
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate NOBINAc ligand affinity for PdII-catalyzed enantioselective C–H activation over reactive β-C–H bonds in ferrocenyl amines†
Devendra
Parganiha
 ,
Raviraj Ananda
Thorat
,
Ashwini Dilip
Dhumale
,
Yagya Dutt
Upadhyay
,
Raushan Kumar
Jha
,
Saravanan
Raju
and
Sangit
Kumar
*
,
Raviraj Ananda
Thorat
,
Ashwini Dilip
Dhumale
,
Yagya Dutt
Upadhyay
,
Raushan Kumar
Jha
,
Saravanan
Raju
and
Sangit
Kumar
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-Pass Road, Bhopal, Madhya Pradesh 462066, India. E-mail: sangitkumar@iiserb.ac.in
First published on 13th December 2024
Abstract
Ferrocenyl amines as directing groups for C–H activation have limitations as they are prone to undergo oxidation, allylic deamination, and β-hydride elimination. The fundamental challenge observed here is the competition between the desired C–H activation versus the vulnerable β-C–H bond activation of amines and fine-tuning of a suitable oxidant which avoids the oxidation of the β-C–H bond and ferrocene. Herein, the potential of an axially chiral NOBINAc ligand is revealed to implement the enantioselective PdII-catalyzed C–H activation process of ferrocenyl amines. Mechanistically, the affinity between the NOBINAc ligand and sulfonate group of amine facilitated by the Cs+ cation plays an impressive role in the desired reaction outcome via an enhanced substrate ligand affinity. This approach resulted in a Pd-catalyzed enantioselective C–H activation, the first intermolecular annulation, and alkenylation of ferrocenyl amines with allenes and olefins, leading to ferrocene fused tetrahydropyridines and alkenylated ferrocenyl amines with up to 70% yields and 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er.
:1 er.
1 Introduction
The journey of ferrocene began in the 1950s, and it quickly garnered attention due to its outstanding electronic and structural characteristics.1,2 It exhibits a highly reversible one-electron redox event and the intriguing ability to exhibit planar chirality, positioning it as an esteemed framework for asymmetric transformations in academia, industry, materials chemistry, and medicinal chemistry (Scheme 1, part a).2 In the last few decades, ferrocene has evolved into one of the most extensively researched and developed systems for synthesizing planar chiral molecules. Consequently, several conventional organolithiums and recently developed TM-catalyzed methodologies have been employed to construct planar chiral ferrocenes.3 However, conventional and or TM-catalyzed methodologies demand multi-step synthetic routes and oxidation-protected directing groups namely tert- and sec-amides,4tert-amines,5 oxazolines,6 and aldehyde.7 This limits the construction of complex molecular architecture, as tethered directing groups8 are difficult to remove or have difficulty associated with further derivatization of directing groups and requisite complex ligands for high enantioselectivity. Our group has been working on the C–H activation of ferrocene.9 Recently, we embarked on enantioselective C–H activation strategies and preparation of chiral ferrocene molecules from ferrocene amides having an acidic proton.9d Enantioselective C–H activation and subsequent functionalization in the substrates having activated β-CH bonds adjacent to acidic proton containing directing groups namely –CH2NHR, –CH2OH, and –CH2CO2H are difficult as these functional groups are prone to undergo dehydrogenation, oxidation and undesired β-CH activation (Scheme 1, part b).10 Moreover, in the case of ferrocene, it becomes more difficult to prevent oxidation as compared to other aromatics due to its lower redox potential. Therefore, conventional organo-lithiation routes or TM-catalyzed methodologies could not be employed on such substrates.3 On the other hand, ferrocene having –CH2NHR groups offers numerous opportunities for complex molecular architecture. For example, these molecules can be easily annulated or readily transformed into diverse functionality after C–H activation due to the presence of reactive –CH2NHR groups. Palladium catalysis offers a more accessible approach for highly enantioselective C–H activations.11 Therefore, various ligands have been developed for PdII-catalysis, solving various fundamental challenges associated with enantioselective C–H activations.11 Here ligands play a crucial role in regulating the selectivity and expediting the C–H activation process involving various interactions.12 | ||
| Scheme 1 Application of ferrocene-based molecules, challenges, and rational design for enabling amines as directing groups for annulation and alkenylation. | ||
In continuation of our work on the methodology development and our long-continued interest in ferrocene,2b,c,e we were curious to investigate catalytic enantioselective C–H activation in the ferrocenyl amines having acidic protons which offers diverse possibilities. We envisioned that developing a Pd-catalytic pocket, with the addition of a suitable chiral ligand, could circumvent the interaction of Pd with an activated β-CH bond. Consequently, it could lead to the desired enantioselective C–H activation of ferrocenyl amines (Scheme 1, part c).
Here, we have devised a catalytic pocket by utilizing a NOBINAc ligand that underwent enantioselective C–H activation with an allene coupling partner to afford chiral ferrocene-fused pyridines with up to 99:1 er. Moreover, the developed PdII-catalyzed C–H activation has been reacted with olefins to enable 1,2-alkenylated ferrocenyl amines up to 98:2 er. Control experiments and DFT calculations have also been carried out to understand the role of cation interaction in NOBINAc ligand-enabled Pd-catalyzed C–H activation.
2 Results and discussion
We initiated our study with ferrocenyl amine 1a to explore enantioselective C–H activation by using a Pd(OAc)2 catalyst, chiral ligand, and CuO oxidant in the presence of an additive CsF in THF (see Scheme 2, ESI, Tables S1–S6†). | ||
| Scheme 2 Screening of the ligand and optimization of the reaction conditions. aReaction conditions: 1a (0.05 mmol), 2a (0.1 mmol), Pd(OAc)2 (0.004 mmol), ligand (0.014 mmol), CsF (0.1 mmol), CuO (0.05 mmol), THF (1 ml), air, T °C, 18 h. bCrude yield of 3a is determined by 1H NMR with CH2Br2 as an internal standard. cer of 3a was determined by HPLC analysis. dIsolated yield of 3a and enantioselectivity when the reaction was carried out in dry air. | ||
Fascinated by the distinct regio- and chemo-divergent one to three carbon synthons possibility and further functionalization opportunity offered by allenes,13 we set to explore allenes (2a–2j) as a coupling partner for enantioselective C–H activation followed by the annulation reaction. Initially, mono-protected amino acid (MPAA) ligands were tried for enantioselective C–H activation of ferrocenyl-sec-amine 1a with allene 2a. Monoanionic (L–X) MPAA ligands L1–L5 led to poor yield (13–16%) of annulated ferrocene fused tetrahydropyridine 3a along with moderate 81:19–87:13 er enantioselectivity (Scheme 2). Ligand L5 yielded the best results among ligands L1–L5, achieving a 14% yield and moderate 87:13 er (Scheme 2). Meanwhile, di-anionic (X–X) derivatized MPAA L6 yielded 17% of 3a with a significant loss in enantioselectivity (69:31 er). Furthermore, in the presence of MPAA ligands (L1–L6), the excessive formation of a side product, ferrocene carboxaldehyde, was observed in the reaction, presumably due to the background oxidation and β-hydride elimination from the ferrocenyl-sec-amine 1a by its undesired interaction with the PdII catalyst. Interestingly, during the screening of axially chiral binaphthyl-derived ligands L7–L10, NOBINAc ligand , developed by Gulias and co-workers,12e,14 offered a good 94:6 er and moderate yield (57%) of ferrocene fused tetrahydropyridine 3a. Furthermore, screening of reaction conditions by modifying NOBINAc to N-Boc group L11 and NOBINAc to –OMe group L12 afforded enantioselectivity 92:8 er and 53:47 er with low 51 and 10% yields, respectively.
To our delight, NOBINAc ligand L10, under dry air atmospheric conditions, offered a 70% yield with an excellent 99:1 er of 3a (see ESI, Tables S1–S5†). Moreover, using S-NOBINAc L10 afforded opposite enantiomer (Sp)-3a in nearly the same yield and er. The absolute configuration of both the enantiomers of 3a and their structures have been studied by single crystal XRD (Scheme 2). After the optimization of the reaction conditions, the applicability of the developed reaction methodology was explored regarding a variety of allenes (Scheme 3).
 | ||
| Scheme 3 Substrate scope of allene derivatization. aIsolated yields of 3a–3j were obtained using (R)-NOBINAc unless otherwise stated. ber of the product was determined using an HPLC analysis. cer of the product obtained by using (S)-NOBINAc. dE:Z was determined by 1H NMR. | ||
α,α-Diaryl substituted symmetrical allenes 2b–2d with electron-donating and withdrawing substituents reacted smoothly under NOBINAc L10-accelerated reaction conditions, leading to chiral tetrahydropyridines 3b–3d in 62–68% yields and 95:5–99:1 er. Furthermore, α,α-unsymmetrical allenes 2f–2j also underwent the enantioselective annulation reaction to afford respective tetrahydropyridines 3f–3j in 86:14–97.5:2.5 er and better E:Z selectivity up to (1:10) and (1:5), whereas substitution at the phenyl ring lowers the E:Z selectivity as phenyl-substituted tetrahydropyridines 3g and 3j were obtained in a poor E:Z up to 1:2. Alkyl-substituted allenes 2e and 2i offered alkyl-substituted tetrahydropyridines 3e and 3i in nearly the same yields, but with relatively lower er (89:11 and 86:14 er).
It seems that the trifluorosulfonyl (–NHTf) group not only facilitates C–H activation but also helps in preventing oxidation15 of secondary amines through secondary cationic interactions (vide infra). Next, various ferrocenyl amines 1b–1f having the possibility of potential secondary cationic interactions under the developed NOBINAc L10 accelerated enantioselective C–H activation have been explored (Scheme 4). To our delight, ferrocenyl amines 1b–1e having methyl, ortho-nitrophenyl, 3,5-difluorophenyl, and para-methyl-phenyl sulfonyl groups also underwent enantioselective C–H-annulation leading to diverse N-substituted chiral tetrahydropyridines 3k–3z ranging yields of 28–58% with 86:14 to 99:1 er, whereas nosyl and SO2CH3 protected amines yielded ferrocene fused tetrahydropyridines 3t–3y in relatively lower yields 28–42% with 86:14 er to 97:3 er (Scheme 4). N-Acetyl-protected ferrocenyl-sec-amine 1f failed to afford any annulated ferrocene fused pyridine.
 | ||
| Scheme 4 Substrate scope with regard to ferrocenyl amines. aIsolated yields of 3k–3z were obtained using (R)-NOBINAc unless otherwise stated. ber of the product was determined using an HPLC analysis. cer of the product obtained by using (S)-NOBINAc. dE:Z was determined by 1H NMR. | ||
Furthermore, styrene and activated olefin coupling partners, which could react through π-interaction with the in situ formed proposed metallacycle, were also explored under the NOBINAc-accelerated reaction conditions (Scheme 5). Gratifyingly, ethyl acrylate underwent an enantioselective C–H activation reaction to provide dehydrogenative Heck-coupled ferrocenyl acrylate 4a in 52% yield and 91:9 er under the optimized reaction conditions. Next, a series of substituted acrylates were coupled with ferrocenyl amines 1c and 1d to afford respective chiral 1,2 alkenylated ferrocenyl amines 4b–4h in 94:6 to 98:2 er under enantioselective NOBINAc accelerated reaction conditions.
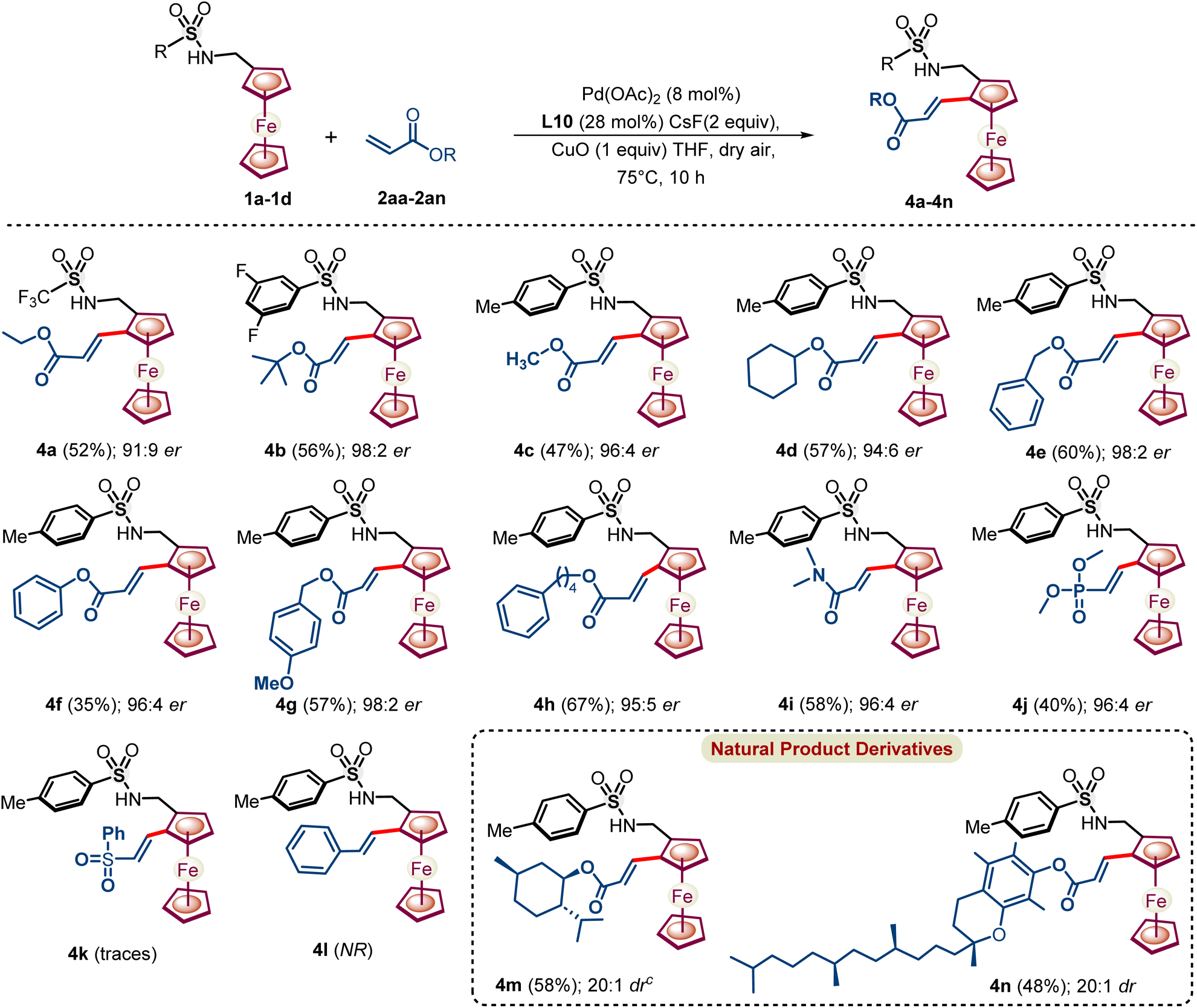 | ||
| Scheme 5 Substrate scope with regard to olefins. aIsolated yields of 4a–4n. bThe er was determined by HPLC. cdr was determined by 1HNMR. | ||
Furthermore, not only acrylates, but also N,N-dimethylacrylamide and dimethyl vinylphosphonate coupled enantioselectively with ferrocenyl amines to provide respective chiral 1,2-alkenylated ferrocenyl amines 4i–4j in good 96:4 to 96:4 er, respectively. However, (vinylsulfonyl)benzene reacted sluggishly to afford traces of 4k and unactivated styrene failed to react with ferrocenyl amine under the optimized reaction conditions.
Next, acrylate consisting of natural product moiety L-menthol and α-tocopherols reacted with excellent diastereoselectivity to afford natural product derivatized ferrocenyl amines in >20:1 dr. We have also tested the alkynes, phenylacetylene and 1-phenyl-1-propyne, in place of allenes as coupling partners under the optimized conditions. Phenylacetylene failed to afford any coupled product with ferrocenyl amine 1a under the reaction conditions and 1-phenyl-1-propyne afforded only traces of the coupled product which was confirmed by mass spectrometry from the reaction mixture.
Next, we have shown that the sulfonyl protection at the amine backbone can be eliminated from synthesized chiral tetrahydropyridine 3a, affording unprotected tetrahydropyridine 5a with a 70% yield and 94:6 er (Scheme 6A).
 | ||
| Scheme 6 Post derivatization and application of ferrocene fused chiral amine as a catalyst. Reaction conditions: (a) 3a (0.05 mmol), red Al (0.5 mmol) in dry toluene. (b) 4b (0.05 mmol), Cs2CO3 (0.1 mmol) in DMF at 110 °C for 6h. (c) Chalcone (0.1 mmol), PhSH (0.25 mmol), 5a (0.005 mmol). | ||
Furthermore, amine 4b can be readily oxidized into a versatile aldehyde group leading to chiral 1,2-alkenylated ferrocene carboxaldehyde 6a in 55% yield and 98:2 er which has also been reduced into chiral ferrocenyl alcohol 7a in 96% yield and 98:2 er (Scheme 6B). The utility of the synthesized chiral ferrocene fused amine 5a has been tested as a catalyst for the enantioselective thia-Michael reaction with chalcone providing 8a with up to 80% yield and 65:35 er (Scheme 6C).
2.1 Mechanistic investigation
In pursuit of an understanding of efficient work function in NOBINAc catalysis, control experiments were performed, where the cation from the envisioned catalytic cycle was deliberately removed using water and 18-crown ether (Scheme 7). The addition of water and 18-crown ether under the standard optimized conditions shows a significant reduction in both yield and enantioselectivity [(A) conditions a and b, Scheme 7]. On the other hand, in the absence of a cation (Scheme 7A, condition c), the reaction leads to only traces of annulated product 3a without any er. | ||
| Scheme 7 Understanding of the NOBINAC work function in the presence of Cs+. (A) Control experiment. (B) 19F NMR experiment. (C). Ligand acceleration experiment. (D). DFT computation for favorable TS1 and unfavorable transition state TS2. aThe optimization of the proposed structures derived from 1a, PdII, S-NOBINAc, and Cs+ and the energies were obtained at a DFT-B3LYP/LANL2DZ level of theory (for details, see ESI, pp. S101–S111†). | ||
Furthermore, other organic cationic sources exemplified by TBAF and TBAB were also tried under optimized reaction conditions; however, the reaction outcome was poor and provided nearly the same result (yields and enantioselectivity) for these organic cation sources (Scheme 7A, condition d). Additionally, NOBINAc ligand L12 has –OH protection with a methyl group to avoid the possible cationic interaction leading to erosion of er (57:43 ervs. 98:2 er; Scheme 7A, condition e). This outcome indicates that the OH group is necessary for NOBINAc for an efficient work function.
Next, 19F NMR was carried out on the reaction of 1a, Pd(OAc)2, NOBINAc, and various cations (Cs2CO3, Na2CO3, and Li2CO3). A new peak at −76.07 ppm corresponding to CF3 of 1a emerges and increases to a maximum with the increase of the size of the cations (Li+, Na+, and Cs+). The only mixture of Cs2CO3 and 1a does not lead to any new peak in the 19F NMR spectra. This may suggest that Cs+ cation enhances the interaction among substrate 1a, NOBINAc ligand L10, and PdII (for details, see Scheme 7B and ESI, p. S99†).
Furthermore, the rate of the reaction in the presence and absence of NOBINAc L10 was monitored by 19F NMR with respect to 3f, which suggests that the NOBINAc L10 accelerates the reaction by 2.2 fold (for details, see Scheme 7C and ESI, p. S101†).
Next, DFT computations were carried out to shed light on high enantioselective discrimination. The structural optimization reveals that the Cs+ ion is away from the lower Cp ring in the favorable TS1, whereas, in unfavorable TS2, the Cs+ ion is close to the Cp ring. A relative energy difference of 10.45 kcal mol−1 was realized between favorable TS1 and unfavorable TS2 (Scheme 7D). Thus, based on the evidence for the initial CMD step and similar previously reported literature precedence,13e,g a catalytic cycle has been proposed (Scheme 8).13f,h,i The NOBINAc ligand and ferrocenyl amine 1a seem to undergo substitution with Pd(OAc)2 in the presence of a CsF enabling Pd(II)amidate, which, upon the interaction of Cs+ with the sulfonyl group and NOBINAc, leads to the generation of a chiral catalytic pocket in palladium intermediate I.12e
 | ||
| Scheme 8 Catalytic cycle for C–H annulation. | ||
Here the cationic interaction may dictate the vacant dsp2-hybrid orbital of PdII (intermediate shown in Scheme 1) towards the desired enantiotopic C–H bond and avoid interaction with the β-C–H bond to circumvent β-hydride elimination. Furthermore, enantio-determining C–H activation occurs via the concerted metalation deprotonation (CMD) process which leads to palladacycle II.13f Subsequently, allene insertion takes place into palladacycle II to form palladacycle III.13f Furthermore, migratory insertion would lead to intermediates IV and V which upon reductive elimination could afford ferrocene fused tetrahydropyridine 3a with concomitant release of the catalyst.13b,f,i
In the developed NOBINAc-catalyzed annulation reaction, Z-regioselectivity was obtained across the double bond in the ferrocenyl tetrahydropyridines. Z-selectivity over the E-regioisomer was understood by using DFT computations (Fig. 1). The optimized Z-isomer is more stable by 1.51 kcal mol−1 than the isolated E-regioisomer. Furthermore, palladacycle III seems to be the key intermediate with respect to unsymmetrical allenes to determine relative E,Z-selectivity (Fig. 1). Here, the respective Z-intermediate in which the phenyl substituent of unsymmetrical allene is away from the Cp ring shows lower energy by 3.45 kcal mol−1 than the respective E-intermediate (Fig. 1).
 | ||
| Fig. 1 DFT computation studies to determine relative Z- and E-isomer selectivity (for details, see ESI, pp. S105–S120†). | ||
3 Conclusion
In summary, we have addressed a fundamental problem associated with amine substrates having activated β-CH bonds adjacent to the acidic proton (–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2NR) for the desired enantioselective CH activation by a meticulously designed Pd-catalytic pocket. Here, the NOBINAc ligand and ferrocenyl amine enable PdII-catalyzed highly enantioselective C–H activation. Furthermore, it has been observed that the amines having sulfonyl groups are crucial for high enantioselective induction. The subsequent annulation and alkenylation enable the development of an efficient methodology for synthesizing chiral ferrocene-fused tetrahydropyridine and 2-alkenylated ferrocenyl amines. The sulfonyl-protecting group can be effortlessly removed to provide chiral ferrocenyl amine, which shows the application as a chiral catalyst. Similarly, 2-alkenylated ferrocenyl amine has been oxidized into respective ferrocene carboxaldehyde which is a versatile group for further derivatization into alcohols. So far the initial results have allowed us to explore the introduction of coupling partners within allenes and acrylates. Future investigations are underway to explore the feasibility of introducing coupling partners through transmetallation or oxidative addition, enabling ferrocenyl amine-derived arylation and heteroatom insertion reactions.
2NR) for the desired enantioselective CH activation by a meticulously designed Pd-catalytic pocket. Here, the NOBINAc ligand and ferrocenyl amine enable PdII-catalyzed highly enantioselective C–H activation. Furthermore, it has been observed that the amines having sulfonyl groups are crucial for high enantioselective induction. The subsequent annulation and alkenylation enable the development of an efficient methodology for synthesizing chiral ferrocene-fused tetrahydropyridine and 2-alkenylated ferrocenyl amines. The sulfonyl-protecting group can be effortlessly removed to provide chiral ferrocenyl amine, which shows the application as a chiral catalyst. Similarly, 2-alkenylated ferrocenyl amine has been oxidized into respective ferrocene carboxaldehyde which is a versatile group for further derivatization into alcohols. So far the initial results have allowed us to explore the introduction of coupling partners within allenes and acrylates. Future investigations are underway to explore the feasibility of introducing coupling partners through transmetallation or oxidative addition, enabling ferrocenyl amine-derived arylation and heteroatom insertion reactions.
Data availability
Detailed synthetic procedures, reaction condition optimization, data characterization, controlled experiments, X-ray diffraction structural analyses, and geometries and energies of theoretically investigated structures have been provided in the ESI.†Author contributions
SK and DP wrote the manuscript. DP, ADD, YDU, and RT performed synthesis and characterization. DP has performed controlled experiments. RJ and SR performed DFT computations. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK acknowledges DST-SERB (CRG/2023/002473), New Delhi, and IISER Bhopal for financial support. DP acknowledges UGC New Delhi (NTA ref. no. 191620241186) for the fellowship. RAT, RJ, and SR acknowledge IISER Bhopal for fellowships. ADD acknowledges DST INSPIRE (IF210534) for the fellowship. SK also acknowledges Mr Pruthviraj Patil (IISER Bhopal) and Mr Sumit Khevariya (IISER Bhopal) for synthesizing alkenylated ferrocenes.Notes and references
- (a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS; (b) G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS.
- (a) G. Mugesh, A. Panda, H. B. Singh, N. S. Punekar and R. J. Butcher, J. Am. Chem. Soc., 2001, 123, 839–850 CrossRef CAS PubMed; (b) G. Mugesh, A. Panda, S. Kumar, S. D. Apte, H. B. Singh and R. J. Butcher, Organometallics, 2002, 21, 884 CrossRef CAS; (c) S. Kumar, H. B. Singh and G. Wolmershauser, Organometallics, 2006, 25, 382 CrossRef CAS; (d) T. Muraoka, K. Kinbara and T. Aida, Nature, 2006, 440, 512–515 CrossRef CAS; (e) S. Kumar, J.-C. P. Helt, J. Autschbach and M. R. Detty, Organometallics, 2009, 28, 3426 CrossRef CAS; (f) G. Mirri, S. D. Bull, P. N. Horton, T. D. James, L. Male and J. H. R. Tucker, J. Am. Chem. Soc., 2010, 132, 8903–8905 CrossRef CAS; (g) D. Astruc, Eur. J. Inorg. Chem., 2017, 2017, 6–29 CrossRef CAS; (h) M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS; (i) O. Bernardo, S. G. Pelayo and L. A. López, Eur. J. Inorg. Chem., 2022, 2022, e202100911 CrossRef CAS.
- (a) M. Tsukazaki, A. Roglans, B. J. Chapell, N. J. Taylor and V. Snieckus, J. Am. Chem. Soc., 1996, 118, 685 CrossRef CAS (more references therein on ortho-lithiation).; (b) Z.-J. Cai, C.-X. Liu, Q. Wang, Q. Gu and S.-L. You, Nat. Commun., 2019, 10, 4168–4175 CrossRef PubMed; (c) S. R. Yetra, Z. Shen, H. Wang and L. Ackermann, Beilstein J. Org. Chem., 2018, 14, 1546–1553 CrossRef CAS PubMed; (d) C. X. Liu, Q. Gu and S.-L. You, Trends Chem., 2020, 2, 737–749 CrossRef CAS; (e) Z.-Z. Zhang, D.-Y. Huang and B.-F. Shi, Org. Biomol. Chem., 2022, 20, 4061–4073 RSC; (f) P. Dolui, A. Verma and A. J. Elias, Directing Group-Assisted C-H Functionalization of Ferrocene and CpCo(C4Ph4) Derivatives, Wiley-VCH, 2022, DOI:10.1002/9783527834242.chf0074; (g) C.-X. Liu, P.-P. Xie, F. Zhao, Q. Wang, Z. Feng, H. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2023, 145, 4765–4773 CrossRef CAS; (h) C.-X. Liu, F. Zhao, Z. Feng, Q. Wang, Q. Gu and S.-L. You, Nat. Synth., 2023, 2, 49–57 CrossRef CAS; (i) T. Carny and R. Sebesta, Synlett, 2024, 35, 165–182 CrossRef CAS.
- (a) Q. Wang, Y. H. Nie, C. X. Liu, W. W. Zhang, Z. J. Wu, Q. Gu, C. Zheng and S.-L. You, ACS Catal., 2022, 12, 3083–3093 CrossRef CAS; (b) Q.-J. Yao, F.-R. Huang, J.-H. Chen and B.-F. Shi, Nat. Commun., 2024, 15, 7135 CrossRef CAS PubMed.
- (a) D. W. Gao, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2016, 138, 2544–2547 CrossRef CAS; (b) Z. J. Cai, C. X. Liu, Q. Gu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 2149–2153 CrossRef CAS; (c) L. Zhou, H. G. Cheng, L. Li, K. Wu, J. Hou, C. Jiao, S. Deng, Z. Liu, J. Q. Yu and Q. Zhou, Nat. Chem., 2023, 15, 815–823 CrossRef CAS PubMed.
- (a) W.-J. Kong, Q. Shao, M.-H. Li, Z.-L. Zhou, H. Xu, H.-X. Dai and J.-Q. Yu, Organometallics, 2018, 37, 2832–2836 CrossRef CAS; (b) X. Kuang, J.-J. Li, T. Liu, C.-H. Ding, K. Wu, P. Wu and J.-Q. Yu, Nat. Commun., 2023, 14, 7698 CrossRef CAS PubMed.
- C.-X. Liu, F. Zhao, Q. Gu and S.-L. You, ACS Cent. Sci., 2023, 9, 2036–2043 CrossRef CAS PubMed.
- Reference for tethered directing groups: (a) M. Ogasawara, S. Watanabe, K. Nakajima and T. Takahashi, J. Am. Chem. Soc., 2010, 132, 2136–2137 CrossRef CAS PubMed; (b) D. W. Gao, Q. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2014, 136, 4841–4844 CrossRef CAS.
- (a) M. Sattar and S. Kumar, Org. Lett., 2017, 19, 5960–5963 CrossRef CAS; (b) R. A. Thorat, S. Jain, M. Sattar, P. Yadav, Y. Mandhar and S. Kumar, J. Org. Chem., 2020, 85, 14866–14878 CrossRef CAS PubMed (other related references therein).; (c) R. A. Thorat, Y. Mandhar, D. Parganiha, K. B. Patil, V. Singh, B. Shakir, S. Raju and S. Kumar, ChemistrySelect, 2023, 8, e202203945 CrossRef CAS; (d) R. A. Thorat, D. Parganiha, S. Jain, V. Choudhary, B. Shakir, K. Rohilla, R. K. Jha and S. Kumar, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-918dp-v3.
- (a) P. L. Theofanis and W. A. Goddard, Organometallics, 2011, 30, 4941–4948 CrossRef CAS; (b) M. B. Li, Y. Wang and S. K. Tian, Angew. Chem., Int. Ed., 2012, 51, 2968–2971 CrossRef CAS; (c) Z. Zhuang and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 12015–12019 CrossRef CAS; (d) V. G. Landge, A. J. Grant, Y. Fu, A. M. Rabon, J. L. Payton and M. C. Young, J. Am. Chem. Soc., 2021, 143, 10352–10360 CrossRef CAS PubMed; (e) Y. Sekiguchi, J. H. Pang, J. S. Ng, J. Chen, K. Watanabe, R. Takita and S. Chiba, JACS Au, 2022, 2, 2758–2764 CrossRef CAS PubMed; (f) R. Keshri, D. Rana, A. Saha, S. A. Al-Thabaiti, A. A. Alshehri, S. M. Bawaked and D. Maiti, ACS Catal., 2023, 13, 4500–4516 CrossRef CAS; (g) D. A. Strassfeld, C. Y. Chen, H. S. Park, D. Q. Phan and J.-Q. Yu, Nature, 2023, 622, 80–86 CrossRef CAS.
- (a) B.-F. Shi, N. Maugel, Y. H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS; (b) D. W. Gao, Y. C. Shi, Q. Gu, Z. L. Zhao and S.-L. You, J. Am. Chem. Soc., 2013, 135, 86–89 CrossRef CAS PubMed; (c) L. Chu, K. J. Xiao and J.-Q. Yu, Science, 2014, 346, 451–455 CrossRef CAS PubMed; (d) Q. J. Yao, P. P. Xie, Y. J. Wu, Y. L. Feng, M. Y. Teng, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2020, 142, 18266–18276 CrossRef CAS; (e) B. B. Zhan, L. Jin and B.-F. Shi, Trends Chem., 2022, 4, 220–235 CrossRef CAS; (f) X. Lv, M. Wang, Y. Zhao and Z. Shi, J. Am. Chem. Soc., 2024, 146, 3483–3491 CrossRef CAS PubMed.
- (a) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. S. Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS; (b) G. R. Genov, J. L. Douthwaite, A. S. K. Lahdenpera, D. C. Gibson and R. J. Phipps, Science, 2020, 367, 1246–1251 CrossRef CAS; (c) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, eabd5992 CrossRef CAS PubMed; (d) Y. Lou, J. Wei, M. Li and Y. Zhu, J. Am. Chem. Soc., 2022, 144, 123–129 CrossRef CAS; (e) J. M. González, X. Vidal, M. A. Ortuño, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2022, 144, 21437–21442 CrossRef; (f) L. Hu, G. Meng, X. Chen, J. S. Yoon, J. R. Shan, N. Chekshin, D. A. Strassfeld, T. Sheng, Z. Zhuang, R. Jazzar, G. Bertrand, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2023, 145, 16297–16304 CrossRef CAS PubMed; (g) N. Goswami, S. K. Sinha, P. Mondal, S. Adhya, A. Datta and D. Maiti, Chem, 2023, 9, 989–1003 CrossRef CAS; (h) M. Kadarauch, D. M. Whalley and R. J. Phipps, J. Am. Chem. Soc., 2023, 145, 25553–25558 CrossRef CAS PubMed; (i) K. Wu, N. Lam, D. A. Strassfeld, Z. Fan, J. X. Qiao, T. Liu, D. Stamos and J.-Q. Yu, Angew. Chem., Int. Ed., 2024, 63, e202400509 CrossRef CAS.
- (a) S. Nakanowatari, T. Muller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15891–15895 CrossRef CAS; (b) B. Cendo, N. Casanova, C. Comanescu, R. G. Fandin, A. Seoane, M. Gulías and J. L. Mascarenas, Org. Lett., 2017, 19, 1674–1677 CrossRef; (c) R. Santhoshkumar and C.-H. Cheng, Asian J. Org. Chem., 2018, 7, 1151–1163 CrossRef CAS; (d) J. Mo, T. Müller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 7719–7723 CrossRef CAS; (e) C. Zhu, J. L. Schwarz, S. Cembellín, S. Greßies and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 437–441 CrossRef CAS; (f) X. Vidal, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2019, 141, 1862–1866 CrossRef CAS PubMed; (g) J. P. Schmidt and B. Breit, Angew. Chem., Int. Ed., 2020, 59, 23485–23490 CrossRef CAS; (h) J. M. González, B. Cendón, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2021, 143, 3747–3752 CrossRef; (i) X. Vidal, J. L. Mascarenas and M. Gulías, Org. Lett., 2021, 23, 5323–5328 CrossRef CAS; (j) F. Panahi, F. Bauer and B. Breit, Acc. Chem. Res., 2023, 56, 3676–3693 CrossRef CAS PubMed; (k) Y. Xu, Y. Lin, S. L. Homölle, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2024, 146, 24105–24113 CrossRef CAS PubMed; (l) F. Panahi and B. Breit, Angew. Chem., Int. Ed., 2024, 63, e202317981 CrossRef CAS PubMed.
- (a) P. Losada, L. Goicoechea, J. L. Mascarenas and M. Gulias, ACS Catal., 2023, 13, 13994–13999 CrossRef CAS; (b) D. H. Dethe, V. Kumar and M. Shukla, Chem. Sci., 2023, 14, 11267–11272 RSC.
- S. Feng, M. Huang, J. R. Lamb, W. Zhang, R. Tatara, Y. Zhang, Y. G. Zhu, C. F. Perkinson, J. A Johnson and Y. S. Horn, Chem, 2019, 5, 2630–2641 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, reaction optimization, spectroscopic data, theoretical analysis and crystallographic information for 2347572 (Rp-3a), 2347573 (Sp-3a), and 2402043 (3f). CCDC 2347572, 2347573, and 2402043. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06867j |
| This journal is © The Royal Society of Chemistry 2025 |