properties including band energies, redox potentials, and exciton diffusion and transport. Much progress has been made in controlled block copolymer self-assembly, but despite their promising properties, reports of conjugated bottlebrushes have been limited, and their self-assembly is relatively unexplored. For the first time, we report the synthesis of conjugated core–shell bottlebrush polymers. These materials contain poly(3-hexylthiophene) (P3HT) and poly(ethylene glycol) (PEG) in either core or shell position. We demonstrate that the use of P3HT as a crystallizable conjugated polymer and PEG as a colloidally stabilizing and disaggregating block facilitates their self-assembly into a number of unique crystalline morphologies with longer conjugation lengths and lower exciton bandwidths relative to analogous diblock copolymers. These include intramolecularly self-assembled segregated bottlebrush polymers, short nanofibers formed by end-on-end stacking of bottlebrush molecules, extremely long >20 μm nanofibers formed exclusively by end-on-end stacking, and >15 μm nanoribbons formed from both end-on-end and side-by-side stacking of bottlebrush polymers.
Introduction
Conjugated polymers have attracted attention due to their potential as light-weight, processable, and low cost semi-conducting materials and their applications in organic electronic devices, sensors, and biomedical imaging.1–4 Such applications, however, require the optimization of properties including band energies, redox potentials, and exciton diffusion and transport that can be enhanced through self-assembly. Crystallization-driven self-assembly (CDSA) is an exothermic process that promotes the π-stacking of conjugated polymers through non-covalent interactions, and the resulting nanostructures exhibit inter- and intrachain π orbital overlap that improves parameters such as charge transport and exciton diffusion.4–6 This is particularly important, given that most amorphous materials show exciton diffusion over 5–10 nm,7 and that long-range transport can be promoted within well-ordered nanostructures through transient delocalization.8–10 Much progress has been made in the CDSA of block copolymers, including the development of nanofibers with all-conjugated blocks, controllable lengths of poly(3-hexylhiophene) (P3HT)-based assemblies reaching 5 μm, and the development of length and width-tunable conjugated nanofibers with low dispersities through “living” CDSA.11–18 However, the self-assembly of conjugated polymers with complex architectures remains underexplored.Bottlebrushes are a class of polymers with densely grafted polymeric side chains that extend from a linear polymeric backbone. Their high side chain density leads to low chain entanglement. As a result, the bottlebrush topology leads to elastomeric properties in the bulk phase, enabling applications as rheological modifiers, elastomers, or elastomeric coatings.19–23 Extended side chain conformations accompanied by low chain entanglements cause bottlebrush polymers to adopt rigid structures with high persistence lengths and cause molecular segregation, which generally precludes self-assembly.23,24 However, bottlebrush interactions can be promoted by copolymerization or functionalization.25,26 Very recently, bottlebrush polymers have led to self-assembled structures including hexagonal platelets, needles, curved rod-like micelles and closed rings through hydrogen bonding interactions.27 They have also been applied to photonic materials through their self-assembly into porous microspheres and due to their tendency to adopt lamellar structures in blends that enable nanopatterning.22,28–30
The synthesis of bottlebrush polymers typically proceeds through one of three methodologies: grafting-to, grafting-from, or grafting-through synthesis. Grafting-from and grafting-to syntheses are practical and convenient, as they consist of polymerizing from initiating functional groups or directly grafting polymers onto linear backbones, respectively. However, these syntheses result in incomplete grafting, limiting bottlebrush properties and performance.31,32 Grafting-through synthesis involves polymerizing macromonomers, thus affording high grafting densities. These are typically carried out by ring-opening metathesis polymerization (ROMP) due to its fast polymerization rate and quantitative monomer consumption. However, degrees of polymerization can be limited due to entropic factors arising from the use of large macromonomers.33–36
Bottlebrush polymers with conjugated polymer side chains strongly aggregate in a disordered manner due to π–π interactions, limiting their performance.37,38 Efforts to manage this aggregation have come in two forms. The Kilbey group synthesized bottlebrush statistical copolymers and block copolymers, containing P3HT and amorphous poly(D,L-lactide) (PLA), that organize into short flexible nanofibers.39 Our group has previously demonstrated that, upon thermal annealing, bottlebrush polymers containing P3HT side chains disaggregate and self-assemble in an end-to-end manner.40 P3HT is often used as a model conjugated polymer due to its processability, mid-range bandgap, and controlled synthesis.41–44 Recently, the synthesis of core–shell bottlebrush copolymers and their assembly into structures including nanobowls, nanowires, and nanotubes has been reported, while, to our knowledge no such polymers have been developed with conjugated blocks.45–47
Herein, we report the synthesis and self-assembly of conjugated core–shell bottlebrush polymers, featuring P3HT and poly(ethylene glycol) (PEG) at either the core or the shell position. P3HT is a crystallizable conjugated block, while PEG is a colloidally stabilizing block. Notably, PEG exhibits suppressed crystallization in densely grafted brush polymers.48,49 The self-assembly of the P3HT-b-PEG core–shell bottlebrush (CSBB) and PEG-b-P3HT inverse core–shell bottlebrush (ICSBB) polymers in binary solvent systems of chloroform/methanol, chloroform/heptane, and benzonitrile/heptane was studied through UV-visible (UV-vis) spectroscopy, grazing incidence wide-angle X-ray scattering (GIWAXS), and transmission electron microscopy (TEM). Variation of both core and shell polymer block positions as well as solvent system resulted in an unprecedented array of morphologies including intramolecularly self-assembled individual CSBB molecules, short nanofibers formed by the end-on-end assembly of CSBB or ICSBB polymers with lengths averaging 50 to 100 nm, long >20 μm nanofibers formed exclusively by linear end-on-end assembly of CSBB molecules, and >15 μm nanoribbons formed by both end-on-end assembly of ICSBB molecules as well as side-by-side stacking (Fig. 1).
 | ||
| Fig. 1 (A) Previous synthesis of P3HT bottlebrushes using a grafting-through methodology, and their end-on-end self-assembly into nanofibers. (B) This work reports the grafting-through synthesis of P3HT-b-PEG core–shell bottlebrush polymers, with P3HT and PEG in either core or shell position, and their intramolecular self-assembly and intermolecular self-assembly into end-on-end and side-by-side stacked nanofibers. Cartoon nanostructures: black = polynorbornene, red = P3HT, blue = PEG. | ||
Results and discussion
Core–shell bottlebrush synthesis
The synthesis of a core–shell bottlebrush polymer with P3HT and PEG as its core and shell blocks, respectively, necessitates the preparation of P3HT with heterobifunctional end groups amenable to coupling with both PEG and a polymerizable small molecule, namely, norbornene. To this end, we have prepared a novel functional Ni(II) external initiator for Kumada catalyst transfer polycondensation (KCTP) bearing a tetrahydropyran (THP) pendant that can be deprotected post-polymerization to reveal a reactive hydroxyl group for further reactions. THP was selected as a robust protecting group that could withstand many conditions, most relevantly lithiation and Grignard reactions.50First, the primary alcohol on (4-bromo-3-methylphenyl)methanol was protected with 3,4-dihydropyran. The bromine atom on the THP-protected product was then subjected to sequential lithiation, Grignard formation, and transmetalation with Ni(dppe)Cl2 to form a THP-protected external initiator for KCTP (Scheme 1A). 31P NMR revealed the formation of both bromine- and chlorine-Ni(THP)(dppe)X adducts (X = Br, Cl) with Ni(THP)(dppe)Br as the major product, consistent with previous Ni external initiator observations (Fig. S1A†).51–53 MALDI-TOF shows two peaks with m/z values consistent with NiBr elimination and rearrangement after ionization and oxidation (Fig. S1B†),51,53,54 further verifying the identity of the external initiator.
