
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetrical and asymmetrical surface structure expansions of silver nanoclusters with atomic precision†
Honglei
Shen‡
a,
Pu
Wang‡
b,
Jiawei
Xu
a,
Ziwei
Fu
a,
Xi
Kang
 *a,
Yong
Pei
*b and
Manzhou
Zhu
*a
*a,
Yong
Pei
*b and
Manzhou
Zhu
*a
aDepartment of Chemistry, Centre for Atomic Engineering of Advanced Materials, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Institutes of Physical Science and Information Technology, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, Anhui 230601, China. E-mail: kangxi_chem@ahu.edu.cn; zmz@ahu.edu.cn
bDepartment of Chemistry, Key Laboratory of Environmentally Friendly Chemistry and Applications of Ministry of Education, Xiangtan University, Xiangtan, Hunan 411105, P. R. China. E-mail: ypei2@xtu.edu.cn
First published on 7th January 2025
Abstract
Controlling symmetrical or asymmetrical growth has allowed a series of novel nanomaterials with prominent physicochemical properties to be produced. However, precise and continuous size growth based on a preserved template has long been a challenging pursuit, yet little has been achieved in terms of manipulation at the atomic level. Here, a correlated silver cluster series has been established, enabling atomically precise manipulation of symmetrical and asymmetrical surface structure expansions of metal nanoclusters. Specifically, the C3-axisymmetric Ag29(BDTA)12(PPh3)4 nanocluster underwent symmetrical and asymmetrical surface structure expansions via an acid-mediated synthetic procedure, giving rise to C3-axisymmetric Ag32(BDTA)12(PPh3)10 and C1-axisymmetric Ag33(BDTA)12(PPh3)11, respectively. In addition, structural transformations, including structural degradation from Ag32 to Ag29 and asymmetrical structural expansion from Ag32 to Ag33, were rationalized theoretically. More importantly, the asymmetrically structured Ag33 nanoclusters followed a chiral crystallization mode, and their crystals displayed high optical activity, derived from CD and CPL characterization. This work not only provides an important model for unlocking the symmetrical/asymmetrical size growth mechanism at the atomic level but also pioneers a promising approach to activate the optical activity of cluster-based nanomaterials.
Introduction
The surface structure expansion of nanomaterials has emerged as a versatile approach to control their morphologies, increase their structural complexity, and enhance their functionalities for downstream applications.1,2 According to the different growth orientations, the surface structure expansion follows symmetrical or asymmetrical growth patterns, laying the foundation for constructing isotropic or anisotropic nanomaterials, respectively.3 While most work on controlling nanomaterial morphology revolves around symmetrical growth, the introduction of asymmetrical factors and thus symmetry breaking has also emerged as a powerful route to enrich nanomaterials with new shapes and complex morphologies as well as unprecedented functionalities.3,4 To date, a variety of symmetry-preserved or symmetry-broken nanocrystals have been reported, together with some insights into their symmetrical or asymmetrical growth mechanisms;5–8 however, most of these mechanisms have been elucidated on the basis of microscopy, and little has been achieved in terms of understanding at the atomic level. The in-depth comprehension of the symmetrical/asymmetrical structure expansion and atomically precise control over growth orientations require precise molecular entities to serve as model nanosystems and accurate molecular tools.Metal nanoclusters are among the most promising nanomaterials for investigating structural evolutions owing to their uniform sizes and precise structures.9–15 Besides, the prominent quantum size effects and discrete electronic energy levels enable cluster-based nanomaterials with structure-dependent physicochemical properties, and the structure–property correlations are accessible at the atomic level.16–28 Several efficient strategies have been exploited to regulate the structures of metal nanoclusters, such as ligand engineering, kernel alloying, counterion regulation, intercluster reactions, etc., involving both the molecular and supramolecular chemistry of these programmable nanomaterials.29–36 Based on these strategies, several cluster cases of surface structure expansion (or size growth) have been reported, while most of these structural manipulations were non-directional and discontinuous.37–43 In addition, the success of controlling the symmetrical/asymmetrical structure expansion of clusters critically relies on the ability to retain/lift the confinement on symmetry by the underlying surface units of the molecular structures; however, achieving this control has long been a challenging pursuit. The attainment of controlled symmetrical or asymmetrical size growth of metal clusters with preserved frameworks is highly desirable for revealing the structural growth mechanism and the corresponding structure–property correlations at the atomic level.
Herein, the continuous size growth, involving both symmetrical and asymmetrical surface structure expansions, has been accomplished for metal nanoclusters with a preserved framework. The acid-mediated preparation in the one-pot silver-cluster synthetic procedure directed the generation of three correlated Ag nanoclusters (Schemes 1 and S1†): (i) the Ag29(BDTA)12(PPh3)4 nanocluster (Ag29 for short, where BDTA represents 3,5-dithiolbenzoic acid) with C3 axial symmetry, (ii) the C3-axisymmetric Ag32(BDTA)12(PPh3)10 nanocluster (Ag32 for short) exhibiting symmetrical surface structure expansion relative to Ag29, and (iii) the C1-axisymmetric Ag33(BDTA)12(PPh3)11 nanocluster (Ag33 for short) displaying asymmetrical surface structure expansion relative to Ag29 and Ag32. The continuous structural transformations were rationalized via ab initio calculations, including the structural degradation from Ag32 to Ag29 and the asymmetrical structural expansion from Ag32 to Ag33. Significantly, the structurally asymmetrical Ag33 nanocluster followed a chiral crystallization mode, and its crystals displayed high optical activity, derived from circular dichroism (CD) and circularly polarized luminescence (CPL) characterization. In vivid contrast, the structurally symmetrical Ag29 and Ag32 nanoclusters were racemic in the crystal state, encompassing equal amounts of S- and R-cluster enantiomers in the crystal lattice. Overall, the findings in this work provide impetus for future experimental and theoretical developments in the symmetrical growth or symmetry breaking of metal nanoclusters.
