
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-resolved and theoretical analysis of Mo-carbene transformations in metathesis of ethylene with 2-butene†
Tatiana
Otroshchenko
 *a,
Aleksandr
Fedorov
a,
Qiyang
Zhang
a,
David
Linke
a,
Jarosław
Handzlik
b,
Mirjam
Schröder
acd,
Björn
Corzilius
acd and
Evgenii V.
Kondratenko
*a
*a,
Aleksandr
Fedorov
a,
Qiyang
Zhang
a,
David
Linke
a,
Jarosław
Handzlik
b,
Mirjam
Schröder
acd,
Björn
Corzilius
acd and
Evgenii V.
Kondratenko
*a
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, D-18059 Rostock, Germany. E-mail: tatiana.otroshchenko@catalysis.de; evgenii.kondratenko@catalysis.de
bCracow University of Technology, Faculty of Chemical Engineering and Technology, ul. Warszawska 24, 31-155 Kraków, Poland
cInstitute of Chemistry, University of Rostock, Albert-Einstein-Str. 27, 18059 Rostock, Germany
dDepartment Life, Light & Matter, University of Rostock, Albert-Einstein-Str. 25, 18059 Rostock, Germany
First published on 8th January 2025
Abstract
Although supported Mo-containing catalysts have been extensively investigated in the metathesis of ethylene with 2-butene to propene, the mechanisms of the formation and transformation of catalytically active Mo-carbenes in the course of the reaction are still not fully understood. The difficulties arise because only a tiny fraction of MoOx species can form Mo-carbenes in situ, making the detection of the latter by spectroscopic means very unlikely. Herein, purposefully designed steady-state and transient experiments including their kinetic evaluation and density functional theory calculations enabled us to elucidate mechanistic and kinetic details of the above reaction-induced processes in the metathesis reaction over a Mo/P/SiO2 catalyst at 50 °C. We established that, in parallel with the desired reaction cycle, molybdacyclobutanes also undergo reversible structural transformations which might be one of the reasons for low steady-state catalyst activity. Based on the results obtained, strategies for controlling the concentration of the inactive species and accordingly catalyst activity have been suggested and experimentally validated.
Introduction
Metathesis of ethylene with 2-butene is a large-scale process used for the on-purpose production of propene, the second most widely produced olefin in the chemical industry.1,2 Catalysts based on oxides of W, Mo or Re dispersed on high-surface-area supports are used for this reaction.3,4 According to the classical mechanism elucidated by Hérisson and Chauvin,5,6 surface metal-carbenes (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH-R, where M is W, Mo or Re), which serve as active sites, react with ethylene or 2-butene to form metallacyclobutane intermediates decomposing to gas-phase propene. The MCH-R species are formed in situ as a result of the interaction of MOx with olefins. Therefore, the knowledge of the mechanisms of MCH-R formation and, especially, undesired transformations under the reaction conditions is very important to control the concentration of the active species and accordingly the propene production through catalyst design or modulating the reaction conditions.
CH-R, where M is W, Mo or Re), which serve as active sites, react with ethylene or 2-butene to form metallacyclobutane intermediates decomposing to gas-phase propene. The MCH-R species are formed in situ as a result of the interaction of MOx with olefins. Therefore, the knowledge of the mechanisms of MCH-R formation and, especially, undesired transformations under the reaction conditions is very important to control the concentration of the active species and accordingly the propene production through catalyst design or modulating the reaction conditions.
It has been shown that the presence of hydroxyl species on the catalyst surface,7–13 the usage of metathesis inactive cocatalysts,14–16 or a certain catalyst treatment (photoreduction in CO followed by treatment in cyclopropane;17 reduction with organosilicon reductants;18 high-temperature treatment in CH4;19 propene adsorption – desorption treatment;9 treatment in methanol;8etc.) facilitate the transformation of MOx into MCH-R. Although the treated catalysts demonstrate high initial metathesis activity, they rapidly deactivate with time on stream and achieve a steady-state activity like their untreated counterparts. The reason for such deactivation seems to be a decrease in the concentration of MCH-R due to their low stability8,17,20 and/or transformation of the metallacyclobutane intermediates into stable inactive complexes as theoretically suggested for ethylene metathesis.21 Because the fraction of MOx species capable of forming MCH-R is extremely low,8,16,22,23 state-of-the-art spectroscopic methods typically do not provide direct insight into the formation and further conversion of carbenes.
The above challenges and fundamental gaps motivated us to perform sophisticated steady-state and transient catalytic tests including their kinetic evaluation as well as density functional theory (DFT) calculations. The Mo/P/SiO2 catalyst, which was introduced in our previous study10 and shows exceptional activity even at 50 °C, was used in the present study. Special emphasis was placed on the analysis of both selective pathways in the classical Chauvin cycle and side reactions of MCH-R. The results obtained provide the basis for improving the catalyst activity through modulating the reaction conditions.
Results and discussion
Structural catalyst properties
The Mo/P/SiO2 catalyst described in ref. 10 was used for kinetic tests at 50 °C in the metathesis of ethylene with 2-butenes using different reaction feeds under conditions free of heat and mass transport limitations. Its unpromoted counterpart, Mo/SiO2, shows too low activity at 50 °C and was not used in the kinetic tests. Nevertheless, important structural properties of these catalysts are presented and discussed for comparative purposes. The UV-vis spectra of both samples are characterized by strong absorption bands at around 250 and 290 nm and shoulders at around 330 and 400 nm (Fig. S1(a)†). These bands can be assigned to the ligand-to-metal charge transfer transitions in tetrahedral and octahedral MoOx species, as well as in crystalline MoO3.13 A straightforward assignment of the band position to the specific Mo-containing species is however complicated due to significant overlapping of the absorption bands. The promoter P seems to lead to a slight decrease in the degree of oligomerization of MoOx species as concluded from the UV-vis edge energy (Eg) determined from the Tauc plot (Fig. S1(b)†).The effect of P on the oligomerization degree of supported MoOx may be explained as follows. The promotion of SiO2 with P leads to the creation of a new type of OH site characterized by a sharp band at 3667 cm−1 (Fig. S1(c),† red dashed line). These sites should be involved in anchoring MoOx because the ratio of the intensity of this band to that at 3747 cm−1 (related to isolated silanols) for Mo/P/SiO2 is lower in comparison with that for P/SiO2. After deposition of MoOx species on the support, a new broad band centred at about 3627 cm−1 appears (Fig. S1(c),† solid lines), which can be assigned to O–H stretching vibrations in molybdenol groups (Mo–OH) involved in hydrogen bonding to neighbouring oxygen or to silanol groups interacting with oxygen atoms of MoOx species.9
The Raman spectra of both Mo/SiO2 and Mo/P/SiO2 are characterized by the bands at 814 and 990 cm−1 originating from MoOx and/or MoO3 species (Fig. S1(d)†).24 The Raman spectrum of Mo/P/SiO2 additionally contains a weak band at 877 cm−1, which can be assigned to the P–O stretching vibrations in phosphates (νsP(OH), νsP(OH)2, and/or νsP(OH)3).25
The chemical nature of P-containing species was further analysed by XPS (Fig. S1(e)†) and 31P NMR (Fig. S1(f)†). A broad XPS peak in the P 2p region, which can be deconvoluted into a doublet (134.9 eV, 135.8 eV), can be assigned to the surface PO43− species.26–28 No features of metal phosphides (binding energy of ∼129.5 eV) can be observed. The 31P MAS-NMR spectrum of the reference P/SiO2 shows four signals at 0.4, −6.1, −10.4 and −23.1 ppm. The first three signals likely belong to single isolated phosphate groups (Si–O–PO–(OH)2), while the last signal indicates the presence of phosphate species with a bridged structure ((Si–O)2–PO–OH).29 For the Mo/P/SiO2 sample, the signal at ∼0 ppm is upfield shifted by 3 ppm and broadened, hinting at contact or pseudo-contact interactions with the metal. The signals at −10.4 and −23.1 ppm remain unchanged in their chemical shift.
In summary, highly dispersed and polymerized tetrahedral and octahedral MoOx as well as crystalline MoO3 are present on the surface of the catalysts. The latter species unlikely participate in the formation of active metal-carbenes23 and therefore should not play any role in the metathesis reaction. From the above-shown data we can conclude that the majority of P exists as monomeric phosphate species which likely participate in the anchoring of non-crystalline MoOx sites.
Catalytic activity in C2H4 – trans-2-C4H8 metathesis
After treatment of the Mo/P/SiO2 catalyst in air at 500 °C for 3 h and cooling down to 50 °C in nitrogen, an industrially relevant C2H4/trans-2-C4H8/N2 = 5/5/1 feed was added. The rate of propene formation increased more than two times from an initial (determined after 7 min on reaction stream) non-zero value with increasing time on stream and reached a steady-state value after about 4 h on stream (black symbols in Fig. 1). The increase may be due to the reaction-induced conversion of MoOx to catalytically active Mo-carbenes. According to some previous studies,10,11 different olefins have different reactivity in this respect. With this in mind, we treated the Mo/P/SiO2 catalyst at 50 °C in trans-2-C4H8, C2H4, or trans-2-C4H8 followed by C2H4 for different durations of time before starting the metathesis reaction at the same temperature. The same Mo/P/SiO2 catalyst sample (initially tested in the metathesis of ethylene with 2-butenes at 50 °C for 10 h and regenerated in air at 500 °C for 3 h) was applied for all experiments.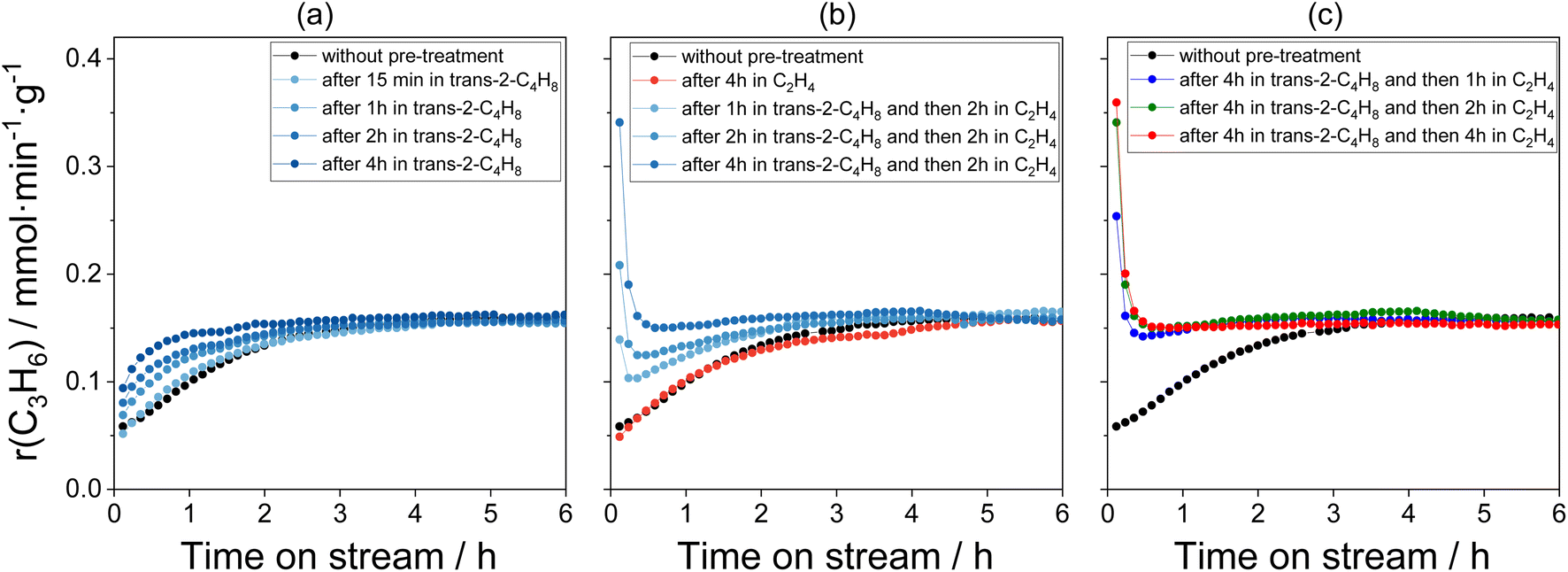 | ||
| Fig. 1 Temporal changes of the rate of propene formation determined over calcined Mo/P/SiO2 (marked in black in all figures) and Mo/P/SiO2 after various treatment procedures: (a) after treatment in trans-2-C4H8 for different durations of time; (b) after treatment in C2H4 for 4 h and after treatment in trans-2-C4H8 for different durations of time followed by treatment in C2H4 for 2 h; (c) after treatment in trans-2-C4H8 for 4 h followed by treatment in C2H4 for different durations of time. Treatments and metathesis reaction were carried out at 50 °C. | ||
The catalyst treatment in trans-2-C4H8 increased the initial rate of propene formation but did not influence the steady-state rate (Fig. 1(a)). Moreover, the time required to reach steady-state operation decreased with increasing treatment time. These changes may be related to an increase in the concentration of Mo-carbenes during the catalyst treatment.
It is worth mentioning that propene and pentenes were observed during the treatment of Mo/P/SiO2 in trans-2-C4H8 (Fig. S2(a)†). Their appearance is more likely due to the metathesis of trans-2-C4H8 with 1-C4H8 formed through isomerization of trans-2-C4H8 which is typical for Mo/SiO2 catalysts.30 According to this reaction, propene and pentene are formed in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. A similar value was determined for the ratio of the formation rates of these olefins during the catalyst treatment in trans-2-C4H8 (Fig. S2(a)†).
:1. A similar value was determined for the ratio of the formation rates of these olefins during the catalyst treatment in trans-2-C4H8 (Fig. S2(a)†).
The catalyst treatment in C2H4 at 50 °C for 4 h did not significantly affect the initial rate of propene formation and its time-on-stream behaviour when compared to the results obtained with the untreated Mo/P/SiO2 (Fig. 1(b)). According to previous DRIFTS and C2H4-TPD data,10 C2H4 hardly interacts with the oxidized catalyst at 50 °C and does not yield any surface hydrocarbon species. Thus, in contrast to the treatment in trans-2-C4H8, the treatment in C2H4 does not result in the formation of Mo-carbenes from MoOx.
The initial rate of propene formation over the catalyst treated first in trans-2-C4H8 and then in C2H4 (Fig. 1(b) and (c)) is significantly higher than that of its untreated counterpart or treated either in trans-2-C4H8 or in C2H4. The highest activity was determined after 4 h treatment in trans-2-C4H8 followed by 4 h treatment in C2H4. It was more than six times higher than the rate determined over the calcined untreated catalyst and more than two times higher than the steady-state value. This high activity, however, decreased rapidly within the first hour on reaction stream regardless of the treatment duration. The time required to reach the steady-state activity increased as the treatment time in trans-2-C4H8 decreased. Noticeably, such behaviour is unlikely related to the presence of phosphorus in the catalyst since similar temporal changes of the rate of propene formation were observed for the unpromoted Mo/SiO2 catalyst (Fig. S3†).
No gaseous products (except traces of C3H6 due to the presence of tiny amounts of trans-2-C4H8 in the line) were observed during the treatment of Mo/P/SiO2 in C2H4 after the treatment in trans-2-C4H8 (Fig. S2(b)†). Such result implies that no products are formed during the interaction of C2H4 with the catalyst pre-activated in trans-2-C4H8 or their concentration is below the detection limit of the on-line GC used.
To check if the propene formation rate can be influenced by short-term treatment of the catalyst in ethylene, we performed a control experiment. The Mo/P/SiO2 sample was first treated in trans-2-C4H8 for 1 h and then in C2H4 for 30 min. Hereafter, the catalyst was exposed to the standard reaction feed (C2H4/trans-2-C4H8/N2 = 5/5/1). After some time on this stream, the catalyst was treated in C2H4 for short intervals (3 min) followed by feeding the reaction mixture. The catalyst activity increased by about 30% after each treatment in C2H4 (Fig. S4†) and then decreased rapidly with increasing time on the metathesis stream.
Determination of the steady-state concentration of Mo-carbenes
Motivated by our previous study,16 we applied the Steady-State Isotopic Transient Kinetic Analysis (SSITKA) method31,32 to verify whether the increase in the rate of propene formation after a two-step treatment of the catalyst in trans-2-C4H8 and then in C2H4 was due to the increase in the concentration of Mo-carbenes. In these tests carried out with untreated Mo/P/SiO2 and its counterpart initially treated in trans-2-C4H8 for 4 h and then in C2H4 for 4 h, we started with a non-labelled reaction feed (C2H4/trans-2-C4H8/Ar) to reach a steady-state operation (after about 20 min on stream). Hereafter, this feed was replaced by a feed containing 13C2H4 (13C2H4/trans-2-C4H8/He/Ar; He was used as an inert tracer). Importantly, the concentrations of ethylene and 2-butene in these feeds were the same. The replacement of 12C2H4 by 13C2H4 in the reaction feed did not lead to a change in the rate of propene formation (confirmed by on-line GC measurements). Thus, a kinetic isotope effect does not play a significant role because of the small difference in the atomic masses of 12C and 13C. The concentration of 13C12C2H6 increased with time on stream after switching to the labelled feed. The normalized (with respect to the steady-state signals) responses of 13C12C2H6 and He (inert tracer) obtained in the SSITKA tests are shown in Fig. 2. Based on the theory of the SSITKA method,31,32 the surface residence time (τP) of Mo-carbenes leading to propene was calculated from the area between the transient responses of He and 13C12C2H6 (Fig. 2, Table S1†). Their concentration (NP) was obtained from eqn (1). It is noteworthy that the fraction (NP divided by the total amount of Mo) of MoOx resulting in Mo-carbenes does not exceed 0.1% (Table S1†).| NP = τP × r(C3H6), | (1) |
 | ||
| Fig. 2 Normalized responses of 13C12C2H6 and He (inert tracer) as a function of relative time (time after the switch of non-labeled feed to 13C-labeled feed) obtained for (a) calcined untreated Mo/P/SiO2 and (b) Mo/P/SiO2 sequentially treated in trans-2-C4H8 at 50 °C for 4 h and in C2H4 at 50 °C for 4 h. | ||
The NP value determined for the untreated Mo/P/SiO2 is more than seven times lower than that for the Mo/P/SiO2 treated first in trans-2-C4H8 and then in C2H4. We used these values and the rate of propene formation over these catalysts to calculate the turnover frequency of propene formation (TOF) according to eqn (2).
| TOF = r(C3H6)/NP | (2) |
No obvious difference was found (Table S1†). Thus, the catalyst treatment in trans-2-C4H8/C2H4 seems to increase the concentration of Mo-carbenes but not to create other highly reactive Mo-carbenes.
Catalyst activity in the presence of 1-C4H8
We also performed the metathesis reaction in the presence of 5 vol% of 1-C4H8 to understand if and how this olefin affects the catalyst activity. Propene, pentenes, and trace amounts of hexenes were detected as reaction products through various metathesis reactions shown in eqn (3)–(5).| C2H4 + 2-C4H8 ⇄ 2C3H6 | (3) |
| 2-C4H8 + 1-C4H8 ⇄ C3H6 + C5H10 | (4) |
| 1-C4H8 + 1-C4H8 ⇄ C2H4 + C6H12 | (5) |
The temporal changes of the rates of formation of propene, pentenes, and hexenes are shown in Fig. 3(a), (b), and (c) respectively, together with the corresponding data obtained in the test performed without co-fed 1-C4H8. All rates in the presence of 1-C4H8 gradually increased with time on stream during the first hour on reaction stream and started to decrease after about 4 h on stream. The initial rate of propene formation was about four times and the final rate (after about 10.5 h on stream) was more than two times higher than the corresponding rates determined in the absence of 1-C4H8. According to eqn (3)–(5), propene is formed via metathesis reactions of C2H4 with trans-2-C4H8 and trans-2-C4H8 with 1-C4H8. Knowing the rate of pentene formation (Fig. 3(b)), we can estimate the contribution of the butene metathesis (eqn (4)) to the formation of propene. Since the reaction gives propene and pentene in the molar ratio of 1:1, the rate of propene formation should be equal to the rate of pentene formation. By subtracting this value from the total rate of propene formation, we can roughly estimate the rate of propene formation through the metathesis of C2H4 with trans-2-C4H8 (Fig. 3(a), open blue circles). In comparison with the control test without co-fed 1-C4H8, the obtained initial rate is higher, and the induction period characterized by an increase in this rate with time on stream is shorter. This result implies that 1-C4H8 accelerates the formation of Mo-carbenes active in the metathesis of C2H4 with trans-2-C4H8.
 | ||
| Fig. 3 Temporal changes of the rates of formation of (a) propene, (b) pentenes, and (c) hexenes determined at 50 °C over calcined Mo/P/SiO2 by using reaction mixture C2H4/trans-2-C4H8/N2 = 10/10/2 (the data are marked in black in all figures) and by using reaction mixture C2H4/trans-2-C4H8/1-C4H8/N2 = 10/9/1/2 (the data are marked in solid blue in all figures). Open blue circles in (a) are related to the data obtained after subtracting the contribution of trans-2-C4H8 – 1-C4H8 metathesis in propene formation. | ||
Formation and transformation of Mo-carbenes
According to the classical Chauvin mechanism of the metathesis of ethylene with 2-butenes, the main reaction steps are described by eqn (6) and (7). The rate of propene formation is influenced by the concentration of Mo-carbenes and their intrinsic activity. These species must be formed in situ through the reaction of supported MoOx with fed olefins.11,23,33–35 | (6) |
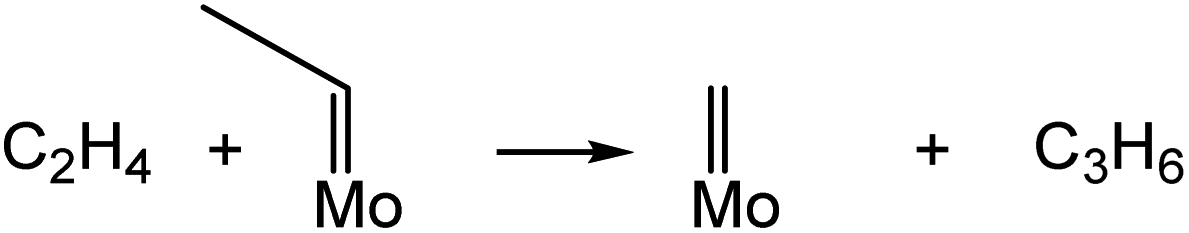 | (7) |
Accordingly, the gradual increase in the rate of propene formation over untreated Mo/P/SiO2 with time on reaction stream (Fig. 1, black symbols) can be related to the increase in the number of active Mo-carbenes. When the rate is extrapolated to zero time on stream, a non-zero value is obtained. Therefore, it is reasonable to assume that there are at least two types of MoOx species differing in their reactivity to yield Mo-carbenes. These species may differ in their molecular structure, location, and/or oxidation state of Mo. The initial activity of Mo/P/SiO2 should be related to the presence of MoOx species (Mo*) that quickly react with trans-2-C4H8 to form Mo-carbenes. Notably, C2H4 is unable to generate the latter species at 50 °C.10 Another type of MoOx species (Mo**) is characterized by a lower reactivity to form Mo-carbenes as reflected by the slow increase in the rate of propene formation with increasing time on stream (Fig. 1, black symbols).
Apart from their formation, the desired Mo-carbenes may be involved in side reactions responsible for the decrease in the rate of propene formation over the catalyst treated initially in trans-2-C4H8 followed by C2H4 (Fig. 1(b) and (c)). As the latter olefin activates the catalyst treated in trans-2-C4H8, we suggest that trans-2-C4H8 may “deactivate” the active Mo-carbenes through the formation of an “inactive” complex that does not participate in the formation of propene. This complex can be a cyclic compound due to the following arguments. The cycloaddition of an olefin molecule to a carbene site, resulting in the formation of a molybdacyclobutane complex, is the first step in the metathesis catalytic cycle.6,36 Theoretical studies of ethylene metathesis over MoOx/Al2O3 (ref. 21, 37 and 38) and MoOx/SiO2 (ref. 36 and 39) have shown that ethylene can react with Mo-methylidene (MoCH2) to give a molybdacyclobutane complex with a trigonal bipyramidal (TBP) geometry. This intermediate can react back to MoCH2 and C2H4 or be converted into its inactive isomer with a square pyramidal (SP) geometry (structure SP1 in Fig. 4). This rearrangement was considered to be one of the reasons for catalyst deactivation during ethylene metathesis.33,39 By analogy, we put forward that such undesired transformations of molybdacyclobutane complexes can also occur in the course of the ethylene – 2-butene metathesis (structures SP2–SP4 in Fig. 4). To check this hypothesis, DFT calculations were performed.
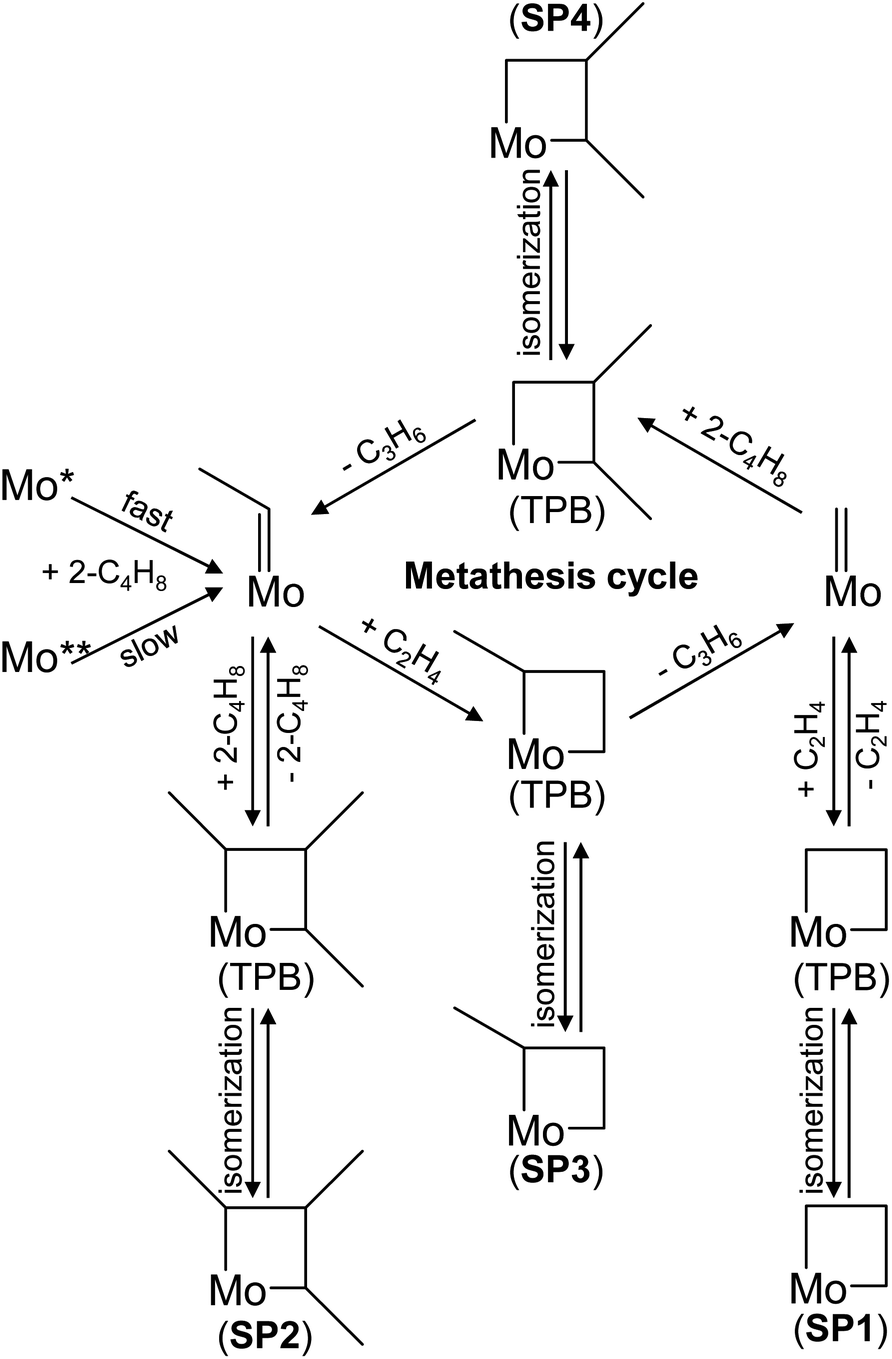 | ||
| Fig. 4 Schematic illustration of Mo-carbene formation from Mo* and Mo** species, metathesis cycle, and the formation of “inactive” complexes (square pyramidal (SP) molybdacyclobutanes). | ||
The calculated pathways of the reaction between MoC2H4 and trans-2-C4H8 are presented in Fig. 5. The formation of the trisubstituted molybdacyclobutane complex 3a with a TBP geometry is endergonic, but the predicted overall activation Gibbs energy for this step is quite low (45 kJ mol−1). The thermodynamically unstable intermediate 3a should rapidly decompose to the reactants in a reverse step with a very low Gibbs energy barrier of 18 kJ mol−1. The isomerization of the TBP molybdacyclobutane 3a to the thermodynamically stable SP molybdacyclobutane 3b is another possibility, requiring a higher energy barrier to overcome, but it still may proceed effectively at 50 °C. The reverse transformation of the SP isomer to the TBP one will occur slower due to a relatively high activation Gibbs energy for this step (74 kJ mol−1). Therefore, the SP molybdacyclobutane 3b can be considered as a resting state of the metathesis process, i.e., the postulated “inactive” complex. The equilibrium constant for its formation from MoC2H4 and trans-2-C4H8 was estimated to be around 8.1.
 | ||
| Fig. 5 Gibbs energy profile (kJ mol−1) at 50 °C for a reaction between syn-Mo(VI) ethylidene species and trans-2-C4H8. | ||
Stable SP molybdacyclobutane complexes can be also formed along the productive metathesis reactions (7) and (6) (Fig. S5 and S6,† respectively). However, the calculated activation barriers for the rearrangement of the monosubstituted and disubstituted SP molybdacyclobutanes to their TBP isomers (60 and 68 kJ mol−1, respectively) are lower than in the case of 3b (74 kJ mol−1) bearing three methyl substituents in the ring. Hence, for the latter the equilibrium shift towards the formation of Mo-carbenes after changing the olefin feed to ethylene will be slower. This is especially seen if compared to the monosubstituted SP molybdacyclobutane formed from ethylene and MoC2H4 (Fig. S5†). This can explain the observed increase of the metathesis activity after final ethylene treatment, in contrast to the standard pretreatment with 2-butene (Fig. 1). Nevertheless, based on the predicted activation Gibbs energies, the SP–TBP interconversion should be much faster than the formation of the active sites from MoOx species.11
Kinetic analysis of the ethylene – 2-butene metathesis
Based on the above-discussed experimental observations we developed a kinetic model considering reaction pathways responsible for the in situ formation of Mo-carbenes as well as for their desired reactions in the course of the metathesis of ethylene and 2-butene and side transformations into inactive intermediates (Table S2†). The obtained kinetic parameters (reaction rate and equilibrium constants) are given in Table S3.† The model was fit to all experimental data from Fig. 1 and 3 simultaneously. It describes the temporal profiles of propene formed over differently treated Mo/P/SiO2 (Fig. 6(a) and S7(a) and (c)†). The profiles of propene and pentenes formed during the experiment performed in the presence of 5 vol% 1-C4H8 were also described by this model (Fig. 6(b)). | ||
| Fig. 6 Experimental and fitting data representing time-on-stream changes of (a) the propene molar fraction during metathesis of ethylene with 2-butene over calcined Mo/P/SiO2 and over that after different treatments, (b) propene and pentene molar fractions during metathesis of ethylene with 2-butene in the presence of 5 vol% 1-C4H8 over calcined Mo/P/SiO2. | ||
The overall comparison of the values of the key kinetic parameters estimated by DFT and data-driven kinetic modelling is presented in Table S4.† The value of equilibrium constant K3 (see Table S3†) describing the formation of an “inactive” complex (trans-2-C4H8 + MoC2H4 ⇄ MoC6H6) was around 35.6 according to the developed kinetic model. This value can be compared with the value of 8.1 obtained from DFT calculations. The agreement is reasonable, taking into account the limited accuracy of the DFT methods and the exponential dependence of the equilibrium constant on the reaction Gibbs energy. It is worth mentioning that the ratio of the rate constants of reactions 2.1 and 2.2 (Table S2, Fig. S5 and S6†), evaluated from the overall Gibbs energy barriers (TBP–SP isomerization step is omitted), is 13, which is in qualitative accordance with the kinetic modelling results (k2.1/k2.2 = 4.5, Table S3†). The conversion of MoC2H41 and ethylene to MoCH26 and propene (Fig. S5†) is predicted by DFT to be about 8 × 103 faster, compared to the formation of the SP molybdacyclobutane 3b from 1 and trans-2-butene (Fig. 5). This estimate also qualitatively agrees with the obtained kinetic parameters (k2.1/k3 ≈ 1750, Table S3†).
Using the developed kinetic model and the obtained kinetic parameters (reaction rate and equilibrium constants) from Table S3,† we calculated the temporal profiles of the “inactive” complex and active Mo-carbenes during the metathesis reaction and various catalyst treatments (Fig. 7 and S7†). On this basis, a major part of MoOx species, which are able to form Mo-carbenes in situ, is present in the form of the “inactive” complex under steady-state conditions. Their fraction with respect to the total amount of activated MoOx is around 73% (Fig. 7(a)). Thus, to increase the production of propene, it is required to transform the “inactive” species to Mo-carbenes. Given the developed kinetic model, this can be achieved by reducing the partial pressure of trans-2-C4H8 to shift the equilibrium of reaction 3 (Table S2†).
 | ||
| Fig. 7 Time-on-stream changes of the fraction of surface species (Mo-carbenes and “inactive” complex) formed from MoOx (a) during metathesis of ethylene with 2-butene over calcined Mo/P/SiO2, (b) during catalyst treatment in trans-2-C4H8 for 4 h followed by metathesis, (c) during catalyst treatment in trans-2-C4H8 for 4 h, then in C2H4 for 2 h followed by metathesis, (d) during metathesis in the presence of 5 vol% 1-C4H8. Fraction of surface species is determined with respect to the total amount of activated MoOx. | ||
The model predicts an enhancement in the rate of propene formation by a factor of 1.5 when the ratio of C2H4/trans-2-C4H8 increases from 1 to 2 (Fig. S8(a)†). This is due to the decrease in the fraction of the “inactive” complex from 73% to 55% in favour of active Mo-carbenes (Fig. S8(b)†). To experimentally check this theoretical conclusion, we performed an additional experiment, in which untreated Mo/P/SiO2 was initially exposed to the reaction mixture C2H4/trans-2-C4H8/Ar = 10/10/2 with a total flow of 22 mL min−1 until a steady-state operation was achieved. Hereafter, this feed was replaced by a feed with more C2H4 and less trans-2-C4H8 (C2H4/trans-2-C4H8/Ar = 13.3/6.7/2). The total feed flow was not changed. As predicted by the model (Fig. S8(a)†), the rate of propene formation increased (Fig. S8(c)†). The activity decreased after switching back to the original feed after 2 h. Thus, the usage of reaction feeds with C2H4/trans-2-C4H8 ratios above the stoichiometric one is beneficial for increasing propene production.
Conclusions
Herein, we provide novel insights into the formation and, particularly, transformation of Mo-carbenes in the metathesis of ethylene with 2-butene to propene over the Mo/P/SiO2 catalyst. There should be at least two types of MoOx species participating in the formation of carbenes but differing in their reactivity in this process. Along with their involvement in the metathesis cycle, Mo-carbenes can be converted into an “inactive” complex formed with the participation of 2-butene. As this side reaction reduces the steady-state concentration of Mo-carbenes, the catalyst cannot achieve its highest possible activity. The fraction of the active and “inactive” species dynamically varies and can be controlled through catalyst treatments in certain olefins or changing the ethylene/2-butene ratio in the reaction mixture. The use of high ratios is beneficial in suppressing the formation of the undesired complexes and opens the possibility for increasing the steady-state metathesis activity of the catalyst.Data availability
The data supporting this article have been included as part of the Supplementary Information.†Author contributions
Conceptualization: T. O. and E. V. K.; investigation: T. O., J. H., and M. S.; formal analysis: A. F. and J. H.; writing – original draft: T. O., A. F., and J. H. All authors discussed the results and gave their approval for the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft (DFG, project OT 586/1-1) is gratefully acknowledged. The authors thank Dr Jana Weiß for performing Raman measurements and Dr Stephan Bartling for performing XPS measurements. J.H. gratefully acknowledges Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2024/016959.Notes and references
- N. Popoff, E. Mazoyer, J. Pelletier, R. M. Gauvin and M. Taoufik, Expanding the scope of metathesis: a survey of polyfunctional, single-site supported tungsten systems for hydrocarbon valorization, Chem. Soc. Rev., 2013, 42, 9035–9054 RSC.
- T. Otroshchenko, G. Jiang, V. A. Kondratenko, U. Rodemerck and E. V. Kondratenko, Current status and perspectives in oxidative, non-oxidative and CO2-mediated dehydrogenation of propane and isobutane over metal oxide catalysts, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- B. Zhang and I. E. Wachs, Identifying the Catalytic Active Site for Propylene Metathesis by Supported ReOx Catalysts, ACS Catal., 2021, 11, 1962–1976 CrossRef CAS.
- T. Otroshchenko, Q. Zhang and E. V. Kondratenko, Room-Temperature Metathesis of Ethylene with 2-Butene to Propene Over MoOx-Based Catalysts: Mixed Oxides as Perspective Support Materials, Catal. Lett., 2022, 152, 2366–2374 CrossRef CAS.
- P. Jean-Louis Hérisson and Y. Chauvin, Catalyse de transformation des oléfines par les complexes du tungstène. II. Télomérisation des oléfines cycliques en présence d'oléfines acycliques, Makromol. Chem., 1971, 141, 161–176 CrossRef.
- Y. Chauvin, Olefin metathesis: The early days (Nobel lecture), Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef PubMed.
- P. Zhao, L. Ye, Z. Sun, B. T. W. Lo, H. Woodcock, C. Huang, C. Tang, A. I. Kirkland, D. Mei and S. C. Edman Tsang, Entrapped Single Tungstate Site in Zeolite for Cooperative Catalysis of Olefin Metathesis with Brønsted Acid Site, J. Am. Chem. Soc., 2018, 140, 6661–6667 CrossRef CAS PubMed.
- K. Amakawa, J. Kröhnert, S. Wrabetz, B. Frank, F. Hemmann, C. Jäger, R. Schlögl and A. Trunschke, Active Sites in Olefin Metathesis over Supported Molybdena Catalysts, ChemCatChem, 2015, 7, 4059–4065 CrossRef CAS.
- K. Amakawa, S. Wrabetz, J. Kröhnert, G. Tzolova-Müller, R. Schlögl and A. Trunschke, In Situ Generation of Active Sites in Olefin Metathesis, J. Am. Chem. Soc., 2012, 134, 11462–11473 CrossRef CAS PubMed.
- T. Otroshchenko, Q. Zhang and E. V. Kondratenko, Enhancing Propene Formation in the Metathesis of Ethylene with 2-Butene at Close to Room Temperature over MoOx/SiO2 through Support Promotion with P, Cl, or S, ACS Catal., 2021, 11, 14159–14167 CrossRef CAS.
- J. Handzlik, K. Kurleto and M. Gierada, Computational Insights into Active Site Formation during Alkene Metathesis over a MoOx/SiO2 Catalyst: The Role of Surface Silanols, ACS Catal., 2021, 11, 13575–13590 CrossRef CAS.
- K. W. Chan, D. Mance, O. V. Safonova and C. Copéret, Well-Defined Silica-Supported Tungsten(IV)–Oxo Complex: Olefin Metathesis Activity, Initiation, and Role of Brønsted Acid Sites, J. Am. Chem. Soc., 2019, 141, 18286–18292 CrossRef CAS PubMed.
- T. Hahn, U. Bentrup, M. Armbrüster, E. V. Kondratenko and D. Linke, The Enhancing Effect of Brønsted Acidity of Supported MoOx Species on their Activity and Selectivity in Ethylene/trans-2-Butene Metathesis, ChemCatChem, 2014, 6, 1664–1672 CrossRef CAS.
- V. Goelden, D. Linke and E. V. Kondratenko, Investigation of the Enhancing Effect of Solid Cocatalysts on Propene Formation in Ethene/trans-2-Butene Metathesis over MoOx/SiO2–Al2O3, ACS Catal., 2015, 5, 7437–7445 CrossRef CAS.
- Q. Zhang, T. Otroshchenko and E. V. Kondratenko, Fundamentals and application potential of the synergy effect between ZnO and Mo/SiO2 for propene production in the metathesis of ethylene and trans-2-butene, Catal. Sci. Technol., 2022, 12, 5210–5216 RSC.
- Q. Zhang, T. Xiao, C. Liu, T. Otroshchenko and E. V. Kondratenko, Performance Descriptors for Catalysts Based on Molybdenum, Tungsten, or Rhenium Oxides for Metathesis of Ethylene with 2-Butenes to Propene, Angew. Chem., Int. Ed., 2023, 62, e202308872 CrossRef CAS PubMed.
- K. A. Vikulov, B. N. Shelimov, V. B. Kazansky and J. C. Mol, Deactivation mechanism of photoreduced and cyclopropane-treated molybdena/silica catalysts for olefin metathesis, J. Mol. Catal., 1994, 90, 61–67 CrossRef CAS.
- V. Mougel, K.-W. Chan, G. Siddiqi, K. Kawakita, H. Nagae, H. Tsurugi, K. Mashima, O. Safonova and C. Copéret, Low Temperature Activation of Supported Metathesis Catalysts by Organosilicon Reducing Agents, ACS Cent. Sci., 2016, 2, 569–576 CrossRef CAS PubMed.
- P. Michorczyk, A. Węgrzyniak, A. Węgrzynowicz and J. Handzlik, Simple and Efficient Way of Molybdenum Oxide-Based Catalyst Activation for Olefins Metathesis by Methane Pretreatment, ACS Catal., 2019, 9, 11461–11467 CrossRef CAS.
- T. Z. H. Gani, Z. J. Berkson, R. Zhu, J. H. Kang, J. R. Di Iorio, K. W. Chan, D. F. Consoli, S. K. Shaikh, C. Copéret and Y. Román-Leshkov, Promoting active site renewal in heterogeneous olefin metathesis catalysts, Nature, 2023, 617, 524–528 CrossRef CAS PubMed.
- J. Handzlik and J. Ogonowski, DFT study of ethene metathesis proceeding on monomeric MoVI centres of MoO3/Al2O3 catalyst: The role of the molybdacyclobutane intermediate, J. Mol. Catal. A: Chem., 2002, 184, 371–377 CrossRef CAS.
- T. Otroshchenko, O. Reinsdorf, D. Linke and E. V. Kondratenko, A chemical titration method for quantification of carbenes in Mo- or W-containing catalysts for metathesis of ethylene with 2-butenes: verification and application potential, Catal. Sci. Technol., 2019, 9, 5660–5667 RSC.
- S. Lwin and I. E. Wachs, Olefin Metathesis by Supported Metal Oxide Catalysts, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- A. Chakrabarti and I. E. Wachs, Molecular Structure–Reactivity Relationships for Olefin Metathesis by Al2O3-Supported Surface MoOx Sites, ACS Catal., 2018, 8, 949–959 CrossRef CAS.
- E. M. Oliveira, M. Rogero, E. C. Ferreira and J. A. Gomes Neto, Simultaneous determination of phosphite and phosphate in fertilizers by Raman spectroscopy, Spectrochim. Acta, Part A, 2021, 246, 119025 CrossRef CAS PubMed.
- I. I. Abu and K. J. Smith, The effect of cobalt addition to bulk MoP and Ni2P catalysts for the hydrodesulfurization of 4,6-dimethyldibenzothiophene, J. Catal., 2006, 241, 356–366 CrossRef CAS.
- I. C. ten Have, E. Valle, A. Gallo, J. L. Snider, M. S. Duyar and T. F. Jaramillo, Development of Molybdenum Phosphide Catalysts for Higher Alcohol Synthesis from Syngas by Exploiting Support and Promoter Effects, Energy Technol., 2019, 7, 1801102 CrossRef.
- J. Kibsgaard and T. F. Jaramillo, Molybdenum Phosphosulfide: An Active, Acid-Stable, Earth-Abundant Catalyst for the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2014, 53, 14433–14437 CrossRef CAS PubMed.
- Y. Maki, K. Sato, A. Isobe, N. Iwasa, S. Fujita, M. Shimokawabe and N. Takezawa, Structures of H3PO4/SiO2 catalysts and catalytic performance in the hydration of ethene, Appl. Catal., A, 1998, 170, 269–275 CrossRef CAS.
- Y.-K. Park, S. J. Kim, N. You, J. Cho, S. J. Lee, J. H. Lee and J.-K. Jeon, MoO3/SiO2 catalysts for double bond migration of 2-butene, J. Ind. Eng. Chem., 2011, 17, 186–190 CrossRef CAS.
- M. Jabłońska, An Application of Steady-state Isotopic-transient Kinetic Analysis (SSITKA) in DeNOx Process, ChemCatChem, 2021, 13, 818–827 CrossRef.
- S. L. Shannon and J. G. Goodwin, Characterization of Catalytic Surfaces by Isotopic-Transient Kinetics during Steady-State Reaction, Chem. Rev., 1995, 95, 677–695 CrossRef CAS.
- C. Copéret, Z. J. Berkson, K. W. Chan, J. de Jesus Silva, C. P. Gordon, M. Pucino and P. A. Zhizhko, Olefin metathesis: what have we learned about homogeneous and heterogeneous catalysts from surface organometallic chemistry?, Chem. Sci., 2021, 12, 3092–3115 RSC.
- K. Yamamoto, K. W. Chan, V. Mougel, H. Nagae, H. Tsurugi, O. V. Safonova, K. Mashima and C. Copéret, Silica-supported isolated molybdenum di-oxo species: formation and activation with organosilicon agent for olefin metathesis, Chem. Commun., 2018, 54, 3989–3992 RSC.
- M. Myradova, A. Węgrzynowicz, A. Węgrzyniak, M. Gierada, P. Jodłowski, J. Łojewska, J. Handzlik and P. Michorczyk, Tuning the metathesis performance of a molybdenum oxide-based catalyst by silica support acidity modulation and high temperature pretreatment, Catal. Sci. Technol., 2022, 12, 2134–2145 RSC.
- J. Handzlik, Theoretical Investigations of Isolated Mo(VI) and Mo(IV) Centers of a Molybdena–Silica Catalyst for Olefin Metathesis, J. Phys. Chem. C, 2007, 111, 9337–9348 CrossRef CAS.
- J. Handzlik and P. Sautet, Active sites of olefin metathesis on molybdena-alumina system: A periodic DFT study, J. Catal., 2008, 256, 1–14 CrossRef CAS.
- J. Handzlik, J. Ogonowski and R. Tokarz-Sobieraj, Dependence of metathesis activity of Mo-methylidene sites on their location on (100) γ-Al2O3—a theoretical study, Catal. Today, 2005, 101, 163–173 CrossRef CAS.
- J. Handzlik, Metathesis Activity and Properties of Mo–Alkylidene Sites Differently Located on Silica. A Density Functional Theory Study, J. Phys. Chem. B, 2005, 109, 20794–20804 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06833e |
| This journal is © The Royal Society of Chemistry 2025 |