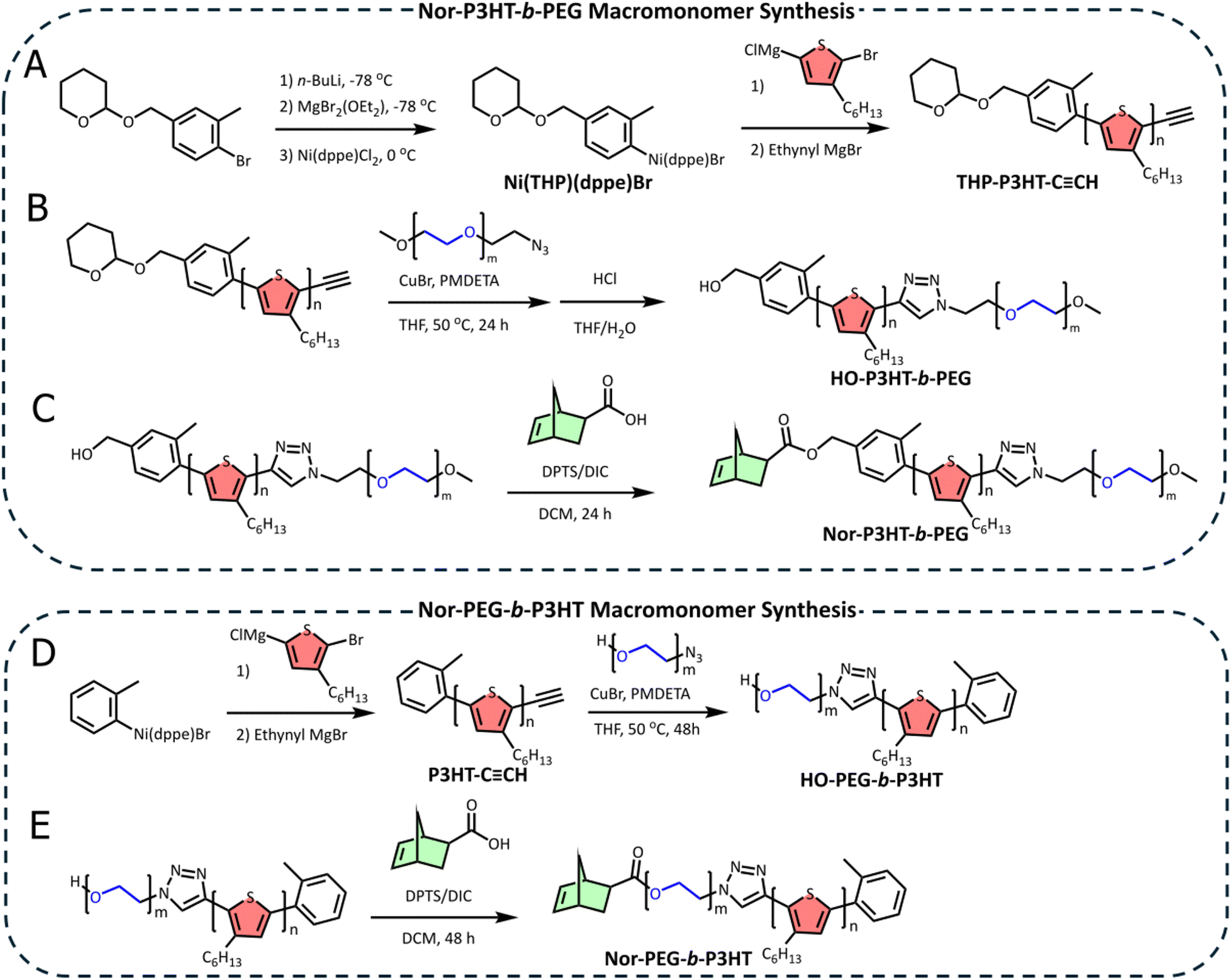 | ||
Scheme 1 (A) Synthesis of Ni(THP)(dppe)Br by the lithiation, Grignard synthesis, and Ni(II) transmetalation, followed by its initiation of thiophene polymerization and ethynyl end-capping to form THP-P3HT-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH. (B) Successive mPEG-N3 “click” coupling and acid-catalyzed THP-deprotection of THP-P3HT-CCH to form HO–P3HT-b-PEG. (C) Synthesis of Nor-P3HT-b-PEG by the Steglich esterification of 5-norbornene carboxylic acid with HO-P3HT-b-PEG. (D) Synthesis of HO-PEG-b-P3HT by “click” coupling HO-PEG-N3 with P3HT-CCH prepared by KCTP initiated by o-tolyl Ni(dppe)Br and end-capped by ethynyl MgBr. (E) Synthesis of Nor-PEG-b-P3HT by the Steglich esterification of 5-norbornene carboxylic acid with HO-PEG-b-P3HT. CH. (B) Successive mPEG-N3 “click” coupling and acid-catalyzed THP-deprotection of THP-P3HT-CCH to form HO–P3HT-b-PEG. (C) Synthesis of Nor-P3HT-b-PEG by the Steglich esterification of 5-norbornene carboxylic acid with HO-P3HT-b-PEG. (D) Synthesis of HO-PEG-b-P3HT by “click” coupling HO-PEG-N3 with P3HT-CCH prepared by KCTP initiated by o-tolyl Ni(dppe)Br and end-capped by ethynyl MgBr. (E) Synthesis of Nor-PEG-b-P3HT by the Steglich esterification of 5-norbornene carboxylic acid with HO-PEG-b-P3HT. | ||
P3HT was synthesized using Ni(THP)(dppe)Br in a typical KCTP procedure with a monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :initiator ratio = 22:1 (Scheme 1A). KCTP was terminated by adding ethynyl magnesium bromide, to form the alkyne-terminated P3HT.551H NMR analysis of the polymer revealed the presence of expected end groups. The alkyne proton is present at 3.56 ppm, while the resonances corresponding to the initiating end are present at 4.81 ppm (O–CH2–Ar), 4.52 ppm (O–CH2–CH2), and 2.53 ppm (CH3–Ar), with methylene splitting resulting from the THP chiral center (Fig. S13†). The synthesized THP-P3HT-CCH was calculated to have a degree of polymerization of eighteen, corresponding to the targeted molecular weight of approximately 3200, whereas GPC analysis resulted in a Mn of 3900 (Fig. S2†). This discrepancy between NMR and GPC-derived molecular weight is expected for rod-like conjugated polymers, and GPC results are known to be up to 1.2 times higher than those from NMR.56
:initiator ratio = 22:1 (Scheme 1A). KCTP was terminated by adding ethynyl magnesium bromide, to form the alkyne-terminated P3HT.551H NMR analysis of the polymer revealed the presence of expected end groups. The alkyne proton is present at 3.56 ppm, while the resonances corresponding to the initiating end are present at 4.81 ppm (O–CH2–Ar), 4.52 ppm (O–CH2–CH2), and 2.53 ppm (CH3–Ar), with methylene splitting resulting from the THP chiral center (Fig. S13†). The synthesized THP-P3HT-CCH was calculated to have a degree of polymerization of eighteen, corresponding to the targeted molecular weight of approximately 3200, whereas GPC analysis resulted in a Mn of 3900 (Fig. S2†). This discrepancy between NMR and GPC-derived molecular weight is expected for rod-like conjugated polymers, and GPC results are known to be up to 1.2 times higher than those from NMR.56
THP-P3HT-CCH was coupled to mPEG-N3 with a molecular weight of 1000 in a copper-catalyzed alkyne–azide cycloaddition (CuAAC) click reaction, and after product isolation, this reaction was followed by THP deprotection with HCl to form HO–P3HT-b-PEG (Scheme 1B). GPC revealed a shift to a Mn of 5000 (Fig. S2†), confirming the successful click reaction. The obtained diblock copolymer also exhibited the expected singlet at 7.85 ppm by 1H NMR, indicating the newly formed triazole group. Deprotection was confirmed by analyzing the methylene singlet at 4.74 ppm, that was no longer split by the THP group. HO-P3HT-b-PEG was then end-functionalized with 5-norbornene carboxylic acid in a Steglich esterification (Scheme 1C). Complete conversion was confirmed by the replacement of the singlet mentioned above at 4.74 ppm with a downfield singlet at 5.16 ppm, indicating the formation of an ester bond. Additionally, the appearance of a multiplet at 6.15 ppm suggests the presence of unsaturated norbornene end group protons (Fig. 2A).
