
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-target macrocycles: pyrogallol derivatives to control multiple pathological factors associated with Alzheimer's disease†
Jimin
Kwak‡
,
Yelim
Yi‡
,
Seongmin
Park
and
Mi Hee
Lim
 *
*
Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: miheelim@kaist.ac.kr
First published on 3rd December 2024
Abstract
Designing multi-target chemical tools is a vital approach to understanding the pathology of Alzheimer's disease (AD), which involves a complex network of pathological factors, such as free organic radicals, amyloid-β (Aβ), and metal-bound Aβ (metal–Aβ). The pyrogallol moiety, known for its ability to lower redox potentials and interact with both Aβ and metal ions, presents a promising framework for this molecular design. Here we show how simple structural variations of pyrogallol can be used to enhance its ability to scavenge free organic radicals and regulate the aggregation of both metal-free Aβ and metal–Aβ. By incorporating multiple pyrogllol units into a macrocyclic scaffold via methylene bridges, we achieve synergistic reactivity against several pathological targets. Our structure–reactivity relationship studies also reveal that the macrocyclic structure noticeably improves antioxidant activity as well as interactions with both Aβ and metal ions, leading to oxidation of Aβ peptides and influencing their conformation and aggregation in both the absence and presence of metal ions. This work demonstrates the potential of simple redox-active structural entities in developing multifunctional chemical reagents that effectively manage the pathological components associated with AD.
Introduction
Alzheimer's disease (AD) is the most common form of dementia, characterized by a decline in cognitive functions, such as memory and language skills. Currently, more than 55 million people worldwide are affected by dementia, with over 10 million new cases emerging every year.1 Numerous efforts to understand the etiology of AD have identified multiple pathological elements, including free radicals,2,3 amyloid-β (Aβ),4–7 metal ions,8,9 and metal-bound Aβ (metal–Aβ),10–13 and their intertwined connections.3,14 The brains of AD patients are particularly vulnerable to oxidative stress, triggered by the overproduction of free radicals, which can damage biomolecules and induce cellular death.9,15 Additionally, Aβ peptides are prone to aggregation, forming neurotoxic aggregates that accumulate as senile plaques (SPs) in AD-affected brains.16,17 High concentrations of metal ions [e.g., Cu(I/II) and Zn(II)] have also been detected in the SPs,18 where they can coordinate to Aβ, subsequently altering their aggregation dynamics.19–21 In particular, redox-active metal ions, such as Cu(I/II), can produce reactive oxygen species through Fenton-like reactions and exacerbate oxidative stress.14 To date, various chemical tools and therapeutic candidates, including antioxidants,3 anti-Aβ drugs,22 and metal chelators,3,23,24 have been developed; however, their effectiveness and safety remain controversial, underscoring the limitations of single-target-based compounds and the need for multi-target-directed chemical reagents to address the multifactorial nature of AD.3,14,25–30To delineate the complex pathologies of AD, the multiple reactivities of some natural products, such as myricetin,26,31 epigallocatechin-3-gallate,32 and baicalein,33 have been identified against free organic radicals, metal-free Aβ, metal ions, and metal–Aβ.34–36 Interestingly, these compounds share a common structure, the pyrogallol moiety, implying its potential in engaging their multifunctional properties. Thus, in this study, we rationally designed a series of pyrogallol-based compounds and evaluated their abilities to scavenge free organic radicals, interact with Aβ and metal ions, and influence the assembly of Aβ in the absence and presence of metal ions, as depicted in Fig. 1. We established the relationship between the chemical structures of these compounds and their reactivities against the pathological factors based on variations in the number of hydroxyl groups on the pyrogallol moiety, arranged in either methylene-linked macrocyclic or monomeric forms. Our findings demonstrate that simple structural modifications onto the pyrogallol group can serve as a foundation for developing versatile chemical reagents to elucidate the intertwined pathologies of AD.
 | ||
| Fig. 1 Multiple pathogenic factors and rationally selected compounds studied in this work. (a) Design rationale of compound 1 as a chemical reagent for targeting and controlling four multiple pathological factors associated to AD, including free radicals, metal-free Aβ (PDB 2LFM37 and 2M4J7), metal ions, and metal–Aβ. (b) Amino acid sequences of Aβ40 and Aβ42. Amino acid residues involved in the metal-coordination and self-recognition sites are highlighted in bold/orange and bold/underline, respectively. (c) Chemical structures of 2–6 selected based on 1's structural variations. The number of hydroxyl groups on the aromatic rings with and without the cyclization was changed, as highlighted in gray and blue, respectively. 1, pyrogallol[4]arene [1,3,5,7(1,3)-tetrabenzenacyclooctaphane-14,15,16,34,35,36,54,55,56,74,75,76-dodecaol]; 2, resorcin[4]arene [1,3,5,7(1,3)-tetrabenzenacyclooctaphan-14,16,34,36,54,56,74,76-octaol]; 3, calix[4]arene [1,3,5,7(1,3)-tetrabenzenacyclooctaphane]; 4, 4-methylpyrogallol (4-methylbenzene-1,2,3-triol); 5, 4-methylresorcinol (4-methylbenzene-1,3-diol); 6, toluene (methylbenzene). | ||
Results and discussion
Rational selection and preparation of pyrogallol derivatives
As outlined in Fig. 1a, chemical and biological properties of pyrogallol are reported to be suitable for targeting and controlling multiple pathological features of AD: (i) three electron-donating hydroxyl groups attached to an aromatic ring characterize the electronic properties of pyrogallol, contributing to its redox potential and antioxidant ability;38,39 (ii) the oxygen (O) donor atoms in these hydroxyl groups can chelate metal ions in a bidentate manner; (iii) the amphiphilicity of pyrogallol facilitates the interaction with Aβ via hydrogen bonding and hydrophobic associations, thereby modulating Aβ aggregation (Fig. 1b).31–33,40 Given these characteristics, we selected the pyrogallol group for our molecular design. We then questioned whether incorporating multiple pyrogallol moieties within a single structural entity could enhance the reactivity towards our desired pathological targets. To maximize pyrogallol's potential, we considered arranging them in a macrocyclic scaffold using methylene bridges. The semi-rigidity driven by a methylene-linked macrocyclic structure allows the molecule to optimize binding configurations and enhance interactions with peptides and proteins.41 Given that the macrocyclization of pyrogallols forms the intramolecular hydrogen bonds within its scaffold, ensuing thermodynamic stability and the ability to adopting various conformations (e.g., cone, chair, and boat),42–44 the macrocyclization of pyrogallol moieties with methylene bridges can further improve its interactions with Aβ species. Consequently, we selected compound 1 for further study, as illustrated in Fig. 1a and c.To identify the involvement of the number of hydroxyl groups on the pyrogallol moieties in compound 1 in its reactivities towards the pathological components, we replaced the pyrogallol moieties in compound 1. As described in Fig. 1c, the incorporation of resorcinol, which contains two hydroxyl groups at the meta position, resulted in compound 2 with relatively increased hydrophobicity compared to compound 1. This increase in hydrophobicity facilitates hydrophobic interactions with Aβ. Furthermore, compound 3 without hydroxyl groups was selected to further enhance its hydrophobic property, thereby improving hydrophobic contacts with Aβ. It should be noted that compound 3 was included in our chemical series to probe the reactivities of the backbone structure for compounds 1 and 2 against our targeted pathological factors.
Additionally, the variation in the number of electron-donating hydroxyl groups on the pyrogallol structure can vary the redox potentials of the compounds as well as their capacity to scavenge free organic radicals. Concurrently, unlike the catechol moieties in the pyrogallol groups of compound 1, the metal-chelation effect can be limited in compounds 2 and 3, which further impacts the reactivity with metal–Aβ. In addition to compounds 1–3, we included their monomeric counterparts, represented by compounds 4–6 (Fig. 1c), to assess the relevance of macrocyclization in directing the versatile reactivities of these compounds against pathological components.