 | ||
| Scheme 1 Schematic illustration of the symmetrical surface expansion from Ag29 to Ag32 nanoclusters and the asymmetrical surface expansion from Ag32 to Ag33 nanoclusters. | ||
Results and discussion
The Ag29, Ag32, and Ag33 nanoclusters were prepared via a one-pot acid-mediated synthetic procedure (Scheme 2; see the ESI† for more details). Specifically, after the reduction of NaBH4, the introduction of different amounts of HCl directed the generation of structure-correlated silver nanoclusters, Ag29, Ag32, or Ag33 with an analogous molecular framework but different surface environments (Tables S1–S4†). Of note, previous studies have demonstrated the pH-dependent formation of metal nanoclusters, demonstrating the importance of an acid–base environment for nanocluster regulation.44,45 For the cluster homologue of Ag29, the Ag29(BDT)12(PPh3)4 nanocluster (Ag29-BDT for short; BDT = 1,3-benzene dithiol) might be unstable in the presence of acid, and we observed that the introduction of HCl inhibited the nanocluster preparation. By comparison, for the BDTA-stabilized silver clusters in this work, due to the existence of carboxyl groups on thiol ligands, the acid sensitivity of BDTA was transmitted to the cluster framework, which provided us an opportunity to precisely control the cluster structures by regulating the amount of HCl among the synthesis.46 In addition, the BDTA-stabilized Ag29 and Ag29-BDT presented the same cluster framework, demonstrating that the introduction of carboxyl functional groups would not significantly change the coordination ability (or the nucleophilicity) of the bidentate thiol ligand. The successful structure determination of this silver cluster series (i.e., Ag29, Ag32, and Ag33) enabled the downstream mechanism elucidation towards the symmetrical and asymmetrical surface structure expansions of metal clusters with atomic precision.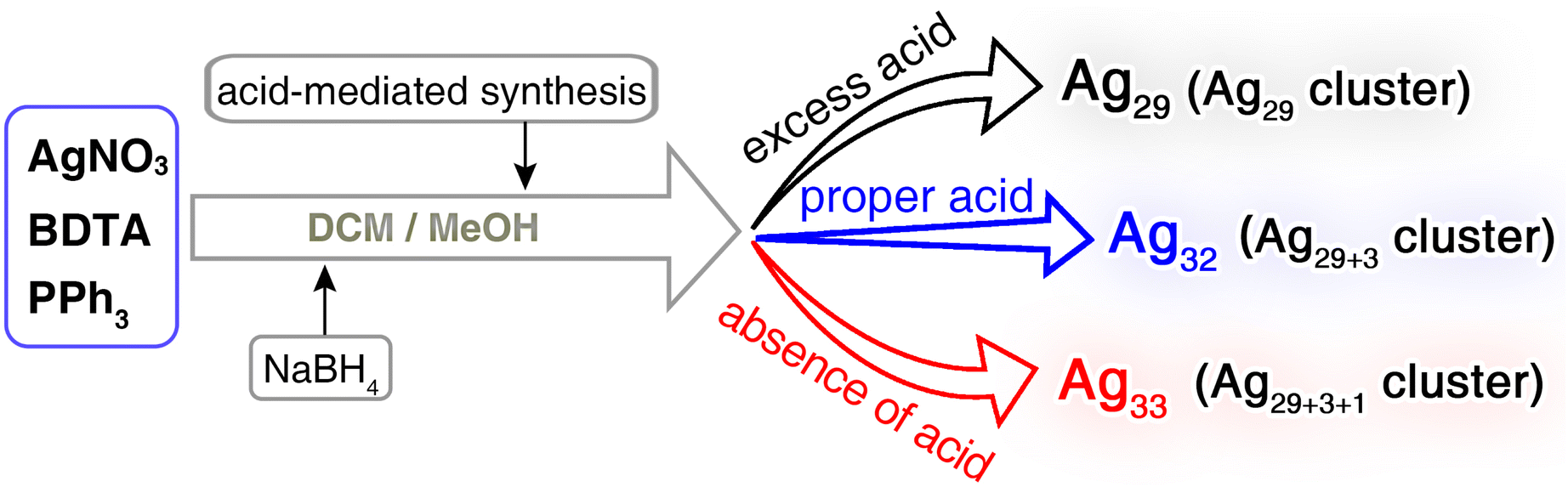 | ||
| Scheme 2 Schematic illustration of the synthetic procedure for Ag29, Ag32, and Ag33 nanoclusters via an acid-mediated synthetic procedure. | ||
The geometric structure of Ag29 was the same as that of Ag29-BDT reported previously,47 except for their different peripheral bidentate thiol ligands (Fig. S1†). Structurally, the Ag29 nanocluster contained an icosahedral Ag13 kernel that was stabilized from four Ag3(SR′)6 units via sharing the bidentate BDTA thiol ligands, where SR′ was half of the BDTA ligand (Fig. 1A–C). Then, the obtained Ag25(BDTA)12 framework was capped by four vertex Ag-PPh3 units to construct the overall structure of Ag29-BDT (Fig. 1D and E). The Ag29 exhibited a highly symmetrical overall structure with four C3 symmetry axes, passing through the vertex Ag-PPh3 unit and the innermost Ag kernel (Fig. 1F).