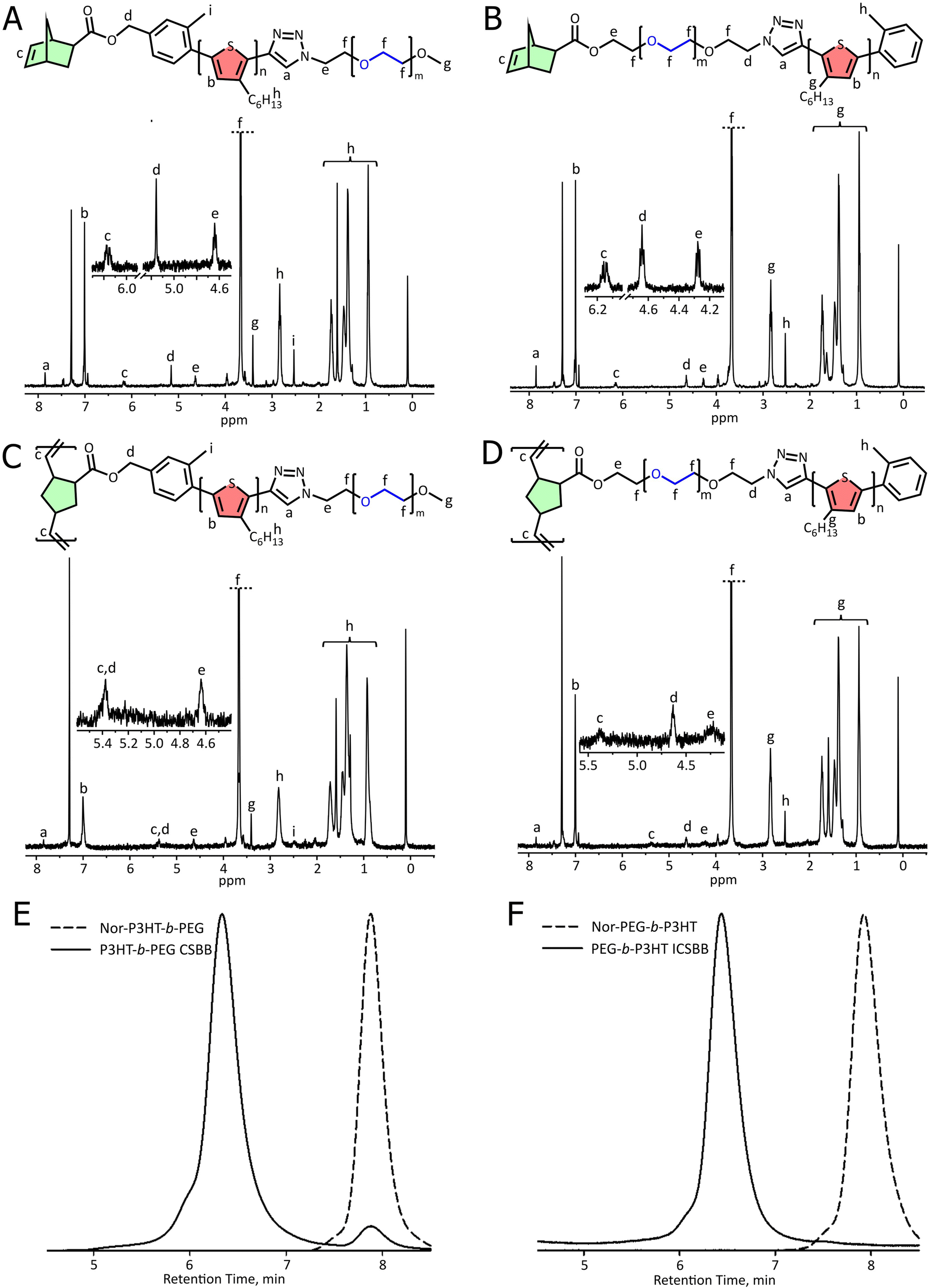 | ||
| Fig. 2
1H NMR characterization of (A) Nor-P3HT-b-PEG, (B) Nor-PEG-b-P3HT, (C) P3HT-b-PEG CSBB, and (D) PEG-b-P3HT ICSBB. GPC characterization of (E) Nor-P3HT-b-PEG (Mn = 5,400, Đ = 1.11) and P3HT-b-PEG CSBB (Mn = 84600, Đ = 1.21) and (F) Nor-PEG-b-P3HT (Mn = 4,800, Đ = 1.11) and PEG-b-P3HT ICSBB (Mn = 70100, Đ = 1.12). | ||
The Nor-P3HT-b-PEG macromonomer was polymerized by ROMP using a Grubbs III catalyst (monomer:initiator ratio = 50:1) to yield the final core–shell bottlebrush polymer (Scheme 2A). The unsaturated norbornene resonances at 6.15 ppm are completely consumed, suggesting a successful reaction (Fig. 2C). GPC revealed a much lower retention time than the macromonomer which corresponds to a Mn of 81600, confirming successful CSBB synthesis (Fig. 2E). This Mn corresponds to 16–20 repeating units along the bottlebrush backbone, when considering molecular weights derived by GPC or NMR.
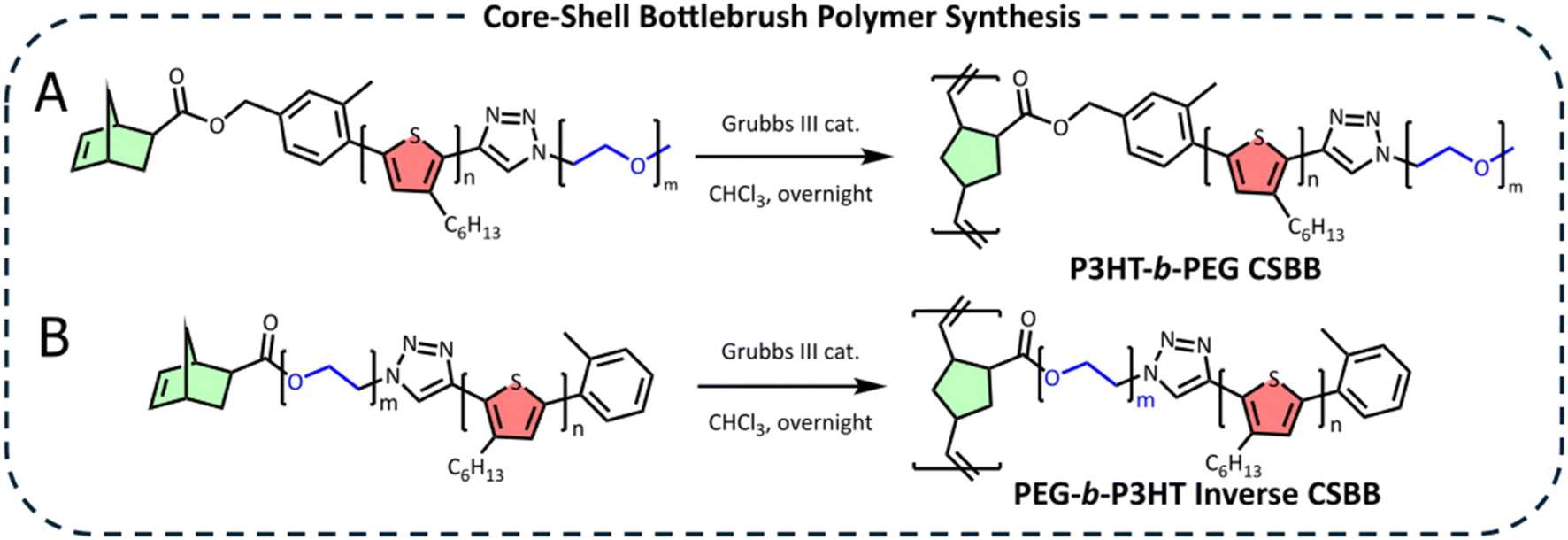 | ||
| Scheme 2 Ring opening metathesis polymerization of (A) Nor-P3HT-b-PEG and (B) Nor-PEG-b-P3HT using a Grubbs III catalyst to form P3HT-b-PEG CSBB and PEG-b-P3HT ICSBB, respectively. | ||
A more conventional synthetic pathway was taken to prepare the inverse core–shell bottlebrush polymer. HO-PEG-N3 was linked to P3HT-CCH in a CuAAC click reaction (Scheme 1D). Then, the product was coupled with norbornene-5-carboxylic acid in a Steglich esterification to yield the Nor-PEG-b-P3HT macromonomer (Scheme 1E). An examination of the 1H NMR spectrum shows that the click reaction was successful due to the appearance of the triazole proton at 7.85 ppm in addition to all relevant PEG and P3HT peaks. The unsaturated norbornene region can be seen by the resonances centered at 6.15 ppm (Fig. 2B). GPC revealed a Mn of 4,800, consistent with the coupling of P3HT with PEG. This macromonomer was polymerized using a Grubbs III catalyst to yield the PEG-b-P3HT ICSBB (monomer:initiator ratio = 50:1) (Scheme 2B). The obtained ICSBB exhibited all relevant PEG and P3HT resonances by 1H NMR, which appear to have slightly broadened, as well as the complete consumption of the unsaturated norbornene resonances at 6.15 ppm (Fig. 2D). GPC indicantes a shift to a much lower retention time than the macromonomer, corresponding to a Mn of 70100 (Fig. 2F).
Core–shell bottlebrush photophysical properties
Upon the successful synthesis of the P3HT-b-PEG CSBB and PEG-b-P3HT ICSBB, solvent-induced self-assembly experiments were carried out and monitored by UV-vis spectroscopy. Both polymers, in addition to a HO-P3HT-b-PEG diblock copolymer, which was used as a control, were dissolved in chloroform or benzonitrile, and self-assembly was induced by adding 0–90% v/v poor solvent. Benzonitrile and chloroform are both good solvents for PEG. They have similar Hansen solubility parameter (HSP) values of 17.2 and 19.0 MPa1/2, respectively, yet the higher polarity component for benzonitrile than in chloroform (9.0 vs. 3.1 MPa1/2), leads it to be a marginal solvent for P3HT, while chloroform remains a good solvent for it.11,57,58 Methanol and heptane were chosen as crystallization-inducing poor solvents.Typically, large HSP differences between methanol and P3HT induce precipitation in the latter alongside uncontrolled aggregation.51,59 PEG blocks in P3HT copolymers have been shown to confer colloidal stability while promoting P3HT π-stacking.17,57,60 Contrarily, short alkane solvents, such as heptane or hexane, act as poor solvents for PEG and marginal solvents for P3HT, as they can solvate the hexyl side chains and solubilize smaller MW P3HT. Combined with the narrow surface energy difference between these solvents and chloroform, chloroform/heptane mixtures are expected to result in controlled crystallization and long nanofiber growth, whereas benzonitrile/heptane polarity difference and pre-nucleation by benzonitrile would result in a larger population of smaller nanostructures.57,59,61
In our initial experiments, self-assembly of polymers in chloroform was induced by methanol. It was visually apparent from the solutions that the core–shell bottlebrush underwent a much more significant bathochromic shift relative to the diblock copolymer due to its deeper red/purple colour, while retaining colloidal stability at all solvent ratios. On the other hand, the diblock copolymer showed signs of precipitation at >60% MeOH (Fig. S3†). In pure chloroform, both diblock and CSBB were fully solvated, and had similar λmax values of 442 and 444 nm, respectively (Fig. 3A and D). Notably, the transition of both polymers into their self-assembled states occurred at roughly 50% methanol for the diblock copolymer and 38% methanol for the CSBB (Fig. 4A).
 | ||
| Fig. 3 UV-visible absorption spectra for the self-assembly of the P3HT-b-PEG diblock copolymer in (A) chloroform/methanol, (B) chloroform/heptane, and (C) benzonitrile/heptane, absorption spectra for P3HT-b-PEG CSBB self-assembly in (D) chloroform/methanol, (E) chloroform/heptane, and (F) benzonitrile/heptane, Absorption spectra for PEG-b-P3HT ICSBB self-assembly in (G) chloroform/methanol, (H) chloroform/heptane, and (I) benzonitrile/heptane, All spectra are normalized except for (G) in which the sample precipitates. | ||
 | ||
| Fig. 4 Plots of UV-visible spectroscopy derived λmax values for polymers as a function of (A) methanol in chloroform solutions, (B) heptane in chloroform solutions, or (C) heptane in benzonitrile solutions. Plots of A0−0/A0−1 values for polymers as a function of (D) methanol in chloroform solutions, (E) heptane in chloroform solutions, or (F) heptane in benzonitrile solutions as a volume percent. Black = P3HT-b-PEG CSBB, blue = P3HT-b-PEG diblock copolymer, red = PEG-b-P3HT ICSBB. | ||
This self-assembly can be explained by the densely grafted PEG-b-P3HT chains in close proximity within the bottlebrush architecture, causing the P3HT blocks to aggregate readily. More significantly, the extended side chain conformation afforded by the bottlebrush architecture promotes a much deeper bathochromic shift, and therefore a narrower bandgap,62 relative to the diblock copolymer, despite bearing the same P3HT and PEG blocks. The much longer P3HT conjugation length in the CSBB is reflected by a λmax of 515 nm in the self-assembled state, a 50 nm increase relative to the self-assembled diblock copolymer which exhibits a λmax of 465 nm.
The difference in photophysical properties imposed by polymer architecture can also be seen in the vibronic structure for the self-assembled polymers. Vibronic shoulders at 603 and 543 nm were assigned as the A0−0 and A0−1 transitions, respectively, which correspond to thiophene π-stacking, while the ratio of these shoulders reveals the extent of inter- or intrachain exciton coupling.59,63,64 The A0−0/A0−1 ratio for the P3HT-b-PEG CSBB and the diblock copolymer reach similar values of approximately 0.43, indicating H-type aggregation and predominant interchain exciton coupling in both samples (Fig. 4D),58,65 while the lower A0−0 and A0−1 intensities in the diblock copolymer imply a lesser overall degree of π-stacking, which is reasonable considering its aggregation-induced colloidal instability. It is also interesting to note that whereas the CSBB has a sharp increase in its vibronic shoulders with the addition of methanol, the diblock copolymer has a smoother transition to plateau that reflects individual unimer-to-aggregate addition in this system (Fig. S4†). Conversely, the ICSBB does not benefit from colloidal stability in the chloroform/methanol solvent system due to bearing PEG in the core position, and it precipitates upon the addition of methanol, reflected in the noise and scattering seen in its absorbance spectra (Fig. 3G), and highlighting the importance of polymer block position.
Aggregation experiments were carried out by the addition of heptane, a poor solvent for PEG and a marginal solvent for P3HT, to polymer solutions in chloroform and yielded different results than those for the chloroform/methanol binary blend. First, the onset of aggregation was delayed to 77% poor solvent addition for both the CSBB and ICSBB due to the larger P3HT block relative to PEG (Fig. 4B), while no significant self-assembly was observed for the analogous diblock copolymer (Fig. 3B). Upon full self-assembly, the CSBB achieved a λmax of 513 nm, similar to the chloroform/methanol system, while the ICSBB exhibited a bathochromic shift to 472 nm. This intermediate performance can be ascribed to the less conformationally extended P3HT chains due to their positions at PEG chain ends. It should also be noted that, while the ICSBB precipitated with methanol addition, heptane addition colloidally stabilizes the system due the P3HT shell position. Upon heptane-induced self-assembly, the CSBB reached a A0−0/A0−1 value of 0.66, while that of the ICSBB is 0.43, indicating H-type aggregation for both bottlebrush polymers. On the other hand, the CSBB does not precipitate, but it is meta-stable, with apparent precipitates forming after 24–48 hours. While the two core–shell bottlebrush polymers significantly improved their photophysical properties upon heptane-induced self-assembly, no change in λmax and only minimal vibronic structure was observed for the P3HT-b-PEG diblock copolymer.
In benzonitrile/heptane binary blends, both of which are marginal solvents for P3HT, a bathochromic shift to 491 nm was observed for the P3HT-b-PEG CSBB and 475 nm for the PEG-b-P3HT ICSBB and diblock copolymer (Fig. 4C). While still significant, the relatively minor bathochromic shift observed for the CSBB may be attributed to the partial solvation of both benzonitrile and heptane components. However, this was accompanied by larger vibronic shoulders, indicating more prominent π-stacking and crystallinity, especially for the CSBB (Fig. 3C, F and I). This agrees with GIWAXS-derived azimuthal angles, where both bottlebrush polymers drop cast from benzonitrile/heptane exhibit the lowest FWHM, corresponding to highest crystallinity among the solvent systems (Fig. S6†). The benzonitrile/heptane binary blend again yielded H-aggregates with A0−0/A0−1 values of 0.73, 0.51, and 0.67 for the CSBB, ICSBB, and DB, respectively, indicating higher J-type character and therefore more intrachain exciton coupling relative to the other systems. Notably this result is most prominent in the CSBB in which P3HT chains are shielded by PEG blocks in the bottlebrush shell position and least prominent in the ICSBB in which P3HT and PEG blocks are in opposite positions.
Overall, all polymers undergo H-type aggregation in each binary solvent system, wherein the heptane systems confer slightly more J-type character, and thus intrachain exciton coupling. Notably, the free exciton bandwidth, which decreases with higher conjugation lengths and improved intrachain order,66 is lowest for the CSBB and highest for the ICSBB in all solvent systems (Fig. S5†). This agrees with the observed bathochromic shift trends in which the CSBB exhibits the most pronounced shifts across every solvent system (Fig. 4A–C), indicating the long conjugation lengths and enhanced ordering promoted by the bottlebrush architecture. Increased intrachain order is likely a consequence of both P3HT occupying the core position, which promotes chain extension, and PEG in the shell position, which confers colloidal stability and prevents random aggregation.
Core–shell bottlebrush self-assembly
Polynorbornene is expected to have a contour length of 0.53–0.81 nm per repeating unit,40 giving an approximate expected length of 10.6–16.2 nm for the P3HT-b-PEG CSBB with 20 norbornene repeating units. Bottlebrush polymers with 3 kDa P3HT side chains are reported to have widths of 13–14 nm.40 As a result, these CSBBs are expected to have approximately 1:1 dimensions and thus be square shaped by TEM, as the PEG block does not confer any contrast in electron microscopy. Interestingly, when self-assembled with the addition of methanol, P3HT-b-PEG CSBB samples were observed as individual bottlebrush polymers by TEM. An analysis of P3HT-b-PEG CSBB molecules by TEM reveals a length of 18 ± 4 nm, and a width of 13 ± 3 nm, which agrees with the expected dimensions of single bottlebrush molecules (Fig. 5A and D). This implies that self-assembly induced by methanol is an entirely intramolecular process between adjacent polythiophene chains on individual CSBBs, which results in well-ordered lamellar and π-stacking, as evidenced by drop cast GIWAXS samples (Fig. S6A†).67,68 This is only made possible by the disaggregating PEG blocks in the bottlebrush shell position. These intramolecularly self-assembled single bottlebrushes exhibit long P3HT conjugation lengths and strong interchain exciton coupling due to their unique architecture (Fig. 4A and D).
 | ||
| Fig. 5 TEM images of P3HT-b-PEG CSBB self-assemblies in (A) chloroform/methanol, (B) benzonitrile/heptane, and (C) chloroform/heptane. Histograms with (D) chloroform/methanol self-assembly nanoparticle sizes, (E) benzonitrile/heptane self-assembly lengths and (F) widths, and (G) chloroform/heptane self-assembly widths. TEM images of PEG-b-P3HT ICSBB self-assemblies in (H) chloroform/methanol, (I) benzonitrile/heptane, and (J) chloroform/heptane (with lacey carbon structure in background). Histograms with (K) chloroform/methanol self-assembly nanoparticle sizes, (L) benzonitrile/heptane self-assembly lengths and (M) widths, and (N) chloroform/heptane self-assembly widths. TEM images were analyzed using ImageJ and >50 measurements were made for each histogram. | ||
When self-assembly is induced in CSBB solutions in chloroform by heptane, a poor solvent for PEG in the bottlebrush shell position, well-ordered and long >20 μm nanofibers were formed with widths of 17 ± 3 nm (Fig. 5C, G and S7†). Notably, the nanofiber widths are consistent with the theoretical and measured values for single bottlebrushes obtained by methanol-induced self-assembly, indicating that the nanofibers are approximately 1 CSBB molecule wide while being >1000 CSBB molecules long. The positioning of PEG within these bottlebrushes is a likely reason for the prevention of random aggregation and side-by-side stacking. By GIWAXS, P3HT within these nanostructures exhibit a similar overall degree of lamellar and π-stacking compared to the chloroform/methanol self-assemblies (Fig. S6A†). Nanofibers formed by chloroform/heptane self-assembly are meta-stable, being stable for about 24–48 hours on the benchtop before visible precipitate formation. This can also be deduced from the scattering observed in their absorbance spectra (Fig. 3E). In addition to the poorly solvated shell segment, their lengths may be a contributing factor to this intermediate stability.
Benzonitrile has been shown to induce the crystallization of P3HT into nanofiber-like micelles, particularly after heating and slow cooling cycles, which can promote processes such as living CDSA and dopant-induced self-assembly.11,58 In the case of our CSBBs, dissolution in pure benzonitrile results in a darker red solution relative to chloroform, which was revealed to occur due to a bathochromic shift by UV-vis spectroscopy (Fig. 3F), whereas TEM only showed single CSBB molecules, suggesting a similar internal self-assembly process as with the chloroform/methanol system. Unlike methanol and heptane, benzonitrile can colloidally stabilize the CSBB system without chloroform solutions. However, further self-assembly of CSBB polymers in benzonitrile induced by the addition of heptane resulted in nanofiber formation (Fig. 5B), similar to the chloroform/heptane system, albeit with significantly shorter lengths of 79 ± 35 nm and widths of 11 ± 2 nm (Fig. 5E and F). These lengths correspond to 4–6 bottlebrushes per nanofiber, on average. Approximately 4% of the structures within the measured population are consistent with single molecules.
The longest nanofiber measured was 186 nm long, corresponding to 10–14 CSBB molecules stacked end-on-end. The shorter lengths of these nanofibers may be attributed to poor solvent mixing on the molecular level due to the disparate HSP space, particularly the polarity component δp, which is 0 and 9.0 for heptane and benzonitrile, respectively.57 Additionally, P3HT core nucleation is induced in the pure benzonitrile system prior to the addition of heptane, which may also prohibit long nanofiber growth. This may lead to a high overall degree of crystallinity within these nanofibers, as determined from azimuthal angle analysis of the (200) peak in GIWAXS (Fig. S6B†), but ordering along the π-stacking direction is also less well-defined, compared to CSBB self-assembly in the other two solvent systems (Fig. S6A†). As with the chloroform/methanol system, these nanofibers are completely colloidally stable, with no signs of precipitation observed, possibly due to the shorter length of these nanofibers compared to the ones formed in the chloroform/heptane system.
Adding methanol to PEG-b-P3HT ICSBB solutions in chloroform causes immediate precipitation, as was also seen by the immense scattering and noise in their absorbance spectra (Fig. 3G). This is a result of shell position P3HT in the ICSBB, which is very poorly solvated by methanol. By TEM, they are shown to collapse into aggregates with no defined structure due to this poor solvation (Fig. 5H and K). However, when self-assembly is induced by the addition of heptane to ICSBB in chloroform, colloidal stability is retained, and long ribbon-like nanofibers are observed by TEM (Fig. 5J and S8†). Unlike P3HT-b-PEG CSBB, where P3HT is internal and shielded by the PEG shell block, the ICSBB features P3HT in the shell position, and individual bottlebrushes can therefore stack both end-on-end and side-by-side upon self-assembly, thus promoting nanoribbon-like morphologies.
These nanoribbons feature very long >15 μm lengths and have widths of 98 ± 51 nm (Fig. 5J, N and S8†). The narrowest structure in the measured population is about 28 nm wide, corresponding to approximately 2 ICSBB widths, while the widest is 270 nm across, corresponding to 15–20 ICSBB molecules stacked side-by-side (Fig. S8†). As with the previous bottlebrush polymer, self-assembly in a benzonitrile/heptane solvent system resulted in significantly shorter nanofibers with average lengths and widths of 59 ± 22 nm and 11 ± 1 nm, respectively (Fig. 5I, L and M). The inhibition of longer growth, again, may be a result of both poor molecular mixing of benzonitrile and heptane due to their disparate polarities and the premature nucleation of P3HT in pure benzonitrile before the initiation of further self-assembly with heptane. Notably, this also results in a lack of side-by-side stacking as observed for the ICSBB self-assemblies in chloroform/heptane. Similar to the CSBB samples, ICSBBs drop cast from benzonitrile/heptane were the most crystalline among the solvent systems (Fig. S6D†), and they also featured the most prominent third order (300) lamellar stacking and (010) π-stacking peaks between all bottlebrush systems (Fig. S6C†).
Conclusions
Overall, this work demonstrates the synthesis, photophysical properties, and unique self-assembly behaviour of conjugated bottlebrush polymers and their controlled synthesis with two different core–shell topologies. The synthesis of the P3HT-b-PEG CSBB was achieved by preparing a novel THP pendant-bearing external initiator for KCTP, and following the polymerization, using techniques including CuAAC click chemistry, Steglich esterification, and ROMP. The PEG-b-P3HT ICSBB was synthesized using similar techniques to link P3HT-CCH and norbornene carboxylic to heterobifunctional HO-PEG-N3, followed by ROMP. A broad range of unique morphologies were attained by self-assembly that we ascribe to the core–shell bottlebrush architecture. We found that P3HT blocks within CSBB polymers can be self-assembled intramolecularly in chloroform/methanol due to the presence of a disaggregating and colloidally stabilizing PEG block in the shell position. The extended conformations of core-position P3HT chains in these polymers led to significantly larger bathochromic shifts, longer conjugation lengths, and lower exciton bandwidths upon self-assembly compared to both the ICSBB as well as the P3HT-b-PEG diblock copolymer control. Self-assembly of CSBBs in chloroform/heptane resulted in end-on-end assembly of exceptionally long >20 μm nanofibers with similar bathochromic shifts and lower exciton bandwidths.
Long one-dimensional structures were also observed for the ICSBB in chloroform/heptane, albeit with additional side-by-stack stacking on the order of a few bottlebrush molecules resulting from the intermolecular interactions of P3HT in the bottlebrush shell position. Short CSBB and ICSBB nanofibers were obtained using benzonitrile/heptane, albeit with lower exciton bandwidths than other solvent systems, indicating better intrachain order. These were also shown to be the most crystalline by GIWAXS. To the best of our knowledge, this work represents the self-assembly of nonlinear conjugated polymers into previously unreported structures, and it demonstrates the morphological control that can be attained because of binary solvent system choice and topological variation in bottlebrush polymers. We believe that the self-assembly and photophysical properties of these bottlebrush polymers make them promising candidates for applications involving doping and energy transfer, and we plan to investigate this in future work.
Data availability
All relevant data are presented in the article and ESI.†Author contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript. V. L. and D. S. conceptualized the study. V. L., A. B., N. M., and H. T carried out experiments and analysis. V. L. provided formal analysis, the original draft, and data curation. D. S. provided project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSERC of Canada, the Canadian Foundation for Innovation, and the Ontario Research Fund. V. L. would like to thank Prof. Helen Tran for assistance with UV-vis absorption spectroscopy experiments, Drs Nimrat K. Obhi, Garion E.J. Hicks, Hui Wen Yong, and Harrison A. Mills for helpful feedback and discussions, and Alexander Lotocki for providing 3D graphics used in the TOC.References
- S. Kang, G.-H. Kim and S.-J. Park, Conjugated Block Copolymers for Functional Nanostructures, Acc. Chem. Res., 2022, 55, 2224–2234 CrossRef CAS.
- H. Lin, H. Bai, Z. Yang, Q. Shen, M. Li, Y. Huang, F. Lv and S. Wang, Conjugated polymers for biomedical applications, Chem. Commun., 2022, 58, 7232–7244 RSC.
- H. Sirringhaus, 25th Anniversary Article: Organic Field-Effect Transistors: The Path Beyond Amorphous Silicon, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Emerging applications for living crystallization-driven self-assembly, Chem. Sci., 2021, 12, 4661–4682 RSC.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Functional nanoparticles through π-conjugated polymer self-assembly, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- G. E. J. Hicks, S. Li, N. K. Obhi, C. N. Jarrett-Wilkins and D. S. Seferos, Programmable Assembly of π-Conjugated Polymers, Adv. Mater., 2021, 33, 2006287 CrossRef CAS PubMed.
- O. V. Mikhnenko, P. W. M. Blom and T.-Q. Nguyen, Exciton diffusion in organic semiconductors, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- A. J. Sneyd, D. Beljonne and A. Rao, A New Frontier in Exciton Transport: Transient Delocalization, J. Phys. Chem. Lett., 2022, 13, 6820–6830 CrossRef CAS.
- S. Prodhan, S. Giannini, L. Wang and D. Beljonne, Long-Range Interactions Boost Singlet Exciton Diffusion in Nanofibers of π-Extended Polymer Chains, J. Phys. Chem. Lett., 2021, 12, 8188–8193 CrossRef CAS.
- X.-H. Jin, M. B. Price, J. R. Finnegan, C. E. Boott, J. M. Richter, A. Rao, S. M. Menke, R. H. Friend, G. R. Whittell and I. Manners, Long-range exciton transport in conjugated polymer nanofibers prepared by seeded growth, Science, 2018, 360, 897–900 CrossRef CAS.
- T. Fukui, J. D. Garcia-Hernandez, L. R. MacFarlane, S. Lei, G. R. Whittell and I. Manners, Seeded Self-Assembly of Charge-Terminated Poly(3-hexylthiophene) Amphiphiles Based on the Energy Landscape, J. Am. Chem. Soc., 2020, 142, 15038–15048 CrossRef CAS PubMed.
- M. Vespa, Z. M. Hudson and I. Manners, Homogeneous and Segmented Nanofibers with a Conjugated Poly[3-(2′-ethylhexyl)thiophene] Core via Living Crystallization-Driven Self-Assembly, Macromolecules, 2024, 57, 1509–1520 CrossRef CAS.
- M. Vespa, L. R. MacFarlane, Z. M. Hudson and I. Manners, Crystallization-driven self-assembly of poly(3-hexylthiophene)-b-poly(2,5-bis(2-ethylhexyloxy)p-phenylene), a π-conjugated diblock copolymer with a rigid rod corona-forming block, Polym. Chem., 2024, 15, 1839–1850 RSC.
- H. Kim, J. Lee, S.-H. Hwang, N. Yun, S. Park and T.-L. Choi, Highly Efficient Preparation of Length and Width-Controllable Donor–Acceptor Nanoribbons via Polymerization-Induced Crystallization-Driven Self-Assembly of Fully Conjugated Block Copolymers, J. Am. Chem. Soc., 2024, 146, 20750–20757 CrossRef CAS.
- S. Park, S.-Y. Kang, S. Yang and T.-L. Choi, Independent Control of the Width and Length of Semiconducting 2D Nanorectangles via Accelerated Living Crystallization-Driven Self-Assembly, J. Am. Chem. Soc., 2024, 146, 19369–19376 CrossRef CAS.
- S.-H. Hwang, S.-Y. Kang, S. Yang, J. Lee and T.-L. Choi, Synchronous Preparation of Length-Controllable 1D Nanoparticles via Crystallization-Driven In Situ Nanoparticlization of Conjugated Polymers, J. Am. Chem. Soc., 2022, 144, 5921–5929 CrossRef CAS.
- K. Narasimha, S. K. Albert, J. Kim, H. Kang, S. Kang, J. Park, J. Park and S.-J. Park, Charge-Transfer-Induced Self-Assembly of Doped Conjugated Block Copolymer Nanofibers, ACS Macro Lett., 2023, 12, 382–388 CrossRef CAS.
- J. Kim, W. Chung, D. Kim, J. Kang, C. F. Grandes Reyes, J. Jeong and K. T. Kim, Semi-conductive micellar networks of all-conjugated diblock and triblock copolymer blends, Chem. Commun., 2023, 59, 3578–3581 RSC.
- T. S. Laws, W. K. Ledford, K. Kurtz, V. Oyanedel-Craver, T. Terlier, W. C. Tucker, N. C. Dickenson, T. S. Ramotowski, S. M. I. Kilbey and G. E. Stein, Fouling Resistance of Brush-Modified Elastomers, ACS Appl. Mater. Interfaces, 2023, 15, 28636–28648 CrossRef CAS PubMed.
- D. P. Keane, T. Kolozsvary, B. McDonald and R. Poling-Skutvik, Bottlebrush Midblocks Promote Colloidal Bridging of Telechelic Polymers, ACS Macro Lett., 2024, 13, 1304–1310 CrossRef CAS.
- A. Chremos and P. E. Theodorakis, Impact of intrinsic backbone chain stiffness on the morphologies of bottle-brush diblock copolymers, Polymer, 2016, 97, 191–195 CrossRef CAS.
- X. Li, B. Xia, Y. Song, M. Zheng, C. Huang, Y. Li and D.-P. Song, Light-Controlled Interfacial Self-Assembly of Bottlebrush Amphiphiles toward Highly Tunable Photonic Materials, Macromolecules, 2024, 57, 7915–7925 CAS.
- W. F. M. Daniel, J. Burdyńska, M. Vatankhah-Varnoosfaderani, K. Matyjaszewski, J. Paturej, M. Rubinstein, A. V. Dobrynin and S. S. Sheiko, Solvent-free, supersoft and superelastic bottlebrush melts and networks, Nat. Mater., 2016, 15, 183–189 CrossRef CAS.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Cylindrical molecular brushes: Synthesis, characterization, and properties, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS.
- S. J. Dalsin, T. G. Rions-Maehren, M. D. Beam, F. S. Bates, M. A. Hillmyer and M. W. Matsen, Bottlebrush Block Polymers: Quantitative Theory and Experiments, ACS Nano, 2015, 9, 12233–12245 CrossRef CAS.
- K. Kawamoto, M. Zhong, K. R. Gadelrab, L.-C. Cheng, C. A. Ross, A. Alexander-Katz and J. A. Johnson, Graft-through Synthesis and Assembly of Janus Bottlebrush Polymers from A-Branch-B Diblock Macromonomers, J. Am. Chem. Soc., 2016, 138, 11501–11504 CrossRef CAS.
- K. Zhang, S. Chen and J. Zhu, Programmable Reconfiguration of Supramolecular Bottlebrush Block Copolymers: From Solution Self-Assembly to Co-Crystallization-Assistant Self-Assembly, Angew. Chem., Int. Ed., 2024, e202408730 CAS.
- Y. Aviv, E. Altay, J. Rzayev and R. Shenhar, Assembly of Bottlebrush Block Copolymers and Nanoparticles in Ultrathin Films: Effect of Substrate–Copolymer Interaction on the Nanocomposite Morphology, Macromolecules, 2021, 54, 6247–6256 CrossRef CAS.
- D. F. Sunday, L. J. Richter, L. Q. Flagg, R. Li, J. G. Murphy, P. A. Beaucage and E. Gann, Role of Bottlebrush Additives on the Structure of Block Copolymers in the Bulk and Thin Films, Macromolecules, 2024, 57, 6616–6624 CrossRef CAS.
- L.-C. Cheng, K. R. Gadelrab, K. Kawamoto, K. G. Yager, J. A. Johnson, A. Alexander-Katz and C. A. Ross, Templated Self-Assembly of a PS-Branch-PDMS Bottlebrush Copolymer, Nano Lett., 2018, 18, 4360–4369 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O'Reilly, Getting into Shape: Reflections on a New Generation of Cylindrical Nanostructures' Self-Assembly Using Polymer Building Blocks, J. Am. Chem. Soc., 2019, 141, 2742–2753 CrossRef CAS PubMed.
- K. Zhao, M. Li, H. Geng, Z. Gao, X. Zhang, K. P. C. Sekhar, P. Zhang and J. Cui, Synthesis of Antifouling Poly(ethylene glycol) Brushes via “Grafting to” Approach for Improved Biodistribution, Biomacromolecules, 2024, 25, 6727–6736 CrossRef CAS PubMed.
- H. V.-T. Nguyen, Q. Chen, J. T. Paletta, P. Harvey, Y. Jiang, H. Zhang, M. D. Boska, M. F. Ottaviani, A. Jasanoff, A. Rajca and J. A. Johnson, Nitroxide-Based Macromolecular Contrast Agents with Unprecedented Transverse Relaxivity and Stability for Magnetic Resonance Imaging of Tumors, ACS Cent. Sci., 2017, 3, 800–811 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, A Practical and Highly Active Ruthenium-Based Catalyst that Effects the Cross Metathesis of Acrylonitrile, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS.
- Y. Xia, J. A. Kornfield and R. H. Grubbs, Efficient Synthesis of Narrowly Dispersed Brush Polymers via Living Ring-Opening Metathesis Polymerization of Macromonomers, Macromolecules, 2009, 42, 3761–3766 CrossRef CAS.
- S. E. Blosch, M. Alaboalirat, C. B. Eades, S. J. Scannelli and J. B. Matson, Solvent Effects in Grafting-through Ring-Opening Metathesis Polymerization, Macromolecules, 2022, 55, 3522–3532 CrossRef CAS.
- S. Ahn, D. L. Pickel, W. M. Kochemba, J. Chen, D. Uhrig, J. P. Hinestrosa, J.-M. Carrillo, M. Shao, C. Do, J. M. Messman, W. M. Brown, B. G. Sumpter and S. M. I. Kilbey, Poly(3-hexylthiophene) Molecular Bottlebrushes via Ring-Opening Metathesis Polymerization: Macromolecular Architecture Enhanced Aggregation, ACS Macro Lett., 2013, 2, 761–765 CrossRef CAS PubMed.
- C. D. Heinrich and M. Thelakkat, Poly-(3-hexylthiophene) bottlebrush copolymers with tailored side-chain lengths and high charge carrier mobilities, J. Mater. Chem. C, 2016, 4, 5370–5378 RSC.
- S. Ahn, J. Nam, J. Zhu, E. Lee and S. Michael Kilbey, Solution self-assembly of poly(3-hexylthiophene)–poly(lactide) brush copolymers: impact of side chain arrangement, Polym. Chem., 2018, 9, 3279–3286 RSC.
- N. K. Obhi, C. N. Jarrett-Wilkins, G. E. J. Hicks and D. S. Seferos, Self-Assembly of Poly(3-hexylthiophene) Bottlebrush Polymers into End-On-End Linear Fiber Morphologies, Macromolecules, 2020, 53, 8592–8599 CrossRef CAS.
- T. M. Swager, 50th Anniversary Perspective: Conducting/Semiconducting Conjugated Polymers. A Personal Perspective on the Past and the Future, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS.
- T. P. Kaloni, P. K. Giesbrecht, G. Schreckenbach and M. S. Freund, Polythiophene: From Fundamental Perspectives to Applications, Chem. Mater., 2017, 29, 10248–10283 CrossRef CAS.
- J. R. Reynolds, B. C. Thompson and T. A. Skotheim, Conjugated polymers: perspective, theory, and new materials, CRC Press, 2019 Search PubMed.
- H.-N. Choi, H.-S. Yang, S. Park, T.-L. Choi and I.-H. Lee, Combining Two Powerful Living Polymerizations Using Rationally Designed Bifunctional Initiators for Versatile Synthesis of Conjugated Rod–Coil Block Copolymers, Macromolecules, 2024, 57, 8050–8058 CAS.
- D. Pal, J. B. Garrison, Z. Miao, L. E. Diodati, A. S. Veige and B. S. Sumerlin, Nanobowls from Amphiphilic Core–Shell Cyclic Bottlebrush Polymers, Macromolecules, 2022, 55, 7446–7453 CrossRef CAS.
- K. Huang and J. Rzayev, Well-Defined Organic Nanotubes from Multicomponent Bottlebrush Copolymers, J. Am. Chem. Soc., 2009, 131, 6880–6885 CrossRef CAS PubMed.
- J. Zhang, X. Yuan, Z. Wu and L. Ren, Synthesis and self-assembly of high grafting density core–shell bottlebrush polymer with amphiphilic side chain, React. Funct. Polym., 2024, 202, 105997 CrossRef CAS.
- J. T. Wilk, C. T. Furner, E. W. Kent, M. T. Kelly, B. Zhao and C. Y. Li, Effect of Grafting Density on the Crystallization Behavior of Molecular Bottlebrushes, Macromolecules, 2024, 57, 8487–8497 CrossRef CAS.
- E. Matxinandiarena, M. I. Peñas, B. J. Curole, M. Król, L. Polo Fonseca, J. Ruokolainen, S. M. Grayson, L. Sangroniz and A. J. Müller, Crystallization-Induced Self-Assembly of Poly(ethylene glycol) Side Chains in Dithiol–yne-Based Comb Polymers: Side Chain Spacing and Molecular Weight Effects, Macromolecules, 2024, 57, 4906–4917 CrossRef CAS.
- T. W. Greene, Greene's protective groups in organic synthesis, Wiley-Interscience, 2007 Search PubMed.
- V. Lotocki, E. Grignon, H. A. Mills, S. Ye, A. J. Lough and D. S. Seferos, A SNAr-Active External Initiator that Enables Heterobifunctional Clickable Polythiophenes, Macromol. Chem. Phys., 2024, 225, 2300347 CrossRef CAS.
- S. Ye, S. M. Foster, A. A. Pollit, S. Cheng and D. S. Seferos, The role of halogens in the catalyst transfer polycondensation for π-conjugated polymers, Chem. Sci., 2019, 10, 2075–2080 RSC.
- H. Xu, H. A. Mills, S. Ye and D. S. Seferos, Recyclable terthiophenes for synthesizing precision conjugated oligomers, Polym. Chem., 2024, 15, 3814–3822 RSC.
- E. Y. Osei-Twum, L. A. Litorja, J. Darkwa, L. L. Maisela, A. Lesimple and O. Mamer, Mass spectral behavior of some homoleptic and mixed aryldichalcogenide bis(diphenylphosphino)ferrocenenickel(II), palladium(II), and platinum(II), and bis(diisopropylphosphino)ferrocenepalladium(II) complexes, J. Am. Soc. Mass Spectrom., 2005, 16, 94–99 CrossRef CAS.
- H. A. Bronstein and C. K. Luscombe, Externally Initiated Regioregular P3HT with Controlled Molecular Weight and Narrow Polydispersity, J. Am. Chem. Soc., 2009, 131, 12894–12895 CrossRef CAS.
- J. Liu, R. S. Loewe and R. D. McCullough, Employing MALDI-MS on Poly(alkylthiophenes): Analysis of Molecular Weights, Molecular Weight Distributions, End-Group Structures, and End-Group Modifications, Macromolecules, 1999, 32, 5777–5785 CrossRef CAS.
- A. F. M. Barton, Solubility parameters, Chem. Rev., 1975, 75, 731–753 CrossRef CAS.
- G. E. J. Hicks, R. R. Cranston, V. Lotocki, J. G. Manion, B. H. Lessard and D. S. Seferos, Dopant-Stabilized Assembly of Poly(3-hexylthiophene), J. Am. Chem. Soc., 2022, 144, 16456–16470 CrossRef CAS.
- S.-M. Jin, J. H. Hwang, J. A. Lim and E. Lee, Precrystalline P3HT nanowires: growth-controllable solution processing and effective molecular packing transfer to thin film, CrystEngComm, 2022, 24, 1248–1257 RSC.
- A. C. Kamps, M. Fryd and S.-J. Park, Hierarchical Self-Assembly of Amphiphilic Semiconducting Polymers into Isolated, Bundled, and Branched Nanofibers, ACS Nano, 2012, 6, 2844–2852 CrossRef CAS.
- Y. D. Park, H. S. Lee, Y. J. Choi, D. Kwak, J. H. Cho, S. Lee and K. Cho, Solubility-Induced Ordered Polythiophene Precursors for High-Performance Organic Thin-Film Transistors, Adv. Funct. Mater., 2009, 19, 1200–1206 CrossRef CAS.
- N. Nagahora, S. Yahata, S. Goto, K. Shioji and K. Okuma, 2,5-Diaryltellurophenes: Effect of Electron-Donating and Electron-Withdrawing Groups on their Optoelectronic Properties, J. Org. Chem., 2018, 83, 1969–1975 CrossRef CAS.
- L. Farouil, F. Alary, E. Bedel-Pereira and J.-L. Heully, Revisiting the Vibrational and Optical Properties of P3HT: A Combined Experimental and Theoretical Study, J. Phys. Chem. A, 2018, 122, 6532–6545 CrossRef CAS PubMed.
- F. C. Spano, Modeling disorder in polymer aggregates: The optical spectroscopy of regioregular poly(3-hexylthiophene) thin films, J. Chem. Phys., 2005, 122, 234701 CrossRef PubMed.
- Y. Yuan, J. Shu, P. Liu, Y. Zhang, Y. Duan and J. Zhang, Study on π–π Interaction in H- and J-Aggregates of Poly(3-hexylthiophene) Nanowires by Multiple Techniques, J. Phys. Chem. B, 2015, 119, 8446–8456 CrossRef CAS.
- J. Clark, J.-F. Chang, F. C. Spano, R. H. Friend and C. Silva, Determining exciton bandwidth and film microstructure in polythiophene films using linear absorption spectroscopy, Appl. Phys. Lett., 2009, 94, 163306 CrossRef.
- D. Dudenko, A. Kiersnowski, J. Shu, W. Pisula, D. Sebastiani, H. W. Spiess and M. R. Hansen, A Strategy for Revealing the Packing in Semicrystalline π-Conjugated Polymers: Crystal Structure of Bulk Poly-3-hexyl-thiophene (P3HT), Angew. Chem., Int. Ed., 2012, 51, 11068–11072 CrossRef CAS.
- V. Ho, B. W. Boudouris and R. A. Segalman, Tuning Polythiophene Crystallization through Systematic Side Chain Functionalization, Macromolecules, 2010, 43, 7895–7899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: https://doi.org/10.1039/d4sc06868h |
| This journal is © The Royal Society of Chemistry 2025 |