The selected compounds were synthesized following previously reported procedures42,45 with modifications (for compounds 1–3) and obtained from commercial sources (for compounds 4–6). As presented in Scheme S1a,† compounds 1 and 2 were prepared by a condensation reaction of pyrogallol and resorcinol, respectively, with formaldehyde under basic conditions.42 For the synthesis of compound 3, as shown in Scheme S1b,† the nucleophilic substitution reaction of 3a on (OEt)2POCl in the presence of Bu4NBr yielded compound 3b. Subsequent dephosphorylation of 3b with potassium metal in liquid ammonia afforded compound 3c. Friedel–Crafts dealkylation of 3c with AlCl3 produced the final product 3.45 The characterization of compounds 1–6, as well as intermediates 3b and 3c, is summarized in Fig. S1–S8.†
Redox potentials and antioxidant ability
Since the redox properties of the compounds are associated with their antioxidant ability, the redox potentials of compounds 1–6 were first computed using the previously reported density functional theory (DFT) calculations.46 As depicted in Fig. 2a, the redox potentials [E° values vs. standard hydrogen electrode (SHE)] of the compounds were calculated to decrease as the number of hydroxyl groups on the benzene ring increased. Among the macrocyclic compounds, compound 1 exhibited the lowest redox potential at 1.042 V, with the redox potentials increasing in the order of compounds 2 and 3 with values of 1.084 V and 1.612 V, respectively. Similarly, their monomeric counterparts showed an increase in redox potentials with a decrease in the number of hydroxyl groups on the benzene ring, in the order of 4, 5, and 6, ranging from 1.106–1.835 V. Additionally, the redox potentials of the macrocyclic compounds 1–3 were computed to be lower than those of their monomeric counterparts 4–6. | ||
| Fig. 2 Redox properties of compounds and their ability to quench free organic radicals. (a) Calculated redox potentials (E° vs. SHE), HOMO and LUMO energies, and HOMO–LUMO energy gap of 1–6 and their TEAC values determined by the TEAC assay. The solvation energy in DFT calculations was treated with CH3OH (ε = 32.7). Conditions for the TEAC assay: CH3OH; 25 °C; λabs = 745 nm. an.d., not determined due to the limited solubility or antioxidant activity under our experimental conditions. (b) Isosurface plots (isodensity value = 0.03 a.u.) of their HOMOs. | ||
Second, the redox properties of compounds 1, 2, 4, and 5 were investigated by cyclic voltammetry in CH3CN. All measured compounds displayed irreversible oxidation peaks, as presented in Fig. S9,† making it difficult to obtain the E1/2 values. The anodic peak potentials (Epa) of compounds 1 and 4 were 0.47 V and 0.41 V at 250 mV s−1vs. Ag/Ag(I), respectively, consistent with previously reported values for the pyrogallol moiety.47 For compounds 2 and 5, Epa values of 0.63 V and 0.45 V, respectively, were observed, which aligns with the trend of increasing redox values as the number of hydroxyl groups on the benzene ring decreases. It should be noted that we were unable to obtain cyclic voltammograms of compounds 3 and 6 due to their limited solubility in CH3CN.
We next examined the ability of the compounds to quench free organic radicals by the Trolox equivalent antioxidant capacity (TEAC) assay, which employs the cationic radical form of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (Fig. 2a). Compounds 1, 2, 4, and 5 displayed TEAC values greater than 1.0, relative to the vitamin E analog Trolox,48 indicating that their radical scavenging ability is superior to that of Trolox. Aligning with our DFT calculations, compound 1 demonstrated the highest TEAC value [3.64 (±0.04)] among the compounds, which presents a strong capacity to quench free organic radicals. As expected, compound 6, which has a relatively higher redox potential, showed negligible radical scavenging ability. It should be noted that the TEAC value for compound 3 could not be determined due to its limited solubility in CH3OH.
To better understand the redox potentials and radical scavenging abilities of the compounds, we analyzed their redox activity at the orbital level. The HOMO (highest occupied molecular orbital) and LUMO (lowest occupied molecular orbital) levels are closely linked to the oxidation and reduction potentials, respectively. Additionally, the energy gap between the HOMO and LUMO is associated with the redox potential, as a smaller energy gap correlates with a lower redox potential, influencing a compound's tendency for oxidation or reduction.49 As illustrated in Fig. 2b, compound 1 presented the highest HOMO level (−5.633 eV) among compounds 1–6, with a decrease in the HOMO level observed for compounds 2 and 3, which have fewer hydroxyl groups at the orbital lobes-localized portions. This trend was consistently observed for the monomeric compounds 4–6. The orbital lobes localized at the hydroxyl groups in the HOMO of the compounds suggest that a greater number of electron-donating hydroxyl functionalities on the benzene ring may elevate the HOMO level. In addition, a comparison of the HOMO levels between compounds 1–3 and 4–6 revealed that macrocyclization of the monomeric units tends to reduce the HOMO level. Taken together, an increase in the number of hydroxyl groups on the benzene ring, along with macrocyclization, correlates with the compounds' ability to scavenge free radicals, with corresponding reductions in redox potentials and increment in HOMO levels. Notably, compound 1, which contains multiple pyrogallol moieties in a macrocyclic configuration, highlights the importance of the pyrogallol moieties and their macrocyclization in enhancing the antioxidant ability of these compounds.
Interactions with Aβ and Cu(II)
Possible interactions between the compounds and Aβ were visualized by molecular docking simulations employing the monomeric structure of Aβ40 (PDB 2LFM37) identified by nuclear magnetic resonance spectroscopy in aqueous solutions. As displayed in Fig. 3a, all compounds were situated in a pocket near the N-terminal and α-helical regions of Aβ40. For the compounds containing pyrogallol functionality, compounds 1 and 4 could form hydrogen bonds with Aβ through their hydroxyl groups. Specifically, the hydroxyl moieties of compound 1 acted as hydrogen-bond donors, interacting with the side chains of Arg5 (guanidinium group), Gln15 (amide moiety), Lys16 (amino group), and Glu22 (carboxylic acid), as well as the backbone amide and carbonyl groups between Ser8 and His13. Similarly, the hydroxyl groups in compound 4 formed hydrogen bonds with the guanidinium group of Arg5 and the backbone amide group between His6 and Asp7. Additionally, C–H–π interactions were observed between the benzene rings of compounds 1 and 4 with the side chains of Lys16 and Arg5, respectively. Given that amino acid residues adjacent to the β-turn motifs (Val12–Gln15, Ala21–Asp23),50–52 self-recognition site (Leu17–Ala21),52 and salt bridge (Lys16–Glu22)50,53 are critical for structural transition during the fibrillization of Aβ, therefore, such intermolecular contacts of compounds 1 and 4 with the peptide may affect its conformation and aggregation dynamics. | ||
| Fig. 3 Interactions of compounds with monomeric Aβ40 and Cu(II). (a) Possible interactions of compounds 1–6 with monomeric Aβ40 (PDB 2LFM37) visualized by docking studies. The representative models with the highest binding affinity towards Aβ40 are shown in the figure. Binding energies between compounds 1–6 and Aβ40 are −10.1, −8.8, −7.6, −4.7, −4.3, and −4.0 kcal mol−1, respectively. Other docked models of compounds 1–6 against Aβ40 are presented in Fig. S10.† Hydrogen bonds (2.0–4.0 Å) and C–H–π interactions (3.3–4.1 Å) between compounds and Aβ40 are indicated with dashed lines. O, H, and N atoms are indicated in red, white, and blue, respectively. (b) Electronic absorbance spectra of compound 4 obtained upon addition of Cu(II). Conditions: [compound] = 250 μM (1% v/v DMSO); [Cu(II)] = 250 and 500 μM; H2O; 37 °C. | ||
Although compounds 2 and 5 contain the resorcinol functionality, which has fewer hydroxyl groups than the pyrogallol in compounds 1 and 4, they still interact with Aβ through hydrogen bonding with the side chains of Arg5 and Gln15 and the backbone carbonyl and amide moieties of Ser8–Gly9, Gly9–Tyr10, and Tyr10–Glu11. The additional hydroxyl groups in the macrocyclic scaffold of compound 2 were further involved in hydrogen bonding with Lys16 and Glu22. C–H–π interactions of the benzene rings of compounds 2 and 5 were visualized with Lys16 (for 2) and Gln15 (for 5). Compounds 3 and 6, which lack hydrogen-bond donors, can contact with Aβ via C–H–X (where X = O or N) interactions, such as Arg5 (for 3 and 6), Ser8, Arg5–His6, Tyr10–Glu11, and Gln15 (for 3), and Phe4–Arg5 (for 6). C–H–π interactions were also observed with Phe4 (for 3 and 6), Arg5 (for 3), and Val18 (for 6). Collectively, all compounds can interact with Aβ through various intermolecular interactions, including hydrogen bonding and hydrophobic interactions. Our observations suggest that the macrocyclic compounds (1–3), compared to their monomeric counterparts (4–6), benefit from a relatively large surface area, which may enhance interactions with Aβ. Additionally, a greater number of hydroxyl groups on the benzene ring increases the number of hydrogen-bond donors, making the pyrogallol-containing compounds (1 and 4) more favorable for hydrogen bonding with Aβ compared to the resorcinol-containing compounds (2 and 5) and those without hydroxyl groups (3 and 6). Thus, compound 1, which contains multiple pyrogallol moieties in a macrocyclic scaffold, may preferentially associate with Aβ through a combination of multiple hydrogen bonds and hydrophobic interactions.
We further investigated whether the pyrogallol moiety, including the O donor atoms in its hydroxyl groups, can interact with Cu(II) in aqueous solution by electronic absorption (Abs) spectroscopy. As illustrated in Fig. 3b, the addition of increasing concentrations of Cu(II) to a solution of compound 4 resulted in a gradual increase in Abs at 270 nm, along with the appearance of a broad optical band at 340 nm. Previous studies have reported that such spectral changes can occur when pyrogallol is oxidized into 1,2-benzoquinone-3-ol upon interaction with Cu(II).38,54–57 These observations suggest that the pyrogallol-containing compounds, 1 and 4, can interact with Cu(II). It should be noted that we were unable to quantitatively measure the Cu(II)-binding affinity of compound 4 due to the limited stability of Cu(II)–4 complexes, and the Abs spectrum of compound 1 treated with Cu(II) could not be obtained because of its limited aqueous solubility in the presence of Cu(II).
Impact on the formation of Aβ aggregates in the absence and presence of metal ions
To determine the influence of the compounds on the formation of Aβ aggregates, we monitored the molecular weight (MW) distribution and morphology of the resultant metal-free Aβ and metal–Aβ species by gel electrophoresis with western blotting (gel/western blot) using an anti-Aβ antibody (6E10) and transmission electron microscopy (TEM), respectively (Fig. 4a). Of note, relatively smaller Aβ aggregates (ca. <245 kDa) were analyzed by gel/western blot, while larger species that cannot penetrate the gel matrix were visualized by TEM. As illustrated in Fig. 4b, 5a, and S11a,† upon treatment of compounds 1, 2, and 4, the MW distribution of Aβ40 both in the absence and presence of metal ions was notably changed. When compound 1 was incubated with metal-free Aβ40, the intensity of the bands across the 11–245 kDa range increased, while new bands at 5–48 kDa and around 245 kDa were appeared upon the addition of compound 2 to Aβ40 under metal-free conditions. Compound 4 intensified the bands corresponding to metal-free Aβ40 aggregates with relatively small (5–11 kDa) and large (ca. 245 kDa) MW. In the case of metal–Aβ40 samples, an amplified variation in the MW distribution with compounds 1, 2, and 4 was indicated. Conversely, compounds 3, 5, and 6 showed minimal or no significant reactivity towards both metal-free Aβ40 and metal–Aβ40.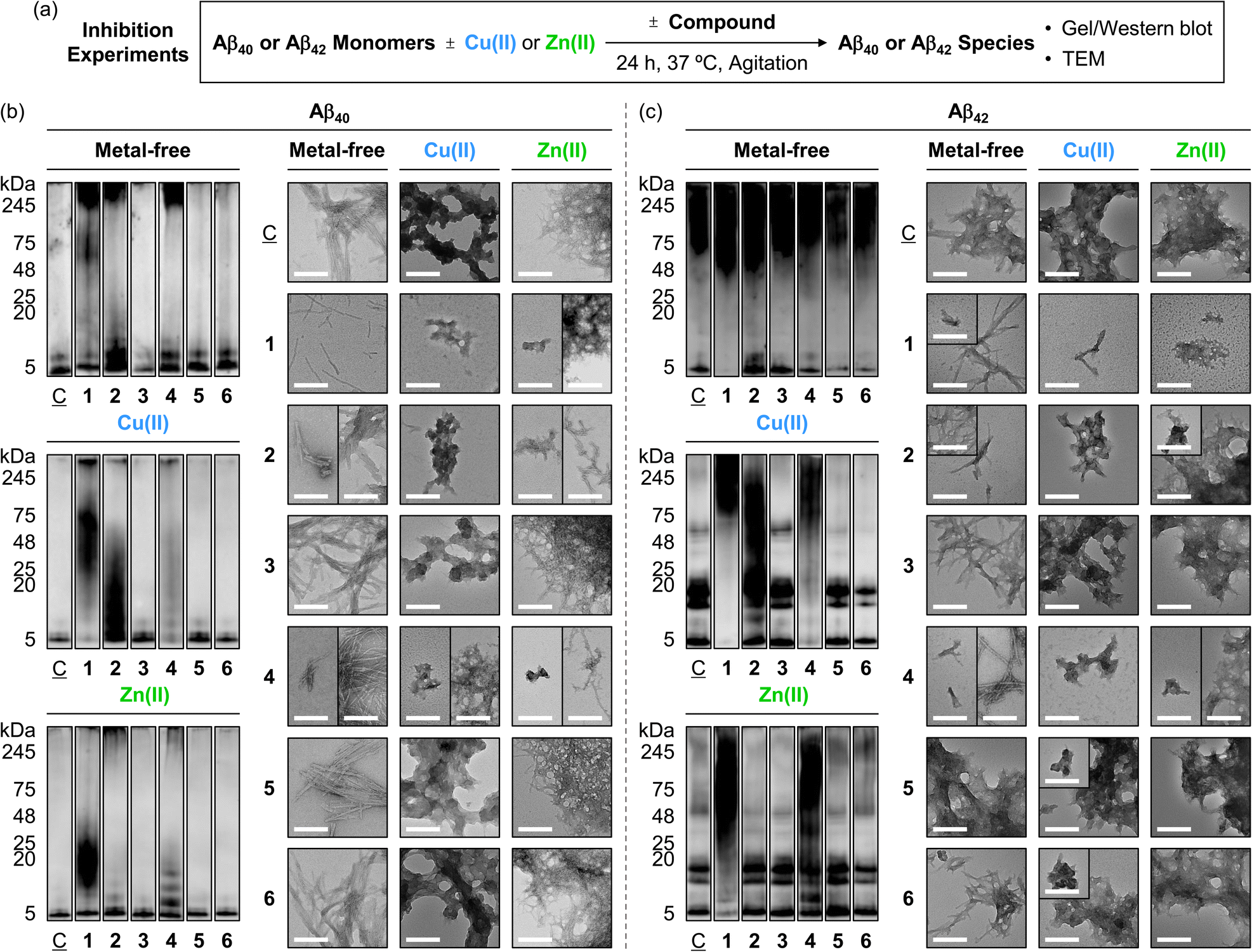 | ||
Fig. 4 Effects of 1–6 on the formation of metal-free and metal-induced Aβ aggregates. (a) Scheme of the inhibition experiments. Analyses of the MW distribution and morphology of the resultant (b) Aβ40 and (c) Aβ42 aggregates by gel/western blot with an anti-Aβ antibody (6E10) and TEM, respectively. Lanes: (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ) Aβ ± Cu(II) or Zn(II); (1) + 1; (2) + 2; (3) + 3; (4) + 4; (5) + 5; (6) + 6. The original gel images are shown in Fig. S11.† Conditions: [Aβ] = 25 μM; [Cu(II) or Zn(II)] = 25 μM; [1, 2, or 3] = 25 μM and [4, 5, or 6] = 100 μM (1% v/v DMSO); 20 mM HEPES, pH 7.4 [for metal-free and Zn(II)-added samples] or pH 6.8 [for Cu(II)-treated samples], 150 mM NaCl; 37 °C; 24 h; constant agitation. Scale bars = 200 nm. ) Aβ ± Cu(II) or Zn(II); (1) + 1; (2) + 2; (3) + 3; (4) + 4; (5) + 5; (6) + 6. The original gel images are shown in Fig. S11.† Conditions: [Aβ] = 25 μM; [Cu(II) or Zn(II)] = 25 μM; [1, 2, or 3] = 25 μM and [4, 5, or 6] = 100 μM (1% v/v DMSO); 20 mM HEPES, pH 7.4 [for metal-free and Zn(II)-added samples] or pH 6.8 [for Cu(II)-treated samples], 150 mM NaCl; 37 °C; 24 h; constant agitation. Scale bars = 200 nm. | ||
 | ||
| Fig. 5 Summary of the (a) gel/western blot and (b) TEM results obtained from the inhibition experiments described in Fig. 4. aThe circle symbol (O) indicates a noticeable effect, compared to the control samples containing Aβ with and without metal ions. bThe dash (–) denotes no significant changes, relative to the control samples. cThe triangle symbol (△) represents relatively mild or less reactivity. | ||
TEM studies further supported the reactivity of compounds 1, 2, and 4 with Aβ40 in the absence and presence of metal ions. Incubation of compound 1 with metal-free Aβ40 produced thinner and shorter fibrils, distinct from the dense fibril bundles observed in compound-untreated metal-free Aβ40. Compounds 2 and 4 also induced the morphological transitions towards Aβ40 under metal-free conditions, showing smaller and chopped fibrils. In the case of Cu(II)- and Zn(II)-associated Aβ40, small amorphous aggregates with and without fibrillary species were visualized upon the addition of compounds 1, 2, and 4, whereas large assemblies of amorphous or filamentous structures were observed in the compound-free metal–Aβ40. The morphologies and sizes of aggregates in Aβ40 samples containing compounds 3, 5, and 6 were similar to those of the compound-free samples, both in the absence and presence of metal ions, as summarized in Fig. 5b.
Furthermore, we assessed the reactivity of compounds towards the aggregation propensity of Aβ42, another major isoform of Aβ, with Aβ40,9 as presented in Fig. 4c, 5a, and S11a.† In the absence of metal ions, compound 1 notably varied the MW distribution of Aβ42 at ca. 5 kDa and 48–75 kDa. Compound 2 increased the intensity of smearing at 5–11 kDa and 48–75 kDa. A slight reduction in signal intensity at ca. 5 kDa was observed upon incubation of compounds 5 and 6 with metal-free Aβ42, while compounds 3 and 4 did not noticeably affect the size distribution under our gel/western blot experiments. TEM investigations showed that shorter fibrils were produced when Aβ42 was treated with compounds 1, 2, 4, and 6 with metal-free Aβ42, in contrast to the longer fibrils observed for compound-untreated and 3-treated samples. Incubation with compound 5 led to the formation of amorphous metal-free Aβ42 aggregates.
For Cu(II)–Aβ42, compounds 1, 2, and 4 modified the MW distribution of the resultant aggregates, relative to that of compound-free sample, to different extent. Compounds 3, 5, and 6 exhibited relatively mild reactivity towards Cu(II)–Aβ42, with shifts in the MW distribution at 48–75 kDa (for 3) and 11–20 kDa/48–75 kDa (for 5 and 6). As shown in the TEM studies, the 1-added Cu(II)–Aβ42 showed chopped filamentous species, while compounds 2 and 4–6 formed smaller aggregates. The 3-treated Cu(II)–Aβ42 sample displayed relatively large-sized aggregates, similar to those observed in the compound-free sample.
The samples of Zn(II)–Aβ42 with compounds 1 and 4 displayed reduced signal intensity at 5–20 kDa, with intense smearing bands larger than ca. 25 kDa, and the formation of new bands at 5–11 kDa and 25–245 kDa, respectively, showing smaller unstructured aggregates. This morphology was similar to those of Zn(II)–Aβ42 incubated with compound 2, which exhibited slightly reduced intensity of smearing bands at 48–245 kDa. Compounds 3 and 5 changed signal intensity at 48–245 kDa and 5–11 kDa. The MW distribution of Zn(II)–Aβ42 was not notably altered by incubation with compound 6. TEM studies revealed that the dense network of fibrils observed in the compound-free Zn(II)–Aβ42 sample remained unchanged in the samples with 3, 5, and 6. Taken together, our inhibition studies elucidate the significant reactivity of pyrogallol-containing compounds (1 and 4) towards both metal-free Aβ and metal–Aβ, implying that pyrogallol moiety acts critical in modulating the aggregation of Aβ with and without metal ions. Additionally, the comparison of macrocyclic compounds (1 and 2) with their monomeric counterparts (4 and 5) supports that the conformation status of the compounds can differ their effects on Aβ aggregation in the absence and presence of metal ions.
Reactivity with preformed Aβ aggregates with and without metal ions
To investigate the ability of compounds to disassemble preformed Aβ aggregates or modulate their further aggregation pathways, metal-free Aβ or metal–Aβ was preincubated for 24 h to form peptide aggregates and the compounds were treated, with an additional 24 h incubation (Fig. 6a). As depicted in Fig. 6b, 7a, and S11b,† compound 1 exhibited weaker band intensity at 4–11 kDa, corresponding to monomeric and dimeric Aβ40, in the absence of metal ions. A slight reduction in band intensity at ca. 5 kDa was observed upon incubation of metal-free Aβ40 with compound 4. Compounds 2, 3, 5, and 6 did not significantly alter the MW distribution of Aβ40 without metal ions. To clarify whether the Aβ40 species resulting from treatment with compound 1 could not penetrate the gel matrix or if 6E10 antibody could not sufficiently recognize the Asp1–Lys16 residues of the resultant Aβ40,58 we additionally performed gel/western blot analysis using anti-Aβ 22–35 antibody, which has an epitope that includes Glu22–Met35.59 The results showed that compound 1 generated gel-permeable Aβ40 aggregates in the range of 11–245 kDa and reduced the amount of monomeric Aβ40 at around 4 kDa. In the TEM images, various sizes of metal-free Aβ40 fibrils, with and without amorphous aggregates, were observed in the 1-, 2-, and 4-added samples. This was different from the filamentous species detected in the samples without compounds and in those added with compounds 3, 5, and 6, as summarized in Fig. 7b. In the case of metal-free Aβ42, the use of 6E10 revealed that compound 1 reduced the signal intensity across the gel lane from 5 kDa to 245 kDa, and compounds 2–6 weakened the band intensity at ca. 5 kDa and 75–245 kDa (Fig. 6c and S11b†). With the anti-Aβ 22–35 antibody, the disappearance of bands at 5–11 kDa along with intensified signals at 48–75 kDa, was observed with the addition of compound 1. TEM analysis revealed that treatment with compounds 1, 2, and 4 resulted in metal-free Aβ42 aggregates, characterized by thinner and shorter fibrils and amorphous species. In contrast, longer fibrils were observed in the compound-free sample as well as those treated with 3, 5, and 6, as schematically depicted in Fig. 7b. | ||
| Fig. 6 Influence of 1–6 on preformed metal-free Aβ and metal–Aβ aggregates. (a) Scheme of the disaggregation experiments. Analyses of the MW distribution and morphology of the resultant (b) Aβ40 and (c) Aβ42 aggregates through gel/western blot with two antibodies [e.g., 6E10 and anti-Aβ 22–35] and TEM, respectively. Lanes: () Aβ ± Cu(II) or Zn(II); (1) + 1; (2) + 2; (3) + 3; (4) + 4; (5) + 5; (6) + 6. The original gel images are shown in Fig. S11.† Conditions: [Aβ] = 25 μM; [Cu(II) or Zn(II)] = 25 μM; [1, 2, or 3] = 25 μM and [4, 5, or 6] = 100 μM (1% v/v DMSO); 20 mM HEPES, pH 7.4 [for metal-free and Zn(II)-added samples] or pH 6.8 [for Cu(II)-treated samples], 150 mM NaCl; 37 °C; 24 h; constant agitation. Scale bars = 200 nm. | ||
 | ||
| Fig. 7 Summary of the (a) gel/western blot and (b) TEM results obtained from the disaggregation experiments described in Fig. 6. aThe circle symbol (O) indicates a noticeable effect, compared to the control samples containing Aβ with and without metal ions. bThe dash (–) denotes no significant changes, relative to the control samples. cThe triangle symbol (△) represents relatively mild or less reactivity. | ||
In the presence of metal ions, the MW distribution of Aβ40 and Aβ42 aggregates incubated with compounds 1, 2, and 4 was significantly varied. In contrast, compounds 3, 5, and 6 exhibited mild or no reactivity against preformed metal–Aβ aggregates in gel/western blot experiments. When we used anti-Aβ 22–35 antibody, in the presence of Cu(II), smearing throughout 5–245 kDa were detected for the 1- and 4-treated sample, different from compound-unadded Cu(II)–Aβ42. For Zn(II)–Aβ42, the aggregates probed by anti-Aβ 22–35 antibody indicated the signal intensity changing at ca. 5 kDa and 75–245 kDa upon the addition of compound 1. Morphological analysis indicated that smaller aggregates with both amorphous and fibrillary characteristics were formed when compounds 1, 2, 3, and 4 were incubated with Cu(II)–Aβ40. In contrast, the compounds-untreated samples displayed a bunch of fibrils, while compounds 5 and 6 also changed the morphology of Cu(II)–Aβ40 aggregates, resulting in amorphous aggregates. For Zn(II)–Aβ40, the samples treated with compounds 1, 2, and 4 showed chopped fibrils mixed with unstructured aggregates, whereas the compound-free sample and those added with compounds 3, 5, and 6 exhibited filamentous species. In the case of metal–Aβ42, the samples incubated with compounds 1, 2, and 4 presented smaller amorphous aggregates with or without thinner fibrils, while the compound-untreated and 3-, 5-, and 6-treated samples displayed relatively larger aggregates.
Collectively, our inhibition and disaggregation studies demonstrate that the pyrogallol-containing compounds (1 and 4) significantly affect the aggregation of Aβ in both the absence and presence of metal ions. These results suggest the importance of pyrogallol functionality, which is responsible for Aβ interactions through a combination of hydrogen bonding, hydrophobic contacts, and metal binding, in directing the reactivity towards metal-free Aβ and metal–Aβ. The distinct reactivities of the macrocyclic compounds (1–3) and their monomeric counterparts (4–6) towards Aβ assembly with and without metal ions are supported by their different degrees of interactions with Aβ and metal ions, depending on the number of hydroxyl groups attached to the benzene ring. Moreover, the methylene-linked macrocyclization of pyrogallol, resorcinol, and benzene units from 4–6 into 1–3 influenced the aggregation of Aβ under both metal-free and metal-present conditions. In particular, the macrocyclic compound 2 notably modified the assembly pathways of metal-free Aβ and metal–Aβ, while its monomeric counterpart 5 displayed minor or no reactivity. These observations imply that the interaction with Aβ amplified by the increased surface area resulting from the macrocyclization of monomeric units, can contribute to the reactivity towards Aβ in the absence and presence of metal ions. Furthermore, compounds 3 and 6 displayed little or no reactivity, indicating that the hydrophilicity of compounds, with the assistance of hydrophobicity, may play a role in determining their ability to modulate the aggregation of Aβ with and without metal ions.
Chemical modifications onto Aβ
To provide the mechanistic details on how metal-free Aβ and Cu(II)–Aβ can be chemically modified by the compounds 1–6, we analyzed the samples of Aβ40 incubated with the compounds, both in the absence and presence of Cu(II) by electrospray ionization-mass spectrometry (ESI-MS). As illustrated in Fig. 8a, metal-free Aβ40 treated with compounds 1, 2, and 4 displayed a new peak at 1449 m/z, with a mass shift of +16 Da from the monomeric Aβ40, corresponding to singly oxidized metal-free Aβ40. Concurrently, we observed chemical transformations of pyrogallol-containing compounds 1 and 4 into pyrogallol quinone or purpurogallin derivatives (Fig. S12†), while Aβ40 oxidation was not detected under anaerobic conditions, as illustrated in Fig. S13.† These observations suggest that O2 plays a role in the oxidation of compounds and the subsequent production of ROS, which can oxidize Aβ.60,61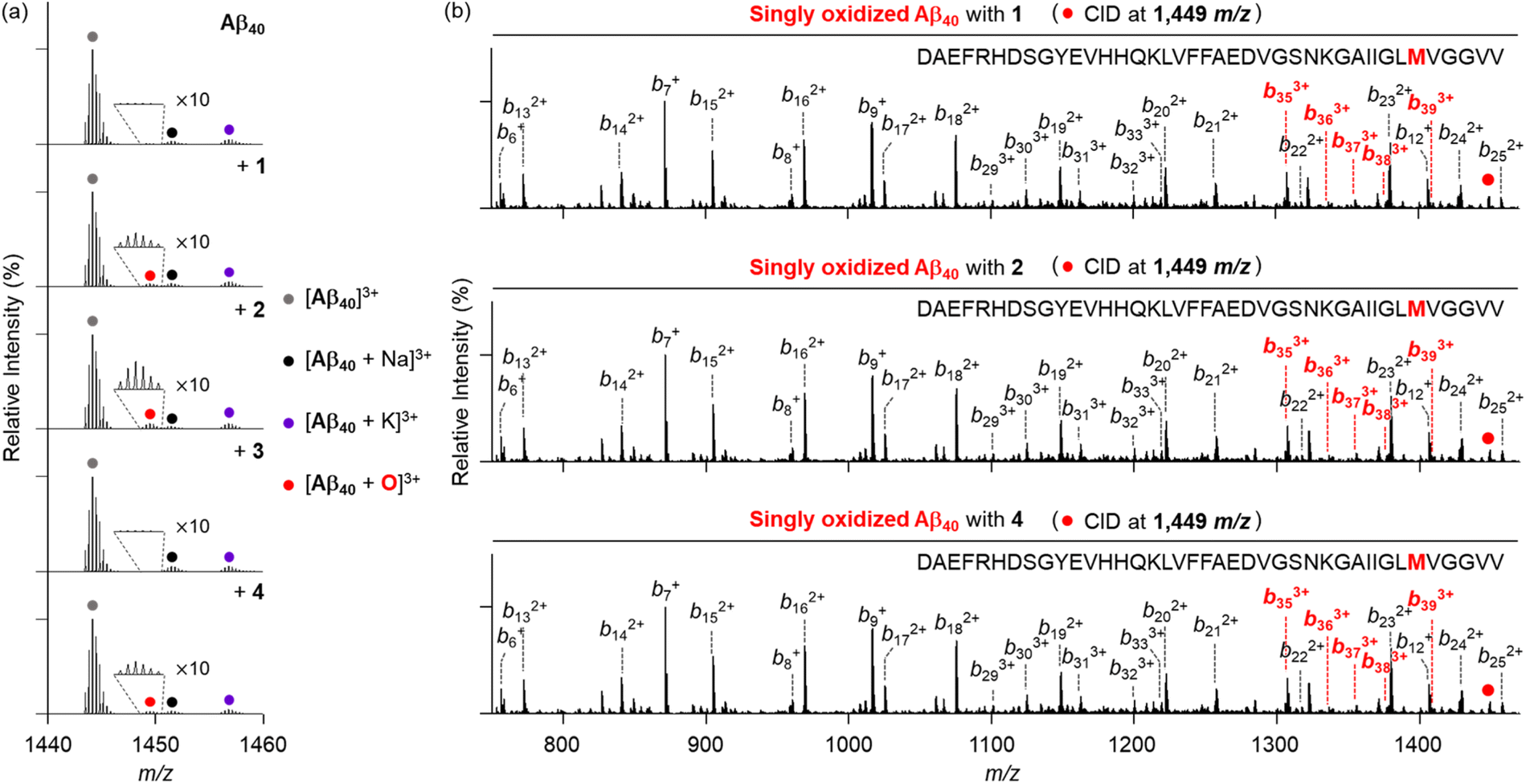 | ||
| Fig. 8 Analyses of metal-free Aβ40 incubated with 1–4 by ESI-MS and ESI-MS2. (a) Mass spectra of +3-charged Aβ40 obtained by treatment with 1–4 under metal-free conditions. (b) ESI-MS2 analyses of singly oxidized Aβ40 produced with 1, 2, and 4. Conditions: [Aβ40] = 25 μM; [compound] = 25 μM (1% v/v DMSO); 20 mM ammonium acetate, pH 7.4; 37 °C; 3 h; constant agitation. The samples were diluted 25-fold with H2O before injection to the mass spectrometer. | ||
To further identify the oxidized amino acid residues in Aβ40, we conducted tandem MS (ESI-MS2). When collision-induced dissociation (CID) energy was applied to the peak at 1449 m/z, as presented in Fig. 8b, oxidized b fragments were observed starting from b35, indicating that Met35 in metal-free Aβ40 can be oxidized by compounds 1, 2, and 4. Given that the oxidation of Met into Met sulfoxide enhances the polarity of Aβ peptides, potentially leading to the destabilization of β-strand structure and disrupting the hydrophobic interactions,33,62,63 such modification can ensue the alternation in Aβ aggregation process. Conversely, this oxidative modification of Aβ40 was not observed with compounds 3, 5, and 6 (Fig. S14†).
As depicted in Fig. 9a, the peak assigned to singly oxidized Aβ40 (1449 m/z) was visualized for Cu(II)–Aβ40 incubated with compounds 1, 2, and 4, while such oxidation was not detected in the absence of O2 (Fig. S15†). As observed in the metal-free samples, the peaks corresponding to the oxidized compounds were also monitored under aerobic conditions (Fig. S12†). The fragmental analysis by applying the CID energy to this peak revealed b fragments starting from b13 in both non-oxidized and oxidized forms (Fig. 9b). This implies that His13, His14, or Met35 in Cu(II)–Aβ40 can be oxidized upon treatment of compounds 1, 2, and 4. Interestingly, an additional peak at 1453 m/z, indicative of doubly oxidized Aβ40, was detected in Cu(II)–Aβ40 added with compounds 1 and 4 (Fig. 9a). Upon application of the CID energy to this peak, as disclosed in Fig. 9c, singly oxidized b fragments smaller than b13 were not monitored, while singly and doubly oxidized b ions larger than b12 and b13, respectively, were shown. This suggests that compounds 1 and 4 can concurrently oxidize two of the amino acid residues: His13, His14, and Met35. In addition to Met, the oxidation of His into 2-oxo-His can impact the aggregation of Aβ. As His engage the ability to participate in hydrogen bonding and π–π stacking through its imidazole ring,64,65 which are involved in the aggregation processes of Aβ, its oxidative transformation can alter the assembly pathways of Aβ. Since His13 and His14 are also responsible for metal coordination to Aβ (Fig. 1b),12,66 the formation of 2-oxo-His can interfere with metal binding to Aβ, resulting in the change in the self-assembly of metal–Aβ. Overall, compounds 1, 2, and 4 can oxidize Aβ peptides in both the absence and presence of Cu(II) in distinct ways, possibly affecting their aggregation profiles to varying degrees.
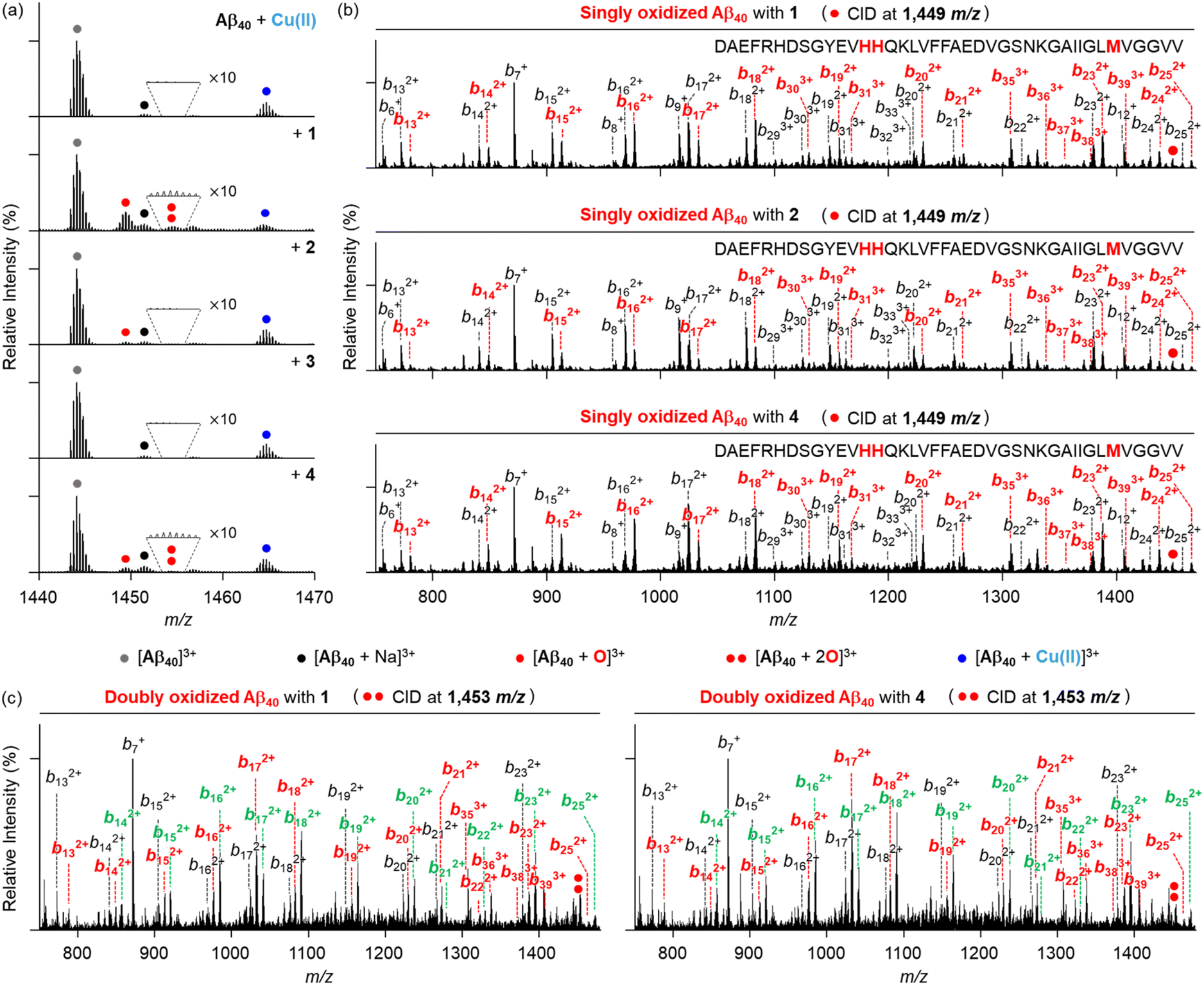 | ||
| Fig. 9 Investigation of Cu(II)–Aβ40 treated with 1–4 by ESI-MS and ESI-MS2. (a) Mass spectra of +3-charged Aβ40 generated by addition of 1–4 in the presence of Cu(II). The number of red dots represents the number of oxygen atoms incorporated into Aβ40 monomer [Aβ40 + nO]3+ (n = 1 or 2). (b) ESI-MS2 analyses of singly oxidized Aβ40 produced by incubation with Cu(II) and 1, 2, or 4. (c) ESI-MS2 spectra of doubly oxidized Aβ40 formed by 1 or 4 with Cu(II). Monooxidized and dioxidized b ions are illustrated in red and green, respectively. Conditions: [Aβ40] = 25 μM; [Cu(II)] = 25 μM; [compound] = 25 μM (1% v/v DMSO); 20 mM ammonium acetate, pH 6.8; 37 °C; 3 h; constant agitation. The samples were diluted 25-fold with H2O before injection to the mass spectrometer. | ||
Conclusions
AD is a complex and multifactorial condition that demands the development of chemical reagents capable of targeting and controlling multiple pathological elements, such as free organic radicals, metal-free Aβ, metal ions, and metal–Aβ. In this study, we introduced a series of rationally selected compounds that exhibit versatile reactivities against these pathological components. These compounds were strategically designed by varying the number of hydroxyl moieties on the pyrogallol group, arranged in either macrocyclic or monomeric forms.Our comprehensive biophysical and biochemical investigations demonstrated that these compounds can effectively quench free organic radicals and modify the aggregation pathways of both metal-free Aβ and metal–Aβ to different degrees. Notably, the macrocyclic compounds (1–3), with the distinct number of hydroxyl groups on the benzene ring, revealed that the increased number of hydroxyl groups lowers the redox potential, enhances interactions with Aβ via multiple hydrogen bonds, and promotes metal binding. These factors contribute to their ability to scavenge free organic radicals, modulate the assembly of both metal-free Aβ and metal–Aβ, and induce oxidative modifications onto the peptides.
Similarly, their monomeric counterparts, 4–6, displayed distinct reactivities towards the pathological targets based on the number of hydroxyl groups. Moreover, the macrocyclization of 4–6 into 1–3 using methylene bridges resulted in reduced redox potentials and maximized interactions with Aβ by expanding the surface area of the semi-rigid compounds. Findings in these studies suggest that simple adjustments in the number of hydroxyl groups and molecular conformations can fine-tune the reactivities of compounds towards the pathological factors associated with AD. In future studies, we will investigate the linear counterparts of compounds 1–3 to determine whether the flexibility or rigidity of molecular structure is critical for their reactivity against our targeted pathological elements. This approach may provide an effective strategy to design multi-target-directed chemical reagents.
More importantly, compound 1, featuring multiple pyrogallol moieties in a macrocyclic structure, exhibited the lowest redox potential among the tested compounds. It demonstrated a strong ability to interact with metal-free and metal-treated Aβ through a combination of multiple hydrogen bonding, hydrophobic interactions, and metal chelation. These properties enabled it to effectively quench free organic radicals and alter the aggregation profiles of both metal-free Aβ and metal–Aβ, inducing oxidative modifications in multiple amino acid residues. It should be noted, however, that the cytotoxicity of compounds 1–3 under our experimental conditions prevented us from assessing their impact on Aβ-induced cytotoxicity (Fig. S16†). Additionally, structural variations of compound 1 with biocompatible functionalities, such as esters and boronic esters/acids, may further enhance its biological applications.67 Collectively, our findings highlight that minor modifications onto the pyrogallol structure can lead to the development of multifunctional chemical reagents with the potential to regulate key pathological features found in AD, providing valuable molecular insights into the disease's complicated pathologies.
Data availability
All experimental details and data supporting the findings of this study are available within the paper and its ESI.† The data are also available from the corresponding authors upon reasonable request.Author contributions
J. K., Y. Y., and M. H. L. designed the research. J. K. and Y. Y. performed the organic synthesis, biochemical assays, Abs spectroscopy, TEM, docking studies, ESI-MS, and cell studies with data analysis. S. P. contributed to DFT calculation. J. K., Y. Y., and M. H. L. wrote the manuscript with input from S. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Research Foundation of Korea (NRF) grants funded by the Korean government {Creative Research Initiative [RS-2022-NR070709 (M. H. L.)]; Global Science Research Center Program [RS-2024-00411134 (M. H. L.)]}. J. K. acknowledges the NRF PhD fellowship grant (RS-2024-00407337). We thank Dr Mingeun Kim for the assistance in measuring cyclic voltammetry.References
- W. M. van der Flier, M. E. de Vugt, E. M. A. Smets, M. Blom and C. E. Teunissen, Nat. Aging, 2023, 3, 494 CrossRef.
- H. J. Forman and H. Zhang, Nat. Rev. Drug Discovery, 2021, 20, 689 CrossRef CAS PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221 CrossRef CAS.
- E. Karran and B. De Strooper, Nat. Rev. Drug Discovery, 2022, 21, 306 CrossRef CAS PubMed.
- F. Panza, M. Lozupone, G. Logroscino and B. P. Imbimbo, Nat. Rev. Neurol., 2019, 15, 73 CrossRef PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147 CrossRef CAS.
- J.-X. Lu, W. Qiang, W.-M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Cell, 2013, 154, 1257 CrossRef CAS PubMed.
- Y. Liu, M. Nguyen, A. Robert and B. Meunier, Acc. Chem. Res., 2019, 52, 2026 CrossRef CAS.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193 CrossRef CAS.
- A. Abelein, Acc. Chem. Res., 2023, 56, 2653 CrossRef CAS PubMed.
- Y. Yi and M. H. Lim, RSC Chem. Biol., 2023, 4, 121 RSC.
- E. Atrián-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 371, 38 CrossRef.
- E. Stefaniak and W. Bal, Inorg. Chem., 2019, 58, 13561 CrossRef CAS PubMed.
- J. Han, Z. Du and M. H. Lim, Acc. Chem. Res., 2021, 54, 3930 CrossRef CAS PubMed.
- M. A. Greenough, J. Camakaris and A. I. Bush, Neurochem. Int., 2013, 62, 540 CrossRef CAS PubMed.
- D. M. Walsh and D. J. Selkoe, Curr. Opin. Neurobiol., 2020, 61, 116 CrossRef CAS.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310 RSC.
- S. Park, C. Na, J. Han and M. H. Lim, Metallomics, 2023, 15, mfac102 CrossRef.
- J.-M. Suh, M. Kim, J. Yoo, J. Han, C. Paulina and M. H. Lim, Coord. Chem. Rev., 2023, 478, 214978 CrossRef CAS.
- I. Zawisza, M. Rózga and W. Bal, Coord. Chem. Rev., 2012, 256, 2297 CrossRef CAS.
- A. K. Sharma, S. T. Pavlova, J. Kim, J. Kim and L. M. Mirica, Metallomics, 2013, 5, 1529 CrossRef CAS PubMed.
- J. Sevigny, P. Chiao, T. Bussière, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O'Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50 CrossRef CAS PubMed.
- J. Kwak, J. Woo, S. Park and M. H. Lim, J. Inorg. Biochem., 2023, 238, 112053 CrossRef CAS PubMed.
- L. M. F. Gomes, J. C. Bataglioli and T. Storr, Coord. Chem. Rev., 2020, 412, 213255 CrossRef CAS.
- M. Ramesh and T. Govindaraju, Chem. Sci., 2022, 13, 13657 RSC.
- S. Park, Y. Yi and M. H. Lim, Bull. Korean Chem. Soc., 2021, 42, 17 CrossRef CAS.
- G. Nam, M. Hong, J. Lee, H. J. Lee, Y. Ji, J. Kang, M.-H. Baik and M. H. Lim, Chem. Sci., 2020, 11, 10243 RSC.
- L. Grcic, G. Leech, K. Kwan and T. Storr, Chem. Commun., 2024, 60, 1372 RSC.
- T. Storr, Can. J. Chem., 2021, 99, 1 CrossRef CAS.
- H.-J. Cho, A. K. Sharma, Y. Zhang, M. L. Gross and L. M. Mirica, ACS Chem. Neurosci., 2020, 11, 1471 CrossRef CAS PubMed.
- A. S. DeToma, J.-S. Choi, J. J. Braymer and M. H. Lim, ChemBioChem, 2011, 12, 1198 CrossRef CAS PubMed.
- S.-J. Hyung, A. S. DeToma, J. R. Brender, S. Lee, S. Vivekanandan, A. Kochi, J.-S. Choi, A. Ramamoorthy, B. T. Ruotolo and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3743 CrossRef CAS.
- S. Park, M. Kim, Y. Lin, M. Hong, G. Nam, A. Mieczkowski, J. Kardos, Y.-H. Lee and M. H. Lim, Chem. Sci., 2023, 14, 9293 RSC.
- P.-G. Pietta, J. Nat. Prod., 2000, 63, 1035 CrossRef CAS.
- L. Mira, M. Tereza Fernandez, M. Santos, R. Rocha, M. Helena Florêncio and K. R. Jennings, Free Radical Res., 2002, 36, 1199 CrossRef CAS.
- C. A. Perez, Y. Wei and M. Guo, J. Inorg. Biochem., 2009, 103, 326 CrossRef CAS.
- S. Vivekanandan, J. R. Brender, S. Y. Lee and A. Ramamoorthy, Biochem. Biophys. Res. Comm., 2011, 411, 312 CrossRef CAS.
- R. Gao, Z. Yuan, Z. Zhao and X. Gao, Bioelectrochem. Bioenerg., 1998, 45, 41 CrossRef CAS.
- S. B. Ozturk Sarikaya, J. Enzyme Inhib. Med. Chem., 2015, 30, 761 CrossRef CAS PubMed.
- S. Kim, D. G. Hyun, Y. Nam, S. J. Shin, D. Im, H. S. Kim, S. H. Leem, H. H. Park, B.-H. Kim, Y. H. Park, E. Cho, W. A. Goddard, D. H. Kim, H. I. Kim and M. Moon, Biomed. Pharmacother., 2023, 168, 115770 CrossRef CAS.
- H. Huang, J. Damjanovic, J. Miao and Y.-S. Lin, Phys. Chem. Chem. Phys., 2021, 23, 607 Search PubMed.
- M. Chwastek and A. Szumna, Org. Lett., 2020, 22, 6838 CrossRef CAS PubMed.
- H. M. Thomas, H. Kumari, J. Maddalena, C. M. Mayhan, L. T. Ellis, J. E. Adams and C. A. Deakyne, Supramol. Chem., 2018, 30, 520 CrossRef CAS.
- C. R. Pfeiffer, K. A. Feaster, S. J. Dalgarno and J. L. Atwood, Cryst. Eng. Comm., 2016, 18, 222 RSC.
- Z. Goren and S. E. Biali, J. Chem. Soc., Perkin Trans., 1990, 1, 1484 RSC.
- M.-H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407 CrossRef CAS.
- V. Ball, J. Electroanal. Chem., 2022, 909, 116142 CrossRef CAS.
- V. J. Forrest, Y.-H. Kang, D. E. McClain, D. H. Robinson and N. Ramakrishnan, Free Radical Biol. Med., 1994, 16, 675 CrossRef CAS PubMed.
- L. Cheng, R. S. Assary, X. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, J. Phys. Chem. Lett., 2015, 6, 283 CrossRef CAS.
- M. R. Elkins, T. Wang, M. Nick, H. Jo, T. Lemmin, S. B. Prusiner, W. F. DeGrado, J. Stöhr and M. Hong, J. Am. Chem. Soc., 2016, 138, 9840 CrossRef CAS.
- F. Hsu, G. Park and Z. Guo, ACS Omega, 2018, 3, 8401 CrossRef CAS.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499 CrossRef CAS.
- A. T. Petkova, R. D. Leapman, Z. Guo, W.-M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262 CrossRef CAS.
- M. J. Sever and J. J. Wilker, Dalton Trans., 2004, 1061 RSC.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Suresh, M. Nethaji, E. Zangrando and D. Das, Inorg. Chem., 2008, 47, 7083 CrossRef CAS.
- M. Mochizuki, S.-I. Yamazaki, K. Kano and T. Ikeda, Biochim. Biophys. Acta, Gen. Subj., 2002, 1569, 35 CrossRef CAS.
- A. Veselinovic and G. Nikolić, Acta Fac. Med. Naiss., 2015, 32, 127 CAS.
- M. Kim, G. Gupta, J. Lee, C. Na, J. Kwak, Y. Lin, Y.-H. Lee, M. H. Lim and C. Y. Lee, Inorg. Chem. Front., 2024, 11, 1966 RSC.
- X. Zhang, Y.-J. Gou, Y. Zhang, J. Li, K. Han, Y. Xu, H. Li, L.-H. You, P. Yu, Y.-Z. Chang and G. Gao, Cell Death Discov., 2020, 6, 113 CrossRef CAS.
- K. Ngamchuea, B. Tharat, P. Hirunsit and S. Suthirakun, RSC Adv., 2020, 10, 28454 RSC.
- R. Gao, Z. Yuan, Z. Zhao and X. Gao, Bioelectrochem. Bioenerg., 1998, 45, 41 CAS.
- A. M. Brown, J. A. Lemkul, N. Schaum and D. R. Bevan, Arch. Biochem. Biophys., 2014, 545, 44 CAS.
- L. Hou, H. Shao, Y. Zhang, H. Li, N. K. Menon, E. B. Neuhaus, J. M. Brewer, I.-J. L. Byeon, D. G. Ray, M. P. Vitek, T. Iwashita, R. A. Makula, A. B. Przybyla and M. G. Zagorski, J. Am. Chem. Soc., 2004, 126, 1992 CAS.
- S.-M. Liao, Q.-S. Du, J.-Z. Meng, Z.-W. Pang and R.-B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef PubMed.
- K. Brännström, T. Islam, L. Sandblad and A. Olofsson, FEBS Lett., 2017, 591, 1167 CrossRef PubMed.
- P. Dorlet, S. Gambarelli, P. Faller and C. Hureau, Angew. Chem., Int. Ed., 2009, 48, 9273 CrossRef CAS.
- M. K. Al-Omari, M. Elaarag, R. M. Al-Zoubi, A. R. Al-Qudimat, A. A. Zarour, E. A. Al-Hurani, Z. E. Fares, L. M. Alkharraz, M. Shkoor, A. D. Bani-Yaseen, O. M. Aboumarzouk, A. Yassin and A. A. Al-Ansari, J. Enzyme Inhib. Med. Chem., 2023, 38, 2220084 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and Fig. S1–S16. See DOI: https://doi.org/10.1039/d4sc06417h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |