
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLate-stage installation and functionalization of alkyl pyridiniums: a general HTE amenable strategy to access diverse aryl alanine containing macrocyclic peptides†
Ahmet
Kekec
*,
Lauren My-Linh
Tran
 ,
Christopher W.
Plummer
and
Dipannita
Kalyani
*
,
Christopher W.
Plummer
and
Dipannita
Kalyani
*
Discovery Chemistry, Merck & Co., Inc., Rahway, New Jersey 07065, USA. E-mail: dipannita.kalyani@merck.com; ahmet_kekec@merck.com
First published on 6th January 2025
Abstract
This manuscript describes a strategy to readily access diverse aryl and homoaryl alanine-containing pharmaceutically relevant macrocyclic peptides. A two-step sequence involving the late-stage installation of the pyridinium functionality on macrocyclic peptides followed by reductive couplings was implemented. These transformations are amenable to microscale high-throughput experimentation (HTE) and enable rapid access to aryl alanine-containing macrocyclic peptides that would otherwise be inaccessible via solid-phase peptide synthesis using commercially available amino acids. Numerous aryl and heteroaryl derivatives can be effectively used in these reactions. In addition, a systematic investigation was undertaken using an “informer” set of macrocyclic peptides which revealed the compatibility of the late-stage diversification with peptides containing diverse side chain functionalities.
Introduction
Over the past two decades, the number of peptide drug approvals has steadily increased.1 This is because peptides enable access to previously undruggable targets by providing exquisite potency and selectivity akin to that of biologics while presenting opportunities for tunability of molecular properties similar to that of small molecules.2 The development of peptide drugs often requires the incorporation of non-canonical amino acids (ncAAs) to enhance cell permeability, oral bioavailability, and protease stability. In fact, several peptide drug therapeutics, including Signifor, MK-0616, and LUNA-18 contain ncAAs (Fig. 1A).3 In particular, aryl alanines are a privileged class of ncAAs that are prevalent in pharmaceuticals due to their ease of incorporation through solid-phase peptide synthesis (SPPS) or genetic code expansion techniques.4 Hence, general methods for efficient incorporation of structurally diverse aryl and heteroaryl alanines into complex peptide scaffolds are highly desirable.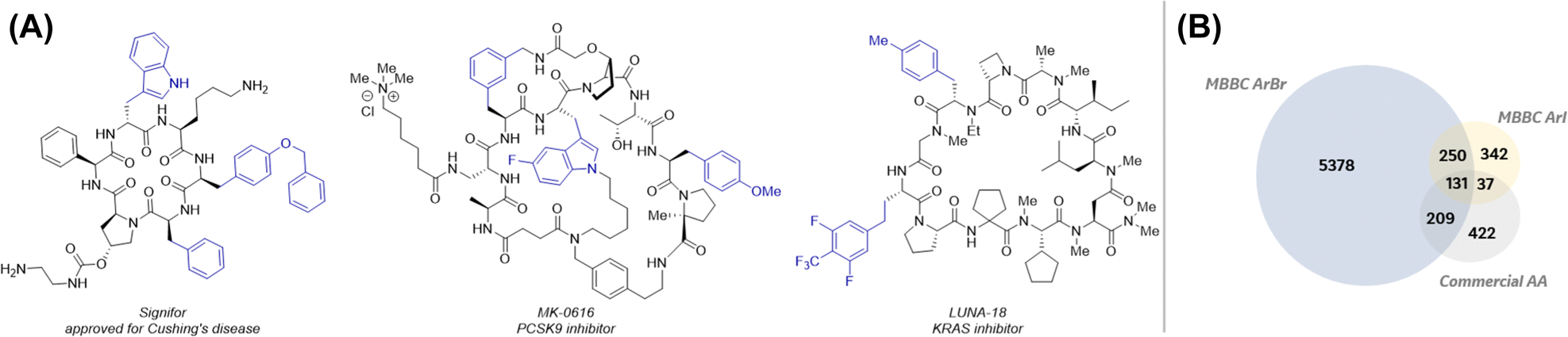 | ||
| Fig. 1 (A) Aryl alanine containing macrocyclic peptide. (B) Comparison of aryl alanine diversity via LSF using ArBr/Arl vs. commercial Fmoc protected aryl alanine amino acid. | ||
The most widely used method to incorporate aryl alanines into peptides is using Fmoc-protected building blocks via SPPS. Notwithstanding the advantages of SPPS, including its amenability with state-of-the-art automated peptide synthesizers, it is limited by the availability of Fmoc-protected amino acids. Furthermore, SPPS to generate peptide analogs bearing a single variable mutation requires the redundant repetitive synthesis of the entire sequence thereby requiring significant quantities of expensive ncAAs. A more attractive approach to aryl alanine-containing peptides would entail the late-stage functionalization (LSF) of peptides5 from a common intermediate using a diverse, large, and readily available pool of aryl halide building blocks. As illustrated in Fig. 1B, >5000 aryl alanines can be uniquely accessed using aryl bromides and iodides from the MSD Building Block Collection (MBBC) that are otherwise commercially unavailable as Fmoc-protected amino acid building blocks. Despite this enormous potential to access a diverse chemical space, sparse examples exist for the robust generation of diverse aryl alanine-containing complex peptides via late-stage arylations using aryl halides.6 Notable examples include the solid-phase decarboxylative arylation of NHP esters embedded in relatively complex long linear peptides (Scheme 1A).6f While this approach represents a significant advance, published examples are largely limited to the use of electron-neutral and electron-deficient aryl bromides and no examples with heteroaryl halides were disclosed. Complementary to this strategy using NHP esters, recently, a collaboration between Merck & Co., Inc., Rahway, NJ, USA and the Watson group at the University of Delaware led to the disclosure of a late-stage arylation of pyridinium containing pharmaceutically relevant macrocyclic peptides (MPs) (Scheme 1B, Strategy A).7b This method was suitable for the synthesis of homologated aryl alanine containing MPs. Key to the success of this method was the stability of the pyridinium moiety during SPPS to enable ready access to the key LSF substrate B. However, this approach was not amenable to the synthesis of aryl alanine-containing MPs largely due to the instability of the corresponding 2,3-diaminopropionic acid (Dap) derived pyridiniums (Scheme 1B, Strategy A, n = 1) during SPPS. Specifically, alkene byproducts resulting from the elimination of the pyridinium group were observed under SPPS Fmoc deprotection conditions. To circumvent this challenge, we envisioned the late-stage introduction of the pyridinium functionality via condensation of the pyrilium salt with a free amine in MP substrates such as C (Scheme 1B, Strategy B). Subsequent reductive couplings of pyridinium-containing MPs with aryl halides would afford the desired MP analogs (E) bearing diverse aryl alanine motifs. While there is no report of late-stage incorporation of pyridiniums in Dap-containing MPs (e.g., C, n = 1), there is a report for the condensation of lysine-containing linear peptides with pyrilium on solid phase to afford the corresponding pyridinium-containing linear peptides (Scheme 1C).8 This report coupled with the generality of the previously reported reductive couplings of pyridinium-containing amino acids, dipeptides, and tripeptides7b lend feasibility to the proposed two-step strategy B in Scheme 1B. Herein, we describe the successful implementation of this two-step strategy representing the first general method for the rapid generation of pharmaceutically relevant complex macrocyclic peptides bearing structurally diverse aryl alanines. Notably, microscale high-throughput experimentation (HTE)9 was imperative for both the rapid optimization of reaction parameters and investigation of the reductive coupling substrate scope.
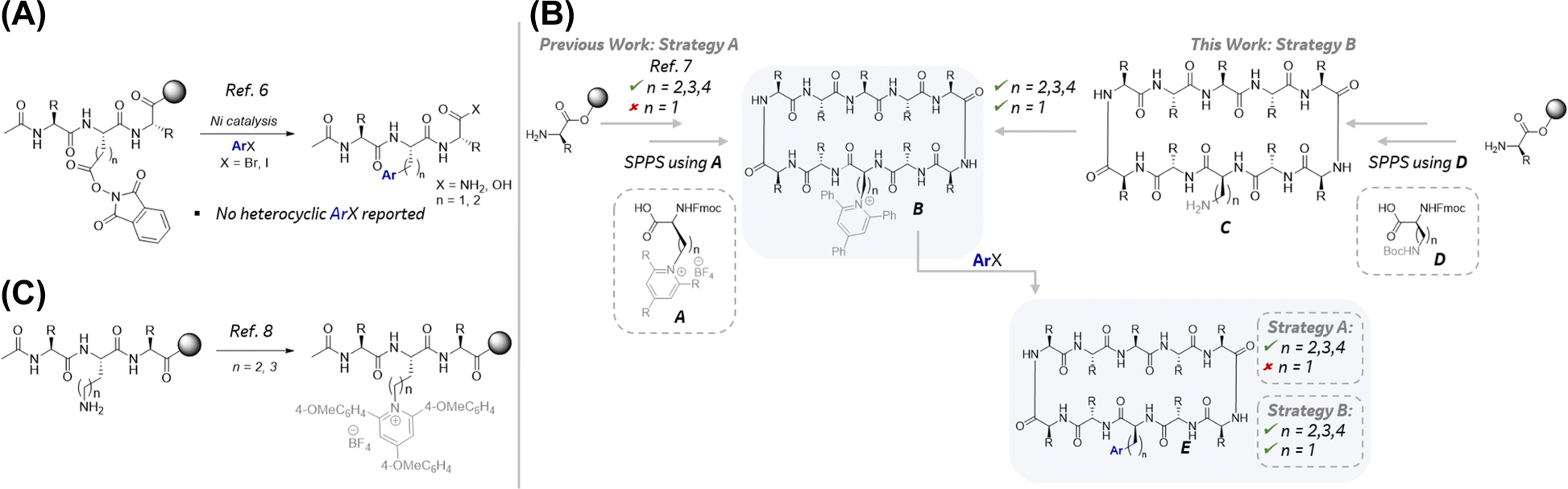 | ||
| Scheme 1 (A) Cross coupling with NHP esters on solid phase. (B) Key innovation. (C) Late stage pyrinidium formation on solid phase bound linear peptides. | ||
Results and discussion
We began our investigations with the synthesis of Gramicidin S-derived macrocyclic peptide pyridinium-containing substrates 1a–1c to evaluate the influence of electronics of the pyridinium moiety on the subsequent reductive cross-couplings (Schemes 2A and 2B). Gramicidin-S analogs are known antibacterial peptides10 and were selected for these studies because: (1) they adopt a β-sheet secondary structure11 and contain β-turns which are prevalent in many peptide hits obtained from mRNA display screening platforms,12 (2) these peptides are comprised of canonical amino acids which are inexpensive and readily accessible, and (3) they contain aryl alanine residues thereby presenting an opportunity to apply the proposed late-stage pyridinium incorporation and reductive coupling (Fig. 1C, Strategy B) in pharmaceutically relevant MPs.13 The desired MPs 1a–1c were readily prepared following the synthetic sequence depicted in Scheme 2A. SPPS followed by resin cleavage, cyclization, and Boc deprotection afforded intermediate INT-1. Gratifyingly, the late-stage condensation of INT-1 with electronically differentiated pyriliums using microwave irridation resulted in the desired pyridinium-containing MPs 1a–1c. The overall yield of 1a–1c over 24-steps starting from SPPS ranged from 35–44% | ||
| Scheme 2 (A) Late stage installation of pyridinium. (B) Optimization of cross coupling. | ||
Having 1a–1c in hand set the stage to investigate the late-stage reductive couplings of these MPs with aryl halides to access the aryl alanine-containing MP. These optimization reactions were conducted using HTE (in 1 mL vials secured in 96-well reaction blocks), using 2 μmol of the MP limiting reagent and 1.5 equivalents of the aryl halide in the presence of a nickel catalyst and a reductant. The reaction efficiency was determined by the product LC area percent (LCAP) in the UPLC-MS chromatograms of the crude reaction mixtures. In addition, the calibrated assay yields of the desired product MP-1-Br-1 was also obtained (See ESI section 3.2†). As shown in Scheme 2B, reaction of MP 1a under previously optimized reductive couplings7b with pyridinium containing MPs led to the desired product MP-1-Br-1 in 42% assay yield (entry 1). Use of 2 vs. 4 equiv. Zn led to comparable yields of product MP-1-Br-1 (entries 1 & 2). Interestingly, the corresponding reactions with electron-rich pyridinium-containing MP 1b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 led to significant increase in the assay yield of product MP-1-Br-1 and use of 4 eq. of Zn was optimal (entries 3 & 4). The use of Mn instead of Zn as a reductant with 1b led to diminished product yields (entries 3 & 4 vs. 5 & 6). Addition of additives such as TBAI did not enhance the product yields (See ESI for details†). To elucidate whether the enhanced yield from the reaction using 1b14 was due to the electronic difference between 1a and 1b, we investigated the reaction of MP 1c which is electronically similar to 1a. As shown in entry 7, the product yield from the reductive coupling using 1c was comparable to that obtained using 1a. These results suggest that the electron rich pyridiniums such as 1b are optimal for these reductive couplings. Careful analysis of the UPLC chromatograms of the crude reaction mixtures revealed that the reaction profile is significantly cleaner with 1bversus1a and 1c (See ESI for details†). Specifically, reactions with 1a and 1c lead to byproducts resulting from reduction of the pyridinium ring or radical addition to the pyridinium ring. Importantly, these byproducts are not observed in the chromatogram for the reaction with 1b suggesting that the enhanced yields with 1b is likely due to a better match between the relative rates of radical generation, radical capture, and oxidative addition steps.7
14 led to significant increase in the assay yield of product MP-1-Br-1 and use of 4 eq. of Zn was optimal (entries 3 & 4). The use of Mn instead of Zn as a reductant with 1b led to diminished product yields (entries 3 & 4 vs. 5 & 6). Addition of additives such as TBAI did not enhance the product yields (See ESI for details†). To elucidate whether the enhanced yield from the reaction using 1b14 was due to the electronic difference between 1a and 1b, we investigated the reaction of MP 1c which is electronically similar to 1a. As shown in entry 7, the product yield from the reductive coupling using 1c was comparable to that obtained using 1a. These results suggest that the electron rich pyridiniums such as 1b are optimal for these reductive couplings. Careful analysis of the UPLC chromatograms of the crude reaction mixtures revealed that the reaction profile is significantly cleaner with 1bversus1a and 1c (See ESI for details†). Specifically, reactions with 1a and 1c lead to byproducts resulting from reduction of the pyridinium ring or radical addition to the pyridinium ring. Importantly, these byproducts are not observed in the chromatogram for the reaction with 1b suggesting that the enhanced yields with 1b is likely due to a better match between the relative rates of radical generation, radical capture, and oxidative addition steps.7
Having the optimal conditions for both the late-stage pyridinium condensation and the subsequent reductive couplings we next explored the generality of this two-step strategy with respect to both the aryl halide and MP substrates leveraging HTE. These studies commenced with investigating the reductive coupling of MP 1b with diverse aryl bromides. The reaction efficiency was determined by the product LCAP in the crude reaction UPLC-MS chromatograms.
As depicted in Scheme 3, this coupling is compatible with 4-substituted aryl bromides bearing varied functional groups at the para position affording the desired products with >20% LCAP which is generally sufficient to provide adequate quantities of products for biological assays. Ketone (Br-3), free alcohols (Br-15), sulfone (Br-10), sulfonamide (Br-4), carboxylic acid (Br-2), urea (Br-17), esters (Br-7), amides (Br-11), aldehydes (Br-20), quinoline (Br-13), oxazole (Br-6), imidazole (Br-22), and oxadiazoles (Br-5) are well tolerated for this coupling. Furthermore, reaction of boronic acid bearing aryl halide Br-8 affords the product which can be subsequently elaborated using Suzuki–Miyaura cross-couplings. These reductive couplings are also compatible with a range of pharmaceutically relevant heteroaryl bromides such as pyridyl (Br-14), pyridazine (Br-19), pyrimidines (Br-9, Br-16, Br-21 and Br-23), indole (Br-18), pyrazole (Br-24) and imidazole (Br-25).15,7a,c
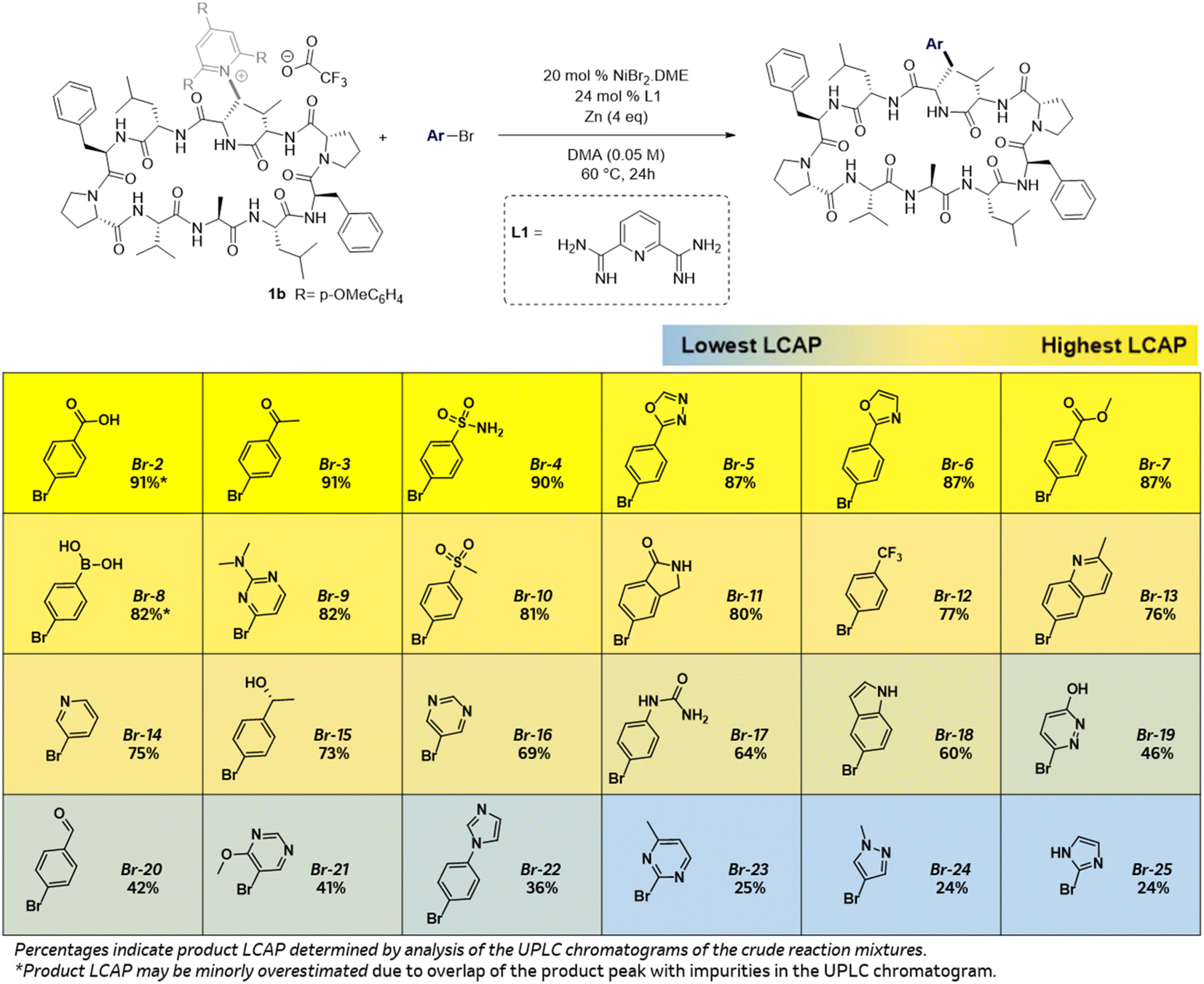 | ||
| Scheme 3 Initial scan of ArBr functional group compatibility. | ||
Encouraged by the generality of this transformation we next leveraged data science methods to objectively select diverse aryl bromides for these reductive couplings (Scheme 4). Specifically, we explored the use of aryl bromides that lead to aryl alanine-containing MPs that cannot be accessed via commercially available Fmoc-protected amino acids (5378 ArBr in blue region in Venn diagram in Fig. 1B). First, these 5378 ArBr were subjected to functional group filtration to remove aryl electrophiles bearing undesired functional groups (See ESI for details†). The resulting >3000 aryl bromides were clustered into 96 groups using the K-Means algorithm after featurization (using RDKit) and dimensionality reduction (using t-SNE). Ninety-six aryl halides, one from each cluster, were selected and subjected to the reductive couplings with 1b using HTE. As shown in Scheme 4, 63/96 of these reactions led to the desired products with >20% LCAP. A number of the low yielding reactions include those with 5-membered hetroaryl or ortho-substituted halides (See ESI for details†).15,7a,c Twelve representative aryl halides that successfully yielded the desired products are shown in Scheme 4 and highlight the impressive functional group compatability of these challenging reductive couplings with macrocyclic peptides.
 | ||
| Scheme 4 96-well plate array for coupling of 1b with 96 ArBr selected via clustering. | ||
To systematically investigate side chain functional group compatibility with this method, reaction of pyridinium-containing MP substrates 1d–1m and 2 were evaluated with 12 aryl halides.16 The 12 ArBr's selected for this study exhibited a broad range of reactivity with 1b (Fig. 2, HTE results, column 1). Such “informer set” libraries are powerful for medicinal chemistry applications for the rapid interrogation of SAR across multiple vectors simultaneously.17 Additionally, it enables the end users to predict whether the observed reactivity and reaction conditions using model substrates (MP 1b, Schemes 3 and 4) would translate to transformations using a diverse range of MP substrates. Gratifyingly, the pyridinium-containing MPs 1d–1m & 2 could be readily accessed using the aforementioned procedure (or a modified procedure, see ESI for details†) for the synthesis of 1b (Scheme 2A). MPs 1d–1j contain functionally diverse amino acids at the position across from the pyridinium functionality in the β-sheet region while 1k contains the pyridinium group as part of the β-turn to probe conformation effects at the coupling site. MP 1l was chosen to illustrate the compatibility of this method for the synthesis of homologated aryl alanines. MP 1m bears a thioether linker which is present in many peptides obtained from mRNA display hit finding campaigns. Finally, 2 represents a MP with the pyridinium moiety on the side chain of a N–Me amino acid. Additionally, 2 has a core structure different from Gramicidin analogs in that it lacks the β-sheet secondary motif.18 Furthermore, 2 contains the core structure of known KRAS inhibitor LUNA-18 (Fig. 1A) and previous studies suggest that the incorporation of different aryl alanines at the position containing the pyridinium functionality influences bioactivity.3c
 | ||
| Fig. 2 Coupling of diverse macrocyclic peptides (R = p-OMeC6H4) with 12 aryl halides. | ||
As shown in Fig. 2, in general, the reaction of MP 1d–1h, 1k, and 1l exhibit reactivity trends similar to that observed with MP 1b. These results demonstrate that the method reported herein is effective for the synthesis of aryl alanine (using 1d–1h, & 1k) and homologated aryl alanine-containing MPs (1l). The low yielding aryl halides include 5-membered heteroaryl halides (Br-112 and Br-60) which are known to be challenging in the context of nickel-catalyzed cross-couplings with alkyl pyridiniums.7b,19 While electron-deficient ortho-substituted halides (e.g., Br-87 and Br-111) afford the products with >20% LCAP, the reactivity is significantly diminished with the relatively more electron-rich ortho-substituted bromide (Br-88). This observation is consistent with the known electronic effects of aryl halides in reductive couplings using alkyl pyridiniums.7b,c,19 The reaction of MP-1i containing trityl-protected histidine exhibited diminished product LCAPs with a few ArBr (e.g., Br-47, Br-1, Br-111 & Br-92) compared to results with MP-1b in part due to the formation of byproducts resulting from the reduction of the pyridinium. This differentiated reactivity between 1b and 1i could be due to the presence of the sterically hindered trityl group in 1i. Similarly, 1k containing the pyridinium in the β-turn region leads to lower product LCAPs with Br-47 and Br-1 relative to the reaction with MP-1b partly due to byproducts arising from the addition of the radical to the pyridinium ring. Substrate 2, containing the pyridinium on the N–Me amino acid side chain also shows lower product LCAP with a few aryl bromides (e.g., Br-47, Br-1, Br-62, Br-111) relative to MP-1b suggesting that small steric differences (N–H versus N–Me) and/or differences in the overall secondary structure can result in differentiated reactivity in these cross-couplings. Nonetheless, the success of these couplings to install N–Me aryl alanines in complex peptides such as 2 is remarkable because the commercial availability of N–Me-Fmoc-protected amino acids is even more limited than their N–H counterparts. Finally, we were delighted to see that the thioether-containing MP-1m also afforded the products in synthetically useful LCAPs for 10/12 aryl halides. This result is particularly significant because previously reported reductive couplings7b with thioether-containing MPs were unsuccessful. Taken together the results in Schemes 3, 4 and Fig. 2 demonstrates the generality of the reductive coupling with respect to both the aryl bromides and the MPs on microscale. To demonstrate the scalability of these couplings, the reactions of select MP pyridinium substrates with Br-1 were conducted on a 28–44 μmol scale (14–22× higher scale than the HTE experiments, Fig. 3). After purification, the products were obtained in 39–58% isolated yields which is more than sufficient for first-tier biological assays. Importantly, consistent with prior reports,19b a slow addition of the pyridinium substrate and Br-1 for the reaction of 1k significantly suppressed the formation of byproducts resulting from the addition of the alkyl radical to the pyridinium ring. As a final demonstration of the applicability of these reductive couplings, Scheme 5 depicts the synthesis of a Gramicidin-S analog. The coupling of bis azide containing MP (3) with Br-1 employing the slow addition protocol afforded the desired product in 41% LCAP and 20% isolated yield with concommitant reductive coupling and azide reduction.
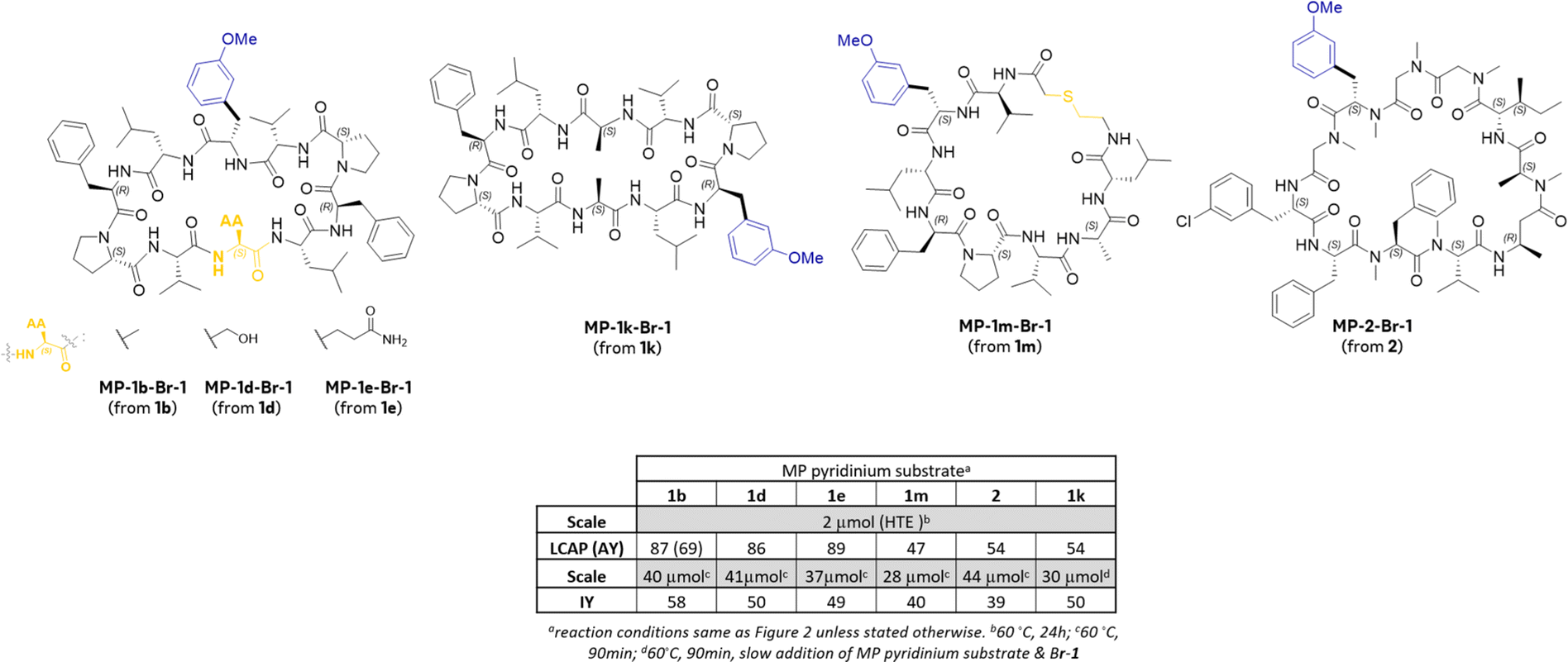 | ||
| Fig. 3 Reaction scalability assessment for coupling of MP pyrinidium with Br-1. | ||
 | ||
| Scheme 5 Synthesis of Gramicidin analog containing free amines. | ||
Conclusion
In summary, this manuscript demonstrates the first general method for the synthesis of pharmaceutically relevant macrocyclic peptides bearing structurally diverse aryl and homoaryl alanines. Efficient late-stage installation of the pyridinium functionality on a macrocyclic peptide was critical to this success. The use of microscale HTE enabled the rapid assessment of the scope of the late-stage reductive couplings with respect to both the aryl bromides and the MP substrate. The remarkable generality of this transformation is illustrated by the numerous aryl and heteroaryl bromides that are compatible with these couplings to enable access to MPs that would be otherwise inaccessible using commercially available Fmoc-protected aryl alanine derivatives. Although this manuscript focusses on LSF on MPs in solution, we anticipate that the strategy outlined herein will apply to the functionalization of linear peptides both in solution and on solid phase. Furthermore, the late-stage pyridinium installation on complex peptides could be followed by other radical cross-couplings (e.g., sp3–sp3 couplings and Giese reactions), beyond the sp2–sp3 reactions described herein. Alternatively, we envision sequential transformations such as Suzuki–Miyaura coupling followed by subsequent pyridinium formation and cross-couplings to enable SAR across multiple vectors on complex peptides. Taken together the late-stage pyridinium installation provides several avenues to access structurally diverse peptides that are rich in ncAAs thereby accelerating the SAR studies for peptide drug discovery.Data availability
The data relevant to the manuscript is included in the ESI.†Author contributions
Ahmet Kekec contributed ideas for experimental design, executed vast majority of the experiments, compiled the ESI† and contributed to manuscript writing. Lauren My-Linh Tran synthesized some macrocyclic peptide substrates. Christopher W. Plummer contributed ideas for experimental design and helped with manuscript writing. Dipannita Kalyani led the manuscript writing and contributed to experimental design.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are grateful to Prof. Mary Watson from the chemistry department at University of Delaware for reviewing this manuscript.Notes and references
- (a) A. Henninot, J. C. Collins and J. M. Nuss, The Current State of Peptide Drug Discovery: Back to the Future?, J. Med. Chem., 2018, 61(4), 1382–1414 CrossRef; (b) T. K. Sawyer, Chapter 1: Renaissance in Peptide Drug Discovery: The Third Wave, Royal Society of Chemistry, 2017, pp. 1–34 Search PubMed; (c) M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Trends in peptide drug discovery, Nat. Rev. Drug Discovery, 2021, 20(4), 309–325 CrossRef PubMed.
- (a) I. Petta, S. Lievens, C. Libert, J. Tavernier and K. D. Bosscher, Modulation of protein-protein interactions for the development of novel therapeutics, Mol. Ther., 2016, 24, 707–718 CrossRef PubMed; (b) L. Nevola and E. Giralt, Modulating protein-protein interactions: The potential of peptides, Chem. Commun., 2015, 51, 3302–3315 RSC; (c) L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Therapeutic Peptides: Current Applications and Future Directions, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef; (d) J. H. Hickey, D. Sindhikara, S. L. Zultanski and D. M. Schultz, Beyond 20 in the 21st century: Prospects and Challenges of Non-Canonical Amino Acids in Peptide Drug Discovery, ACS Med. Chem. Lett., 2023, 14, 557 CrossRef.
- (a) H. Zhang and S. Chen, Cyclicpeptide drugs approved in the last two decades (2001-2021), RSC Chem. Biol., 2022, 3, 18–31 RSC; (b) C. Alleyne, R. P. Amin, B. Bhatt, E. Bianchi, J. C. Blain, N. Boyer, D. Branca, M. W. Embrey, S. N. Ha, K. Jette, D. G. Johns, A. D. Kerekes, K. A. Koeplinger, D. LaPlaca, N. Li, B. Murphy, P. Orth, A. Ricardo, S. Salowe, K. Seyb, A. Shahripour, J. R. Stringer, Y. Sun, R. Tracy, C. Wu, Y. Xiong, H. Youm, H. J. Zokian and T. J. Tucker, Series of Novel and Highly Potent Cyclic Peptide PCSK9 Inhibitors Derived from an mRNA Display Screen and Optimized via Structure-Based Design, J. Med. Chem., 2020, 63(22), 13796–13824 CrossRef PubMed; (c) M. Tanada, M. Tamiya, A. Matsuo, A. Chiyoda, K. Takano, T. Ito, M. Irie, T. Kotake, R. Takeyama, H. Kawada, R. Hayashi, S. Ishikawa, K. Nomura, N. Furuichi, Y. Morita, M. Kage, S. Hashimoto, K. Nii, H. Sase, K. Ohara, A. Ohta, S. Kuramoto, Y. Nishimura, H. Iikura and T. Shiraishi, Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor, J. Am. Chem. Soc., 2023, 145(30), 16610–16620 CrossRef.
- (a) R. B. Merrifield, Solid phase peptide synthesis. i. the synthesis of a tetrapeptide, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef; (b) J. C. Li, T. Liu, Y. Wang, A. P. Mehta and P. G. Schultz, Enhancing Protein Stability with Genetically Encoded Noncanonical Amino Acids, J. Am. Chem. Soc., 2018, 140(47), 15997–16000 CrossRef PubMed; (c) K. Lang and J. W. Chin, Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins, Chem. Rev., 2014, 114(9), 4764 CrossRef CAS PubMed; (d) R. Behrendt, P. White and J. Offer, Advances in Fmoc solid-phase peptide synthesis, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- (a) J. N. deGruyter, L. R. Malins and P. S. Baran, Residue-specific peptide modification: a chemist's guide, Biochemistry, 2017, 56(30), 3863–3873 CrossRef CAS; (b) S. Li, D. Pissarnitski, T. Nowak, M. Wleklinski and S. W. Krska, Merging Late-Stage Diversification with Solid Phase Peptide Synthesis Enabled by High-Throughput On Resin Reaction Screening, ACS Catal., 2022, 12(5), 3201–3210 CrossRef CAS; (c) A. M. Yousif, S. Colarusso and E. Bianchi, Katritzky Salts for the Synthesis of Unnatural Amino Acids and Late-Stage Functionalization of Peptides, Eur. J. Org Chem., 2023, e202201274 CrossRef CAS.
- (a) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, Site-Selective C(sp3)−H Functionalization of Di-, Tri-, and Tetrapeptides at the N-Terminus, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef CAS PubMed; (b) L. Liu, Y.-H. Liu and B.-F. Shi, Synthesis of amino acids and peptides with bulky side chains via ligand-enabled carboxylate-directed γ-C(sp3)−H arylation, Chem. Sci., 2020, 11, 290 RSC; (c) Z. Chen, M. Zhu, M. Cai, L. Xu and Y. Weng, Palladium-Catalyzed C(sp3)−H Arylation and Alkynylation of Peptides Directed by Aspartic Acid (Asp), ACS Catal., 2021, 11, 7401 CrossRef; (d) F. Zhu, W. C. Powell, R. Jing and M. A. Walczak, Organometallic AlaM reagents for umpolung peptide diversification, Chem Catal., 2021, 1, 870 CrossRef PubMed; (e) F. Zhu, E. Miller, W. C. Powell, K. Johnson, A. Beggs, G. E. Evenson and M. A. Walczak, Umpolung AlaB Reagents for the Synthesis of Non-Proteogenic Amino Acids, Peptides and Proteins, Angew. Chem., Int. Ed., 2022, 61, e202207153 CrossRef PubMed; (f) S. Pal, J. Openy, A. Krzyzanowski, A. Noisier and P. t Hart, On-Resin Photochemical Decarboxylative Arylation of Peptides, Org. Lett., 2024, 26(14), 2795–2799 CrossRef.
- (a) N. Shengyang, L. Chun-Xiao, M. Yu, H. Jianlin, W. Yi, Y. Hong and P. Yi, Ni-Catalyzed Deaminative Cross-Electrophile Coupling of Katritzky Salts with Halides via C-N Bond Activation, Sci. Adv., 2019, 5, eaaw9516 CrossRef PubMed; (b) J. C. Twitty, Y. Hong, B. Garcia, S. Tsang, J. Liao, D. M. Schultz, J. Hanisak, S. L. Zultanski, A. Dion, D. Kalyani and M. P. Watson, Diversifying amino acids and peptides via deaminative reductive cross-couplings leveraging high-throughput experimentation, J. Am. Chem. Soc., 2023, 145(10), 5684–5695 CrossRef PubMed; (c) J.-T. Fu, W. Lundy, R. Chowdhury, J. C. Twitty, L. P. Dinh, J. Sampson, Y. – H. Lam, C. S. Sevov, M. P. Watson and D. Kalyani, Nickel-Catalyzed Electroreductive Coupling of Alkylpyridinium Salts and Aryl Halides, ACS Catal., 2023, 13, 9336 CrossRef PubMed.
- J. Openy, G. Amrahova, J.-Y. Chang, A. Noisier and P. Hart, Solid-phase peptide modification via deaminative photochemical Csp3-Csp3 bond formation using Katritzky salts, Chem.–Euro. J., 2022, 28, 39 CrossRef PubMed.
- (a) M. Shevlin, Practical High-Throughput Experimentation for Chemists, ACS Med. Chem. Lett., 2017, 8(6), 601–607 CrossRef PubMed; (b) S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. Eugenio de Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future, Org. Process Res. Dev., 2019, 23(6), 1213–1242 CrossRef; (c) C. L. Allen, D. C. Leitch, M. S. Anson and M. A. Zajac, The Power and Accessibility of High-Throughput Methods for Catalysis Research, Nat. Catal., 2019, 2(1), 2–4 CrossRef.
- (a) Q. Guan, S. Huang, Y. Jin, R. Campagne, V. Alezra and Y. Wan, Recent Advances in the Exploration of Therapeutic Analogues of Gramicidin S, an Old but Still Potent Antimicrobial Peptide, J. Med. Chem., 2019, 62(17), 7603–7617 CrossRef CAS PubMed; (b) S. Pal, U. Ghosh, R. Sankar Ampapathi and T. K. Chakraborty, Recent Studies on Gramicidin S Aanalog Structure and Antimicrobial Activity, Top. Heterocycl. Chem., 2017, 49, 159–202 CAS.
- G. M. J. Schmidt, D. C. Hodgkin and B. M. Oughton, A crystallographic study of some derivatives of Gramacidin S, Biochem. J., 1957, 65, 744 CrossRef CAS PubMed.
- Y. Huang, M. M. Wiedmann and H. Suga, RNA Display Methods for the Discovery of Bioactive Macrocycles, Chem. Rev., 2019, 119(17), 10360–10391 CrossRef CAS PubMed.
- (a) C. Solanas, B. G. de la Torre, M. Fernandez-Reyes, C. M. Santiveri, M. A. Jimenez, L. Rivas, A. I. Jimenez, D. Andreu and C. Cativiela, Therapeutic Index of Gramicidin S is Strongly Modulated by D-Phenylalanine Analogues at the β-Turn, J. Med. Chem., 2009, 52(3), 664–674 CrossRef CAS PubMed; (b) M. Jelokhani-Niaraki, R. S. Hodges, J. E. Meissner, U. E. Hassenstein and L. Wheaton, Interaction of gramicidin S and its aromatic amino-acid analogs with phospholipid membranes, Biophys. J. , 2008, 95(7), 3306–3321 CrossRef CAS PubMed; (c) C. Solanas, B. G. de la Torre, M. Fernandez-Reyes, C. M. Santiveri, M. A. Jimenez, L. Rivas, A. I. Jimenez, D. Andreu and C. Cativiela, Sequence inversion and phenylalanine surrogates at the β-turn enhance the antibiotic activity of gramicidin S, J. Med. Chem., 2010, 53(10), 4119–4129 CrossRef CAS PubMed.
- R. Martin-Montero, V. R. Yatham, H. Yin, J. Davies and R. Martin, Ni-Catalyzed Reductive Deaminative Arylation at sp3 Carbon Centers, Org. Lett., 2019, 21, 2947 CrossRef CAS PubMed.
- The main byproducts for the reactions with low yielding aryl bromides results from the reduction of the carbon-pyridinium C–N bond likely due to a mismatch of rates for the activation of the aryl halide vs. the alkyl pyridinium.
- In place of cysteine containing MP substrate we used the MP 1m containing a thioether which is a mimic of protected cysteine. Disulfide containing substrates were not considered due to the known Zn mediated reduction of disulfides. See the following reference: M. Erlandsson and M. Hallbrink, Metallic Zinc Reduction of Disulfide Bonds Between Cysteine Residues in Peptides and Proteins, Int. J. Pept. Res. Ther., 2005, 11, 261 CrossRef CAS.
- S. D. Dreher and S. W. Krska, Chemistry Informer Libraries: Conception, Early Experience, and Role in the Future of Cheminformatics, Acc. Chem. Res., 2021, 54(7), 1586–1596 CrossRef.
- P. Wadhwai, S. Afonin, M. Ieronimo, J. Buerck and A. S. Ulrich, J. Org. Chem., 2006, 71, 55–61 CrossRef.
- (a) E. C. Hansen, D. J. Pedro, A. C. Wotal, N. J. Gower, J. D. Nelson, S. Caron and D. J. Weix, New Ligands for Nickel Catalysis From Diverse Pharmaceutical Heterocycle Libraries, Nat. Chem., 2016, 8, 1126 CrossRef; (b) J. Liao, C. H. Basch, M. E. Hoerrner, M. R. Talley, B. P. Boscoe, J. W. Tucker, M. R. Garnsey and M. P. Watson, Deaminative Reductive Cross-Electrophile Couplings of Alkylpyridinium Salts and Aryl Bromides, Org. Lett., 2019, 21, 2941 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06837h |
| This journal is © The Royal Society of Chemistry 2025 |