 | ||
| Fig. 1 Structural anatomy of Ag29, Ag32, and Ag33 nanoclusters. (A) The icosahedral Ag13 kernel. (B) The Ag12(BDTA)12 surface structure contains four Ag3(SR′)6 surface units via sharing the bidentate BDTA thiol ligands. (C) The Ag25(BDTA)12 structure. (D) The four Ag1(PPh3)1 vertex units. (E and F) The overall structure of the Ag29(BDTA)12(PPh3)4 nanocluster. (G) The three Ag1(PPh3)2 surface symmetrical expanding units. (H and I) The overall structure of the Ag32(BDTA)12(PPh3)10 nanocluster. (J) The Ag1(PPh3)1 surface asymmetrical expanding unit. (K and L) The overall structure of the Ag33(BDTA)12(PPh3)11 nanocluster. Color legends: orange/light blue/blue spheres, Ag; red/yellow spheres, S; magenta/purple/pink spheres, P; green sphere, O; grey sphere, C. All H atoms are omitted for clarity. | ||
The total structure of the Ag32 nanocluster represented the symmetrical surface structure expansion relative to the Ag29 cluster framework (Fig. S2†). Specifically, three terminal Ag1(PPh3)2 units were symmetrically arranged onto the Ag29 framework by anchoring to the Ag3(SR′)3 ring, resulting in the overall structure of Ag32(BDTA)12(PPh3)10 (Fig. 1G–I). Although the complete structure of Ag32 still followed C3 symmetry (Fig. S2†), symmetry degradation was discovered in contrast to the Ag29 nanocluster. Indeed, due to the introduction of three symmetric Ag1(PPh3)2 units on its surface, the Ag32 nanocluster only showed one C3 symmetry axis that crossed the innermost Ag kernel and a vertex Ag-PPh3 unit.
Continuous size growth from Ag32 gave rise to the generation of the Ag33 nanocluster, which exhibited asymmetrical surface structure expansion relative to the Ag32 cluster framework (Fig. S3†). As shown in Fig. 1J–L, an extra Ag1(PPh3)1 surface unit connected with an inward S atom of the Ag32(BDTA)12(PPh3)10 structure to form the overall structure of Ag33. In the meantime, the coordination sites of the three terminal Ag1(PPh3)2 units on the cluster surface altered to an uneven distribution (Fig. S3†). The combination of two asymmetric factors, i.e., the uneven distributions of (i) Ag1(PPh3)1 and (ii) Ag1(PPh3)2, contributed to the symmetry breaking of the Ag33 nanocluster, making Ag33 asymmetrical with C1 symmetry (Fig. 1L). Collectively, from the aspect of structural evolution, the Ag32/Ag33 nanoclusters could be viewed as the size-growth products of the Ag29 cluster precursor through symmetrical/asymmetrical surface structure expanding patterns. Besides, the Ag32 and Ag33 nanoclusters maintained the original Ag29 structure in terms of their consistent Ag29-containing frameworks and analogous bond lengths. Fig. S4† compares the average lengths of the Ag(icosahedral surface)–Ag(icosahedral surface), Ag(icosahedral surface)–S(shell), Ag(shell)–S(shell), and Ag(vertex)–P(vertex) bonds in Ag29-BDT, Ag29, Ag32, and R/S-Ag33 nanoclusters. The differences in the corresponding bone lengths were all less than 1%, demonstrating the structural integrity of the Ag29 framework among the symmetrical/asymmetrical surface structure expanding processes. In addition, the carboxylated surface endowed the Ag29 nanocluster with an acid-resistant characteristic, and Ag29 showed enhanced stability relative to Ag29-BDT in an acidic environment (Fig. S5†).
Electrospray ionization mass spectrometry (ESI-MS) of the Ag29 nanocluster showed an intense signal at 1779.56 Da in negative mode with characteristic isotopic peaks separated by a m/z of 0.33 Da, corresponding to the chemical formula of [Ag29(BDTA)12]3− (Fig. S6†). In this context, the vertex PPh3 ligands of Ag29 were dissociated entirely because of the high collision energy during the mass detection. The Ag29 nanocluster presented a closed-shell electronic structure with a nominal electron count of eight (i.e., 29(Ag) − 12 × 2(SR) + 3(charge) = 8), consistent with the icosahedral unit in the cluster framework.48 Given the structural consistency among these structure-correlated silver nanoclusters, the Ag32 and Ag33 nanoclusters should manifest the same nominal electron counts as eight. Consequently, the Ag32 cluster molecule would be electrically neutral, while the Ag33 cluster molecule would carry a “+1” charge. However, we failed to observe the characteristic mass signals of the two surface-expanded nanoclusters in ESI-MS (Fig. S7†). In addition, 31P NMR (Nuclear Magnetic Resonance) and elemental analyses were performed to verify the purity and demonstrate the symmetry of such nanoclusters (Fig. S8 and Table S5†).
The structural–property correlations of these structure-correlated silver nanoclusters were investigated. Fig. 2A compares the optical absorptions spectra of the four nanoclusters in the solution state. The DMF solutions of all nanoclusters, including Ag29-BDT, Ag29, Ag32, and Ag33, exhibited almost the same optical absorption spectra with an intense peak at 440 nm and several shoulder bands at 320, 364, and 515 nm (Fig. 2A). Indeed, although Ag29, Ag32, and Ag33 nanoclusters exhibited comparable surface structures, their intrinsic kernel structures were identical as Ag29(BDTA)12(PPh3)4. Such similar absorption spectra demonstrated that the electronic orbits of these silver nanoclusters were primarily constituted by the innermost Ag29 framework, while they were less relevant to the surface Ag-PPh3 stabilizers. The photoluminescence (PL) of Ag29-BDT, Ag29, Ag32, and Ag33 nanoclusters was then measured in the solution state. The normalized PL spectra suggested that both Ag29-BDT and Ag29 emitted at 672 nm; by comparison, the emission of Ag32 red-shifted to 682 nm, while that of Ag33 blue-shifted to 662 nm (Fig. 2B, inset). Under 440 nm excitation, the three BDTA-stabilized nanoclusters showed remarkable PL enhancement relative to Ag29-BDT. Specifically, the Ag32 nanocluster showed a ∼27-fold enhancement in emission intensity compared to Ag29-BDT (Fig. 2B and C), and the PL intensity sequence was Ag32 (PL QY of 25.5%) > Ag29 (PL QY of 5.8%) > Ag33 (PL QY of 3.8%) > Ag29-BDT (PL QY of 0.9%).
 | ||
| Fig. 2 Optical properties of Ag29-BDT, Ag29, Ag32, and Ag33 nanoclusters. (A) Optical absorption spectra of the four nanoclusters in their DMF solutions. (B) Photoluminescence emissions of the four nanoclusters in their DMF solutions. Inset: comparison of the normalized emission wavelengths. (C) Photos of the photoluminescence of the four nanoclusters in DMF. Inset line: changing trends of the PL QY. (D) Optical absorption spectra of the four nanocluster crystals. (E) Normalized photoluminescence emissions of the four nanoclusters in their crystals. (F) Photos of the photoluminescence of the four nanocluster crystals. Inset line: changing trends of the emission wavelength. | ||
The significant PL enhancement of BDTA-stabilized nanoclusters might originate from their intramolecular C–H⋯π and π⋯π⋯π interactions (Fig. S9†), which remarkably restrained the vibrations/rotations of cluster molecules.46 Besides, the introduced Ag-PPh3 surface units in Ag32 and Ag33 further restrained the molecular vibrations/rotations. Accordingly, the energy dissipation of the photo-excited clusters through the radiative transition increased, accounting for the emission enhancement of the BDTA-stabilized silver nanoclusters. Of note, the Ag33 nanocluster with asymmetrical surface structures might exhibit additional intracluster vibrations from the Ag1(PPh3)1 surface asymmetrical expanding unit, resulting in attenuated PL intensity relative to the Ag32 nanocluster. In addition, due to the different intracluster interactions, the energy dissipation pathway of these excited cluster molecules might be affected, and thus their emission wavelengths were slightly different.
The single crystal-based crystals of the four structure-correlated silver nanoclusters exhibited more apparent differences in optical properties (see the ESI† for more details on the preparation of such crystals). The crystals of Ag29-BDT displayed comparable optical absorption spectra to its DMF solution. However, for the three BDTA-stabilized silver nanoclusters, absorption peaks at 532 and 655 nm were observed for their crystals (Fig. 2D), which were optically salient in the corresponding DMF solutions. Besides, the crystals of Ag29-BDT showed an intense emission at 720 nm, while obvious red-shifts were detected for the emissions of BDTA-stabilized nanoclusters (Fig. 2E). In particular, the brightest Ag32 cluster crystals emitted at 790 nm, representing a 70 nm red-shift relative to Ag29-BDT (Fig. 2F). The conspicuous differences in optical absorption (i.e., 655 nm) and emissions of these structure-correlated silver nanoclusters in different forms (crystal and solution) arose from their distinct combinations of the electronic coupling and the lattice-origin, non-radiative decay pathways occurring through electron–phonon interactions.49–51 Indeed, in the solution state, these nanoclusters are more like independent individuals, which are uniformly dispersed in the system. When the cluster molecules spontaneously crystallize, they are fixed within the crystal lattice with intermolecular interactions. This partially affects the electronic structure of the clusters, which in turn influences the photophysical properties of nanoclusters. Besides, the presence of rich carboxyl functional groups might trigger such electron–phonon interactions in cluster crystalline lattices. In addition, these differences between the surface inactive Ag29-BDT and surface-carboxylated Ag29, Ag32, and Ag33 could also be explained in terms of their diverse surface chemistry as well as intra-/inter-molecular interactions.52
Theoretical efforts were made to rationalize the structural transformations among these structure-correlated silver nanoclusters. Considering that the Ag32 nanocluster underwent symmetrical structural degradation to generate Ag29 in an acidic environment (Fig. S10†), we theoretically investigated the corresponding structural transformation mechanism. Here, we simulated the detachment mechanism of an Ag(PPh3)2 group from the Ag32 nanocluster as an example to reveal the structural degradation process. As shown in Fig. 3A and S11,† the H proton reacted with the S site of the Ag3(SR′)3 ring to form the most stable Ag32P10H–C intermediate (marked as Path 3), and the reaction energy was −10.44 eV. This process was beneficial to weaken the interaction between the Ag(PPh3)2 group and the surface Ag3(SR′)3 ring, resulting in an increase of the Ag–S bond length from 2.51 to 2.63 Å. Although another two processes could also effectively weaken the Ag–S bond when the H proton attacked the surface Ag site (marked as Path 1 and Path 2; Fig. S11†), the energies of the formed Ag32P10H+ intermediates (A and B isomers) were 1.14 and 0.90 eV higher than that of the Ag32P10H–C+ isomer, respectively. These results indicated that the H proton was more likely to attack the S site of the Ag3(SR′)3 ring rather than the Ag site of the Ag(PPh3)2 group at the beginning of the degradation process of Ag32. Then, a large number of H protons would continuously attack the Ag site of the Ag(PPh3)2 group, gradually weakening the Ag–S bond until it broke. The DFT calculation results showed that the Ag–S bond length increased from 2.63 Å to 2.88 Å and finally to 3.06 Å under the condition of H protons continuously attacking surface Ag sites. At this time, the Ag(PPh3)2 group has completely detached from the surface of the Ag32 nanocluster, resulting in the formation of the Ag31 cluster. Similarly, the other two Ag(PPh3)2 groups would also separate from the surface of Ag31 with the same detaching mechanism, completing the transformation from the Ag32 to the Ag29 nanocluster.
 | ||
| Fig. 3 Theoretical investigation of the corresponding cluster structural transformations. (A) The intermediate structures during the degradation process of the Ag32 nanocluster under acidic conditions. (B) The proposed two-step transformation of the asymmetrical structural expansion from Ag32 to Ag33+-Exp. | ||
Besides, the asymmetrical structural expansion from Ag32 to Ag33 was simulated. Due to the uneven distributions of the three terminal Ag(PPh3)2 coordination on Ag32 and Ag33 cluster surfaces, we proposed that the structural expansion from Ag32 to Ag33 followed a two-step process (Fig. 3B): (step 1) the isomerization process: the intramolecular transfer of three Ag(PPh3)2 groups on the Ag32 cluster surface from a uniform to a non-uniform distribution induced cluster isomerization, resulting in the formation of an Ag32 isomer; (step 2) the electrophilic process: the reactant Ag(PPh3)1+ species attacked the electrophilic region of the Ag32 isomer and aggregated to form the Ag33 nanocluster. Similarly, we performed the simulation of the migration process of an Ag(PPh3)2 group on the surface as an example to reveal the isomerization transformation of the Ag32 nanocluster. As shown in Fig. S12,† the migration of an Ag(PPh3)2 group of the Ag32 nanocluster from site 1c to 1a was a weakly endothermic process (ΔE = 0.14 eV), and the two transition-state energy barriers during the reaction were 1.29 and 0.42 eV, respectively. In the electrophilic process, the reaction sites of the Ag32 cluster isomer were predicted using an orbital weight dual descriptor (Δf) derived from Fukui functions.53 If Δf(r) > 0, the site was favored for a nucleophilic attack. By comparison, if Δf(r) < 0, the site may be favored for an electrophilic attack. The Ag1(PPh3)1+ reactant received electrons and was characterized by high electrophilicity in this process. By analyzing the orbital dual descriptor of the Ag32 nanocluster, only three surface S sites (1b, 1c, and 3c) have negative Δf values (Fig. S13†), indicating that these sites could donate electrons and were susceptible to electrophilic attacks. Subsequently, we further compared the stability of the three Ag33 isomers. Although the Ag33-1c isomer has the lowest energy, the surface Ag3(SR′)3 structure deformed or even collapsed, as depicted in Fig. S14.† The theoretical structure of the Ag33-1b isomer with a lower energy was consistent with the experimental crystal structure.
Structurally, Ag29 and its derivative Ag32 or Ag33 nanoclusters comprised the uniform Ag29 framework. However, the symmetrical and asymmetrical surface structure expansions endowed the Ag32 and Ag33 nanoclusters with disparate surface environments, which hopefully affected their supramolecular packing and physicochemical properties. The crystal lattices of these structure-correlated silver nanoclusters were compared (Fig. S15†). For the Ag29 nanocluster, the R-Ag29 and S-Ag29 cluster enantiomers coexisted in the crystal lattice in equal amounts, giving rise to a racemic Ag29 crystal (Fig. 4A and S16†). Of note, for a pair of Ag29 enantiomers (Fig. 4A), the peripheral fan-shaped Ag4S6 could be considered as clockwise (the orange one) and anti-clockwise (the blue one) subunits on the nanocluster surface. For differentiating the two cluster enantiomers, the clockwise-packed Ag29 nanocluster was referred to as R-Ag29, while the anti-clockwise-packed one was called S-Ag29. The terms R- and S- of other nanoclusters in this work were determined in the same way. The same racemic packing phenomenon was observed in the Ag32 crystal lattice (Fig. 4B and S17†). Consequently, although the Ag29 and Ag32 crystals presented strong PL intensities, no optical activity of the crystals was detected. In vivid contrast, the surface unsymmetric Ag33 nanocluster entities followed a chiral self-assembled pattern among the crystallization and the R-Ag33 or S-Ag33 cluster enantiomers crystallized in each crystal lattice separately (Fig. 4C, D and S18†), which was reminiscent of the tartaric acid. In contrast, Ag33 and other cluster samples exhibited quenched optical activity in the solution state (Fig. S19†). Accordingly, the chiral characteristic only appeared in the crystal state of the Ag33 nanocluster, and such nanoclusters underwent racemization during the dissolution process. In previous studies, the precise control over the transformation of nanocluster structures has been accomplished by adjusting the concentrations of ligands or metal sources, thereby allowing for the preparation of nanoclusters with specific optical activities.54,55 Herein, the symmetric Ag29 and Ag32 and asymmetric Ag33 nanoclusters formed a structure-correlated cluster series, allowing for precise correlations between cluster structures and photophysical properties.
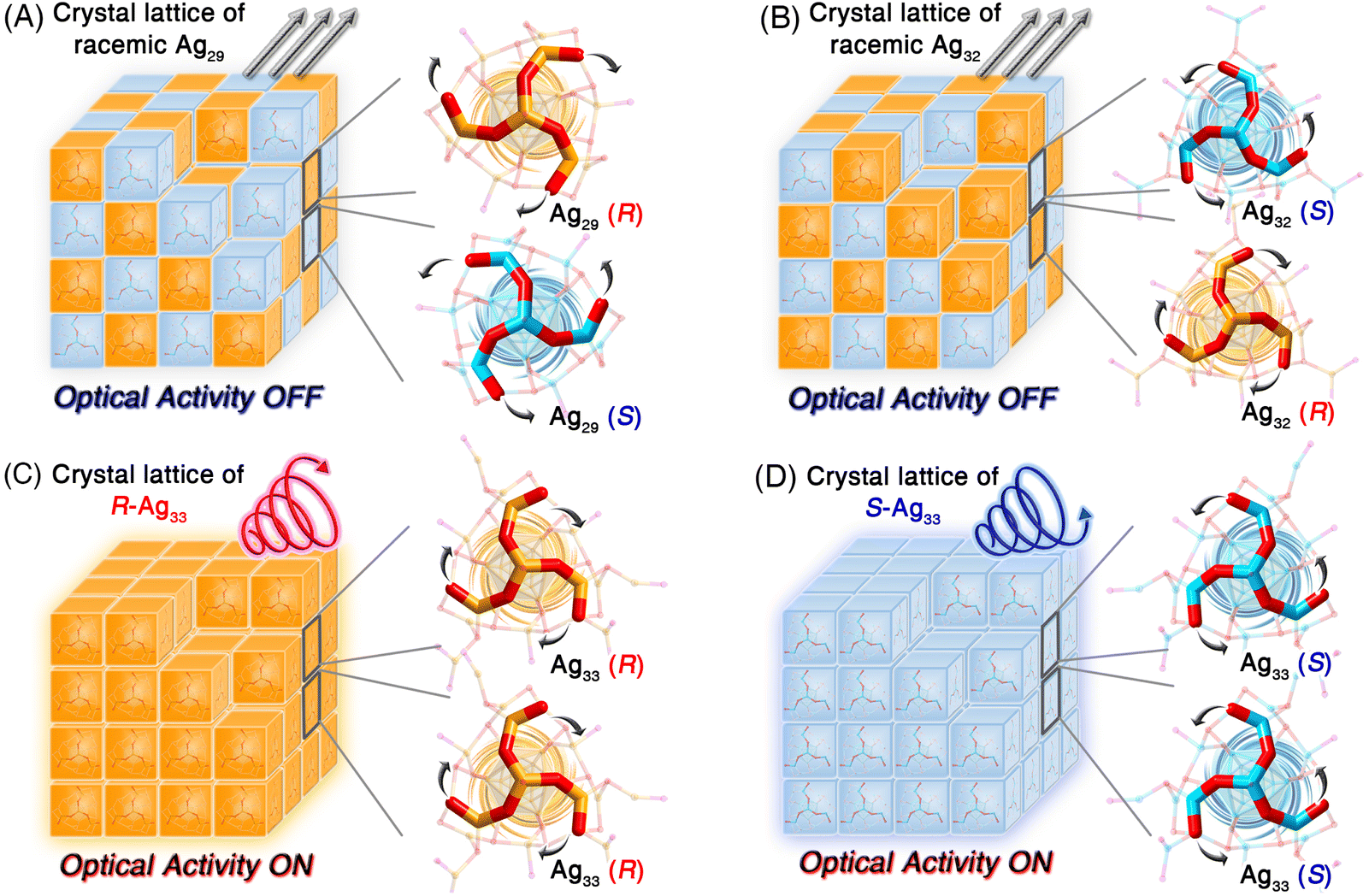 | ||
| Fig. 4 Crystalline packing of Ag29, Ag32, and Ag33 nanoclusters in their crystal lattices. (A) Crystal lattice of the racemic Ag29 nanocluster with no optical activity. (B) Crystal lattice of the racemic Ag32 nanocluster with no optical activity. (C) Crystal lattice of the chiral Ag33 nanocluster (R enantiomer) with optical activity. (D) Crystal lattice of the chiral Ag33 nanocluster (S enantiomer) with optical activity. | ||
The molecule-level and supramolecular-level asymmetric factors (i.e., the unsymmetrical surface expanding and the chiral crystallography packing) exhibited great potential in rendering the Ag33 crystals optically active. In detail, we have collected the Ag33 nanocluster crystals with R- or S-crystalline packing patterns and investigated their optical activity. Specifically, all R-type (or S-type) Ag33 single crystals showed similar CD and CPL signals with comparable peak shapes, while the Ag33 single crystals with different crystalline aggregation types (i.e., R- or S-type) displayed opposite CD and CPL signals, further demonstrating that the optical activity of the Ag33 nanocluster originated from their crystals with a “crystallization-induced spontaneous enantiomer separation” behaviour (Scheme S1†). As shown in Fig. 5A and B, the R-Ag33 and S-Ag33 crystals displayed intense signals at about 340 and 520 nm in CD spectra. Of note, the Ag33 crystalline film exhibits three optical absorption peaks at 350, 532, and 655 nm (Fig. 2D). The absorption peaks at 350 and 532 nm originated from the intrinsic electronic structure of the Ag33 molecule, which were reflected in the CD spectrum. However, the absorption peak at 655 nm of the Ag33 cluster crystal might arise from the distinct combinations of the electronic coupling and the lattice-origin, non-radiative decay pathways occurring through electron–phonon interactions, which might be the possible reason why the 655 nm signal was not reflected in the CD spectrum. Besides, the CPL spectra of R-Ag33 and S-Ag33 crystals showed mirror signals at about 760 nm, demonstrating the apparent optical activity of the Ag33 crystals (Fig. 5C and D). In addition, the crystals of Ag33 nanoclusters showed consistent optical activity by analyzing the CPL results of different points/sides of the crystals or microcrystals (Fig. S20†), demonstrating that the optical activity originated from the nanocluster crystals themselves, i.e., the chiral crystalline packing of the Ag33 nanoclusters in the crystal lattice. In contrast, the Ag29-BDT, Ag29, and Ag32 crystals were optically inactive with no signals in CD and CPL characterization.
 | ||
| Fig. 5 Optical activity test of cluster crystals. (A) Circular dichroism spectra of the four nanoclusters in the crystal state. (B) g factors of the CD results of different cluster crystals. (C) Circularly polarized luminescence spectra of the four nanoclusters in the crystal state. (D) Glum of the CPL results of different cluster crystals. | ||
Conclusion
In summary, the continuous size growth of metal nanoclusters has been accomplished within a preserved framework, including symmetrical surface structure expansion from Ag29 to Ag32 and asymmetrical expansion from Ag32 to Ag33, allowing for nanocluster growth to be meticulously dictated with atomic precision. The symmetrical degradation and the asymmetrical expansion for transforming the geometric structures of nanoclusters were rationalized via ab initio calculations. Besides, the structure-dependent optical properties, including absorption and emission characteristics, of these correlated silver nanoclusters were investigated. More importantly, the molecule-level and supramolecular-level asymmetric factors (i.e., the unsymmetrical surface expanding and the chiral crystallography packing) of the Ag33 nanocluster rendered its crystals highly optically active, derived from CD and CPL characterization. This work presented an important structure-correlated cluster series with controllable symmetrical/asymmetrical surface environments, allowing for some new insights into the cluster structure evolutions.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
H. Shen conceived and carried out experiments. P. Wang performed the theoretical simulation. J. Xu and Z. Fu assisted in the synthesis and optical spectral measurements. X. Kang analyzed the data and wrote the paper and Y. Pei and M. Zhu supervised the project. All authors commented on and agreed on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the financial support of the NSFC (22371003, 22101001, and 22471001), the Ministry of Education, the Natural Science Foundation of Anhui Province (2408085Y006), the University Synergy Innovation Program of Anhui Province (GXXT-2020-053), and the Scientific Research Program of Universities in Anhui Province (2022AH030009). Y. P. acknowledges financial support from the NSFC (22373082). P. W. acknowledges financial support from the Scientific Research Fund of Hunan Provincial Education Department (22B0154).Notes and references
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS.
- Q. N. Nguyen, C. Wang, Y. Shang, A. Janssen and Y. Xia, Chem. Rev., 2023, 123, 3693–3760 CrossRef CAS PubMed.
- M. J. Walsh, W. Tong, H. Katz-Boon, P. Mulvaney, J. Etheridge and A. M. Funston, Acc. Chem. Res., 2017, 50, 2925–2935 CrossRef CAS PubMed.
- S. E. Skrabalak, Acc. Mater. Res., 2021, 2, 621–629 CrossRef CAS.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- C. J. DeSantis and S. E. Skrabalak, J. Am. Chem. Soc., 2013, 135, 10–13 CrossRef CAS.
- J. D. Smith, E. Bladt, J. A. C. Burkhart, N. Winckelmans, K. M. Koczkur, H. M. Ashberry, S. Bals and S. E. Skrabalak, Angew. Chem., Int. Ed., 2020, 59, 943–950 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- M. F. Matus and H. Häkkinen, Nat. Rev. Mater., 2023, 8, 372–389 CrossRef CAS.
- S. Kenzler and A. Schnepf, Chem. Sci., 2021, 12, 3116–3129 RSC.
- W. Jing, H. Shen, R. Qin, Q. Wu, K. Liu and N. Zheng, Chem. Rev., 2023, 123, 5948–6002 CrossRef CAS PubMed.
- Z. Wang, Y.-J. Zhu, Y.-Z. Li, G.-L. Zhuang, K.-P. Song, Z.-Y. Gao, J.-M. Dou, M. Kurmoo, C.-H. Tung and D. Sun, Nat. Commun., 2022, 13, 1802 CrossRef CAS PubMed.
- E. L. Albright, T. I. Levchenko, V. K. Kulkarni, A. I. Sullivan, J. F. DeJesus, S. Malola, S. Takano, M. Nambo, K. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2024, 146, 5759–5780 CrossRef CAS PubMed.
- R.-W. Huang, X. Song, S. Chen, J. Yin, P. Maity, J. Wang, B. Shao, H. Zhu, C. Dong, P. Yuan, T. Ahmad, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2023, 145, 13816–13827 CrossRef CAS.
- L. Fang, W. Fan, G. Bian, R. Wang, Q. You, W. Gu, N. Xia, L. Liao, J. Li, H. Deng, N. Yan and Z. Wu, Angew. Chem., Int. Ed., 2023, 62, e202305604 CrossRef CAS.
- S.-S. Zhang, S. Havenridge, C. Zhang, Z. Wang, L. Feng, Z.-Y. Gao, C. M. Aikens, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2022, 144, 18305–18314 CrossRef CAS.
- Z. Wang, Y. Wang, C. Zhang, Y.-J. Zhu, K.-P. Song, C. M. Aikens, C.-H. Tung and D. Sun, Natl. Sci. Rev., 2024, 11, nwae192 CrossRef.
- S.-S. Zhang, R.-C. Liu, X.-C. Zhang, L. Feng, Q.-W. Xue, Z.-Y. Gao, C.-H. Tung and D. Sun, Sci. China:Chem., 2021, 64, 2118–2124 CrossRef CAS.
- R.-W. Huang, Y.-S. Wei, X.-Y. Dong, X.-H. Wu, C.-X. Du, S.-Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- X. Zhang and H. Xu, Angew. Chem., Int. Ed., 2024, 63, e202317597 CrossRef CAS PubMed.
- C. A. Hosier and C. J. Ackerson, J. Am. Chem. Soc., 2018, 141, 309–314 CrossRef.
- J. Zhao, A. Ziarati, A. Rosspeintner and T. Bürgi, Angew. Chem., Int. Ed., 2024, 63, e202316649 CrossRef CAS.
- L.-J. Liu, F. Alkan, S. Zhuang, D. Liu, T. Nawaz, J. Guo, X. Luo and J. He, Nat. Commun., 2023, 14, 2397 CrossRef CAS PubMed.
- K.-Y. Huang, Z.-Q. Yang, M.-R. Yang, T.-S. Chen, S. Tang, W.-M. Sun, Q. Yao, H.-H. Deng, W. Chen and J. Xie, J. Am. Chem. Soc., 2024, 146, 8706–8715 CrossRef CAS PubMed.
- C.-Y. Liu, S.-F. Yuan, S. Wang, Z.-J. Guan, D.-e. Jiang and Q.-M. Wang, Nat. Commun., 2022, 13, 2082 CrossRef CAS.
- Y. Horita, S. Hossain, M. Ishimi, P. Zhao, M. Sera, T. Kawawaki, S. Takano, Y. Niihori, T. Nakamura, T. Tsukuda, M. Ehara and Y. Negishi, J. Am. Chem. Soc., 2023, 145, 23533–23540 CrossRef CAS.
- A. Baksi, E. K. Schneider, P. Weis, I. Chakraborty, O. Fuhr, S. Lebedkin, W. J. Parak and M. M. Kappes, ACS Nano, 2020, 14, 15064–15070 CrossRef CAS PubMed.
- G. Soldan, M. A. Aljuhani, M. S. Bootharaju, L. G. AbdulHalim, M. R. Parida, A.-H. Emwas, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 5749–5753 CrossRef CAS PubMed.
- Y. Zeng, S. Havenridge, M. Gharib, A. Baksi, K. L. D. M. Weerawardene, A. R. Ziefuß, C. Strelow, C. Rehbock, A. Mews, S. Barcikowski, M. M. Kappes, W. J. Parak, C. M. Aikens and I. Chakraborty, J. Am. Chem. Soc., 2021, 143, 9405–9414 CrossRef CAS PubMed.
- H. Seong, Y. Jo, V. Efremov, Y. Kim, S. Park, S. M. Han, K. Chang, J. Park, W. Choi, W. Kim, C. H. Choi, J. S. Yoo and D. Lee, J. Am. Chem. Soc., 2023, 145, 2152–2160 CrossRef CAS.
- A. Chakraborty, A. C. Fernandez, A. Som, B. Mondal, G. Natarajan, G. Paramasivam, T. Lahtinen, H. Häkkinen and T. Pradeep, Angew. Chem., Int. Ed., 2018, 57, 6522–6526 CrossRef CAS.
- X. Wang, J. Zhao, H. Eliasson, R. Erni, A. S. McKeown Walker and T. Bürgi, J. Am. Chem. Soc., 2023, 145, 27273–27281 CrossRef CAS PubMed.
- R.-Q. Chen, S.-T. Wang, Y.-J. Liu, J. Zhang and W.-H. Fang, J. Am. Chem. Soc., 2024, 146, 7524–7532 CrossRef CAS.
- Q. You, H. Wang, Y. Zhao, W. Fan, W. Gu, H.-L. Jiang and Z. Wu, J. Am. Chem. Soc., 2024, 146, 9026–9035 CrossRef CAS.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS.
- X. Zou, X. Kang and M. Zhu, Chem. Soc. Rev., 2023, 52, 5892–5967 RSC.
- W. Ishii, Y. Okayasu, Y. Kobayashi, R. Tanaka, S. Katao, Y. Nishikawa, T. Kawai and T. Nakashima, J. Am. Chem. Soc., 2023, 145, 11236–11244 CrossRef CAS.
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS PubMed.
- W. W. Xu, Y. Li, Y. Gao and X. C. Zeng, Nanoscale, 2016, 8, 7396–7401 RSC.
- M. S. Bootharaju, C. P. Joshi, M. J. Alhilaly and O. M. Bakr, Chem. Mater., 2016, 28, 3292–3297 CrossRef CAS.
- X. Wei, H. Li, Z. Zuo, F. Song, X. Kang and M. Zhu, J. Am. Chem. Soc., 2023, 145, 13750–13757 CrossRef CAS.
- L. G. AbdulHalim, Z. Hooshmand, M. R. Parida, S. M. Aly, D. Le, X. Zhang, T. S Rahman, M. Pelton, Y. Losovyj, P. A. Dowben, O. M. Bakr, O. F. Mohammed and K. Katsiev, Inorg. Chem., 2016, 55, 11522–11528 CrossRef CAS.
- M. Waszkielewicz, J. Olesiak-Banska, C. Comby-Zerbino, F. Bertorelle, X. Dagany, A. K. Bansal, M. T. Sajjad, I. D. W. Samuel, Z. Sanader, M. Rozycka, M. Wojtas, K. Matczyszyn, V. Bonacic-Koutecky, R. Antoine, A. Ozyhard and M. Samoc, Nanoscale, 2018, 11335–11341 RSC.
- H. Shen, J. Xu, Z. Fu, X. Wei, X. Kang, W. Shi and M. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202317995 CrossRef CAS PubMed.
- L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. Del Gobbo, R. G. AbdulHalim, M. Eddaoudi, D.-e. Jiang and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11970–11975 CrossRef CAS PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Angew. Chem., Int. Ed., 2008, 105, 9157–9162 CAS.
- X.-Y. Dong, H.-L. Huang, J.-Y. Wang, H.-Y. Li and S.-Q. Zang, Chem. Mater., 2018, 30, 2160–2167 CrossRef CAS.
- H. Döllefeld, H. Weller, A. Eychmüller and J. Phy, Chem. B., 2002, 106, 5604–5608 CrossRef.
- J. Zhang, C. Rowland, Y. Liu, H. Xiong, S. Kwon, E. Shevchenko, R. D. Schaller, V. B. Prakapenka, S. Tkachev and T. Rajh, J. Am. Chem. Soc., 2015, 137, 742–749 CrossRef CAS PubMed.
- X. Wei, X. Kang, Z. Zuo, F. Song, S. Wang and M. Zhu, Nat. Sci. Rev., 2021, 8, nwaa077 CrossRef CAS.
- R. Pino-Rios, O. Yañez, D. Inostroza, L. Ruiz, C. Cardenas, P. Fuentealba and W. Tiznado, Angew. Chem., Int. Ed., 2017, 38, 481–488 CAS.
- C. Zhang, W.-D. Si, Z. Wang, C.-H. Tung and D. Sun, Angew. Chem., Int. Ed., 2024, 63, e202404545 CrossRef CAS.
- X.-L. Peng, S. Huang, Y.-J. Zhao, X.-M. Luo, H.-Y. Li, J.-H. Hu, J.-H. Huang and S.-Q. Zang, Sci. Bull., 2024 DOI:10.1016/j.scib.2024.09.047.
Footnotes |
| † Electronic supplementary information (ESI) available: Scheme S1, Fig. S1–S20 and Tables S1–S5. CCDC 2243513, 2243517, 2243519 and 2243542. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06847e |
| ‡ H. S. and P. W. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |