
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Depolymerisation of poly(lactide) under continuous flow conditions†
Sophie
Ellis
 ab,
Antoine
Buchard
*c and
Tanja
Junkers
*a
ab,
Antoine
Buchard
*c and
Tanja
Junkers
*a
aPolymer Reaction Design Group, School of Chemistry, Monash University, 17 Rainforest Walk, Clayton, VIC 3800, Australia. E-mail: tanja.junkers@monash.edu
bDepartment of Chemistry, Institute for Sustainability, University of Bath, Claverton Down, Bath BA2 7AY, UK
cDepartment of Chemistry, University of York, York, YO10 5DD, UK. E-mail: antoine.buchard@york.ac.uk
First published on 12th November 2024
Abstract
Poly(L-lactic acid) (PLLA) is commercially successful bio-based plastic, where end-of-life materials can undergo industrial composting. To create a circular economy, a desirable alternative to composting is chemical recycling to monomer (CRM), where direct depolymerisation to L-lactide can be achieved. CRM of PLLA is typically impeded by thermal decomposition and side reactions, due to the high ceiling temperate (Tc) of PLLA in bulk (>600 °C), which preclude implementation on a large scale, and has led to the development of catalytic strategies, under vacuum or high dilution in high boiling point solvents conditions. In this study, a commercially available Sn(II) catalyst and low boiling point solvents, at a range of temperatures and concentrations, were explored for the CRM of PLLA in a continuous flow process. The solvent THF was found to produce the best results, where up to 92% conversion of lactide could be achieved, with 92–97% selectivity for L-lactide formation at temperatures 150–170 °C. Further, inline monitoring of monomer and polymer concentrations in flow were used to determine the depolymerisation rate coefficient kdepo and the activation energy of kdepo was determined to be 129.4 kJ mol−1.
Introduction
Poly(lactide) (PLA) is a bio-sourced thermoplastic polymer with a versatile range of applications, including packaging, 3D printing and uses in the biomedical sector.1–4 PLA is made up of lactic acid repeat units and can be produced directly from lactic acid or its cyclic dimer, lactide. Lactic acid is typically obtained from the fermentation of starch and has been demonstrated to have a low carbon footprint.5 While not readily degradable in the open environment, PLA is fully compostable under industrial conditions (norm EN13432).6 Other methods for dealing with end-of-life (EOL) PLA, such as mechanical recycling or incineration, ultimately lead to lower quality plastics or loss of molecular complexity resulting in economic losses.7–9 Therefore, an alternative approach to dealing with EOL PLA is desired, in order to achieve true circularity for its use and production.10 Lifecycle assessments of PLA have found that most of the process's energy input is used in production of L-LA from the plant, meaning the greatest energy intensive steps must be repeated if PLA is composted and the starch resource regrown.11,12 PLA can alternatively be degraded to the one of the monomer's precursors, lactic acid, or alkyl lactates.13–15 Whilst this creates a closed loop system, there is still a significant energy input required to transform lactic acid to L-LA, therefore, the direct chemical recycling to the (lactide) monomer (CRM) is most desirable to minimise energy inputs.16 Direct depolymerisation to lactide results in a monomer feedstock which can be repolymerised to the original or modified polymer without loss of quality, allowing indefinite recycling and creating a fully closed-loop cycle.A pathway to chemical recycling of polymers is given by the thermodynamics of a polymerisation reaction. Every polymerisation reaches by definition at some temperature an equilibrium state when the enthalpy term becomes as large as the entropy part in the Gibbs free energy equation.17 The temperature by which this occurs is called the ceiling temperature Tc. Tc is, however, not an absolute, and depends on the monomer concentration, as presence of higher monomer concentrations will favour the forward polymerisation reaction, while absence of monomer favours the reverse depolymerisation. The relationship between the equilibrium monomer concentration [M]eq and the thermodynamic parameters is given in eqn (1).
 | (1) |
Rearrangement of eqn (1) yields the ceiling temperature.17
 | (2) |
If the ceiling temperature of a given polymer is lower than the temperature of decomposition, then raising the polymer above this temperature should favour depolymerisation of the polymer to recover the initial monomer (if the polymer is in its reactive form).17 However, if the ceiling temperature is higher than the decomposition temperature then a mixture of products will be obtained due to competition of depolymerisation and degradation occurring simultaneously.18 Most CRM processes have been demonstrated with new monomers within research, but for already existing commercial plastics this has not yet been fully achieved.19–22L-LA CRM is rather challenging due to the high Tc of PLA when present in bulk (>600 °C).23,24 The elevated temperature required for this process leads to thermal degradation (Td,PLLA ∼ 350 °C) and side reactions, including epimerisation which produces D-lactide and meso-lactide, and elimination reactions which can form acrylic acid.18
However, under vacuum and reactive distillation conditions, Tc can be lowered below the onset of thermal decomposition, and the use of catalysts such as tin(II) 2-ethylhexanoate (Sn(Oct)2) and zinc(II) chloride or zinc acetate (Zn(OAc)2) have also been shown to improve the selectivity towards LA.18,25,26 Enthaler and co-workers have thus successfully depolymerised EOL PLA in bulk under reduced pressure, using Zn(OAc)2 (0.4 mol%) as a catalyst and temperatures of 200–210 °C, producing 98% LA in 6 hours, with around 12% meso-LA content.25 Previous work has shown rates of depolymerisation to be dependent on the degree of polymerisation, where high molar mass PLA have slower depolymerisation rates.27 To overcome this issue, a recent two-step, one-pot process was designed by where high molecular weight PLA first underwent transesterification with a triol to produce a low molecular weight PLA, followed by depolymerisation to L-LA.28 The reaction achieved 92% conversion in ∼3 hours, with >99% L-LA selectivity (5 mbar, 0.01% Sn(Oct)2, 160 °C). A similar concept was applied by Byers and co-workers using a ZnCl2/PEG600 catalyst system.26
In an alternative approach, Odelius and co-workers focussed on lowering the Tc of LA through the use of solvents.29 Increasing the dilution of the system will decrease the Tc by lowering the absolute monomer concentration below the equilibrium concentration, and in low Tc systems simple dilution is enough to drive depolymerisation.30,31 However, in high Tc systems like LA dilution is not enough and the monomer–polymer–solvent interactions were found to be key for suppressing Tc.29 They found Tc of LA to be reduced to as low as 119 °C, allowing for up to 96% recovery of LA at 140 °C (99% L-LA selectivity, DMF, 0.5 M, 10 mol% Sn(Oct)2). However, the solvents explored all had high boiling points (>150 °C), resulting in a low yield of LA isolation (38%).
For CRM of a commercial polymer like PLA to be relevant industrially, the process needs to be scalable, the depolymerised monomer must be easily recoverable, and the process needs to be energy efficient. While energy efficiency can be reached by decreasing the ceiling temperature, effective methods of heating also present ideal solutions. Flow chemistry allows to utilise both factors for a beneficial depolymerisation process. The closed and typically slightly overpressured flow reactors allow for the use of low boiling point solvents for high temperature reactions. At the same time, flow chemistry offers excellent heat transfer capabilities. This typically results in the ability to flash heat a reaction with high temporal control, and with a narrow temperature distribution. It is this narrow distribution that classically allows to carry out flow reactions in widened process windows (usually higher temperatures) with increased yields (as side product reaction pathways are diminished due to the higher precision in temperature).32 This feature makes flow reactors ideal to carry out depolymerisations, as these require high temperatures, precise control and allow for more benign solvents to be used. Further, flow reactors are simple to scale, which makes them highly attractive for industrial application.32 Flow reactors also provide conditions of ideal heat transfer, further minimising energy needs. Despite these obvious advantages, to date, the use of continuous flow for depolymerisation reactions has mainly been focussed on depolymerisation of lignin and oligosaccharides, with no reports on the use of flow conditions for CRM of plastics.33–35 In this work, with the focus of easier lactide recovery in mind, a series of lower boiling point solvents were explored for PLA depolymerisation in continuous flow (Fig. 1). Various solvents were first tested in batch for this purpose, followed by flow application where temperatures well above the boiling points could be applied with ease. Once the best solvent was identified a range of conditions were explored and kinetic analysis carried out. For this, we made use of inline monitoring of the reaction to provide real-time data on the progress of the reactions, which facilitates the optimisation of the reaction, and which allows for direct insights into the reaction without tedious sampling of aliquots for offline analysis.
 | ||
| Fig. 1 Overview of PLA depolymerisation under continuous flow conditions. | ||
Experimental section
Materials
All reactions were performed under an atmosphere of argon using standard Schlenk techniques unless otherwise stated. All reagents were purchased from either Sigma Aldrich, Alfa Aesar or Acros Organics and used without further purification unless specified. All solvents used were anhydrous unless otherwise stated. L-LA was recrystallised in dry toluene three times and stored under argon prior to use. 4-Methylbenzyl alcohol was recrystalised in diethyl ether and stored under argon prior to use.Instruments
1H NMR spectra were recorded on a Bruker 400 MHz instrument and referenced to residual solvent peaks. Coupling constants are given in Hertz. Polymer conversions were determined by 1H NMR spectroscopy. Size-exclusion chromatography (SEC) was carried out on a PSS SECcurity2 GPC system operated by PSS WinGPC software, equipped with an SDV 5.0 μm guard column (50 × 8 mm), followed by three SDV analytical 5.0 μm columns with varying porosity (1000 Å, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å, and 1000000 Å) (50 × 8 mm) and a differential refractive index 3 detectors using THF as the eluent at 40 °C with a flow rate of 1 mL min−1. The SEC system was calibrated using linear narrow polystyrene standards from PSS Laboratories ranging from 682 to 2.52 × 106 g mol−1. In situ IR monitoring of L-LA polymerisations were carried out using a ReactIR 700 (model 701L) with a Micro Flow Cell DS DiComp 50 μL detector. The monitoring software used was iC IR. Kinetic data was extracted from monitoring the asymmetric ester C–O–C stretching vibrations of L-LA at 1240 cm−1 and PLLA at 1185 cm−1 (Fig. 2).36 Peak heights of the absorption spectra were correlated to conversions from samples taken throughout, analysed by 1H NMR spectroscopy. Data analysis in Microsoft Excel allowed for the construction of first-order plots, and thus extraction of the initial reaction rate constant. Flow reactions were carried out using a Vapourtec RS-200 system with a high temperature reactor (HTR, Hastelloy C reactor of 2 mL volume).
000 Å, and 1000000 Å) (50 × 8 mm) and a differential refractive index 3 detectors using THF as the eluent at 40 °C with a flow rate of 1 mL min−1. The SEC system was calibrated using linear narrow polystyrene standards from PSS Laboratories ranging from 682 to 2.52 × 106 g mol−1. In situ IR monitoring of L-LA polymerisations were carried out using a ReactIR 700 (model 701L) with a Micro Flow Cell DS DiComp 50 μL detector. The monitoring software used was iC IR. Kinetic data was extracted from monitoring the asymmetric ester C–O–C stretching vibrations of L-LA at 1240 cm−1 and PLLA at 1185 cm−1 (Fig. 2).36 Peak heights of the absorption spectra were correlated to conversions from samples taken throughout, analysed by 1H NMR spectroscopy. Data analysis in Microsoft Excel allowed for the construction of first-order plots, and thus extraction of the initial reaction rate constant. Flow reactions were carried out using a Vapourtec RS-200 system with a high temperature reactor (HTR, Hastelloy C reactor of 2 mL volume).
 | ||
| Fig. 2 Example of IR absorbances: L-LA (1240 cm−1) and PLLA (1185 cm−1) in an inline depolymerisation and their structures. | ||
Synthesis of PLLA
L-LA (20.0 g, 0.139 mol) was added to a round-bottom flask together with 4-MeBnOH (0.170 g, 0.00139 mol) and Sn(Oct)2 (0.563 g, 0.00139 mol) as catalysts. ROP was carried out under an argon atmosphere at 100 °C for 1 h, after which the reaction was quenched by cooling. The polymer was dissolved in dichloromethane (DCM) and precipitated in n-hexane, of volume at least 10 times the volume of DCM. The precipitation was repeated four times to remove Sn(Oct)2 and unreacted monomers. The purified polymer was dried in a fume hood overnight and thereafter under vacuum (2 days at room temperature). The polymer was stored in a glove box under an argon atmosphere. 90%, Mn 15.8 kDa, ÐM 1.10.Depolymerisation in looped flow mode
In a general looped flow reaction, PLLA (0.72 g, 5 mmol) was added to a dried Schlenk flask under an argon atmosphere followed by 10 mL of anhydrous solvent ([PLLA] = 0.5 M), once the polymer was fully dissolved Sn(Oct)2 was added (0.18 mL, 10 mol%). Under constant stirring and argon supply, the Schlenk flask was the feed input for the Vapourtec R-200 system equipped with a 2 mL Hastelloy high temperature tube reactor and 250 psi back-pressure regulator at the output. This outlet feed fed back into the starting Schlenk flask to create a closed loop system. An in-line IR system was integrated using a Mettler Toledo ReactIR with a micro flow cell (DiComp), reactions were monitored using the iC IR software. The in-line IR was positioned between the Vapourtec HPLC pump and the high temperature tube reactor. Once the reactor was up to temperature (140–170 °C), the flow rate was set to 1 mL min−1, initially the output was set to waste until the PLLA had flushed through the reactor where it was then fed back into the feed solution. Samples were taken intermittently to cross-check IR signals; reactions were run until a steady state was observed. A looped flow approach with a semi-batch reservoir was chosen to allow for flexibility in the study on reaction times. A linear reactor would present considerable limitations with regard to the maximum reaction time accessible. With the looped flow, flow rates can be chosen to be fairly high (reducing resident time distribution issues) while allowing for extended reaction times. Still, the chosen design features all advantages typically associated with flow chemistry, that is flash heating, efficient heat and mass transfer as much as continuous operation. The reactor setup already features the ability to scale the reaction up (by increasing the semi-batch and the continuous flow reactor compartment volume), which can be extended further by feeding PLA into the semi-batch reservoir while removing monomer via distillation or other means.Results and discussion
Looped flow depolymerisations
 | ||
| Fig. 3 Inline IR traces of PLLA depolymerisation under looped flow conditions ([PLLA] 0.5 mol L−1; Sn(Oct)2 10 mol%; 150 °C). PLLA shown in solid lines, L-LA shown in dashed lines. Dioxane (red), DCE (yellow), THF (dark blue). | ||
When depolymerisation was tested under batch conditions, only 3% lactide was achieved ([PLLA] 0.5 mol L−1, Sn(Oct)2 10 mol%, 80 °C, 3 h), highlighting the benefits of employing a looped flow system over a traditional batch type. While a direct comparison may appear unfair due to the large temperature difference, it is noteworthy to add that a batch system simply does not allow for more elevated temperatures as solvents would evaporate almost instantaneously. Further, pressurised batch reactors are associated with considerably higher risk profiles. Hence, this comparison presents a classical advantage of using flow reactors, that is the shift of the operation window, which in itself then leads to the process intensification and increase in yield.
The SEC data shows both a reduction in peak intensity and molecular weight of the polymer with a slight increase in ÐM as the reaction progressed. When molecular weight is plotted against conversion (Fig. S6†), a linear relationship can be seen; this linear relationship indicates that depolymerisation likely occurs through an unzipping mechanism, where the –OH propagating chain-ends undergo back-biting to reform the cyclic monomer. These results are consistent with another paper which found depolymerisation in solvated conditions to proceed through the same mechanism.29
The fraction of meso-LA in the lactide mixture was investigated using the CH3 signals found in the NMR spectrum. Levels of meso-LA formation (Table S2†) followed the same trends seen for rates: THF produced the highest levels of meso-LA, reaching a final content of 3.6% (initial meso-LA content 0.86%). Conversely, meso content found in depolymerisation in DCE showed fluctuations, resulting in a final meso-LA content of 1.7%, whereas the use of dioxane showed both low conversions and low meso-LA formation. Work by Odelius and co-workers had also previously found solvent choice to have an impact on levels of meso-LA formation in batch, where DMSO reduced the selectivity of L-LA formation compared to DMF (meso-LA 3.5%, DMF; meso-LA 13.2%, DMSO; 140 °C, 0.5 mol L−1, Sn(Oct)2 10 mol%).29 Nonetheless, despite the higher meso content obtained with THF as the reaction solvent, THF is considered a greener option due to its lower toxicity and relatively better environmental impact.37 Moreover, through process optimisation it may be possible to control levels of epimerisation seen. THF was therefore selected as solvent for further work.
 | ||
| Fig. 4 Inline IR traces of PLLA depolymerisation under looped flow conditions ([PLLA] 0.5 mol L−1; Sn(Oct)2 10 mol%; THF). PLLA shown in solid lines, L-LA shown in dashed lines. 170 °C (yellow), 160 °C (red), 150 °C (dark blue), 140 °C (blue). | ||
Positively, when using THF as the reaction solvent the formation of acrylic acid could not be detected at any temperature. It was found meso-LA contents typically increased with the raised reaction temperatures (Table S3†), however, the level of meso-LA content remained relatively low, where a maximum final meso content of 4.3% was obtained when the reaction temperature was 160 °C at 0.5 mol L−1.
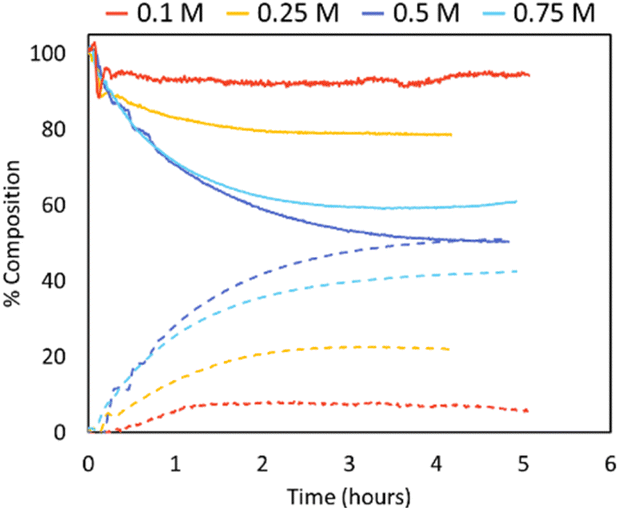 | ||
| Fig. 5 Inline IR traces of PLLA depolymerisation under looped flow conditions (varied concentrations, Sn(Oct)2 10 mol%, THF, 150 °C). PLLA shown in solid lines, L-LA shown in dashed lines. 0.1 M (red), 0.25 M (yellow), 0.5 M (dark blue), 0.75 M (blue). | ||
Depolymerisations were repeated using the same Sn(Oct)2 concentration throughout (0.06 mol L−1; Fig. 6). When the catalyst concentration remained constant, the trends with initial polymer concentration became as expected, with lower initial PLA concentrations increasing the extent of depolymerisation. For example, reducing concentration from 0.75 mol L−1 to 0.1 mol L−1 increased LA production from 47% to 88%, respectively.
 | ||
| Fig. 6 Inline IR traces of PLLA depolymerisation under looped-flow conditions (varied concentrations, Sn(Oct)2 0.06 M, THF, 150 °C). PLLA shown in solid lines, L-LA shown in dashed lines. 0.1 M (red), 0.25 M (yellow), 0.5 M (dark blue), 0.75 M (blue). | ||
 | ||
| Fig. 7 Arrhenius plots of PLLA using 0.06 mol L−1 Sn(Oct)2. 0.1 M (130.4 kJ mol−1, A = 1.57 × 1014 s−1); 0.25 M (Ea = 128.4 kJ mol−1, A = 6.04 × 1013 s−1); 0.5 M (Ea = 130.2 kJ mol−1, A = 3.21 × 1013 s−1); 0.75 M (Ea = 128.8 kJ mol−1, A = 2.39 × 1013 s−1). | ||
Conclusions
Previous works reported the successful use of high boiling point solvents to lower the Tc of PLLA, towards an efficient and selective CRM process.29 Our work explored the use of a continuous flow system that allowed the use of low-boiling point solvents for PLLA CRM, in order to aid in later monomer recovery. We successfully identified THF as a suitable solvent under a range of temperatures. Depolymerisation conversions reached up to 92% in 1.2 h ([PLLA] = 0.1 mol L−1, [Sn(Oct)2] = 0.06 mol L−1, THF, 170 °C, 5% meso-LA). While this is an example where a high catalyst loading was used, depolymerisations under conditions with 10% of catalyst also result in high crude monomer yields, for example 82% in 3 h ([PLLA] = 0.5 mol L−1, [Sn(Oct)2] = 0.05 mol L−1, THF, 170 °C, 3.7% meso-LA). While we did not recycle the catalyst in this present work, it is possible to continuously run the reactor without quenching of the reaction, giving rise to potential upscaling of the process. Also the use of immobilised catalysts are in principle possible. Construction of an Arrhenius plot revealed activation energy of the reaction to be 129.4 ± 1.0 kJ mol−1, and meso-LA content of the recovered monomer was between 3–6% depending upon conditions used. These results showcase the potential of continuous flow processes to facilitate the chemical recycling of PLA back to monomer, under fast rates and mild conditions. Continuous flow chemistry presents itself here as an enabling technology that allows to shift the process window into conditions that are not conventionally accessible to solution-based batch processes. We envision that in future studies it will also be possible to use the same strategy for depolymerisation of other polyesters, for example polycaprolactone.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
SE: investigation, data curation, formal analysis, visualisation, writing – original draft. AB and TJ: supervision, conceptualisation, resources, writing – final draft.Conflicts of interest
AB has received funding and is collaborating on other topics with TotalEnergies Corbion, a PLA manufacturer.Acknowledgements
Research funding from the University of Bath and Monash University (studentship to SE), and from the Royal Society (UF/160021 and URF\R\221027: fellowship to AB) is acknowledged in support of this study. Further the authors are grateful for funding via the Australian Research Council (DP240100121 and LE210100100).References
- N. Saito and K. Takaoka, New synthetic biodegradable polymers as BMP carriers for bone tissue engineering, Biomaterials, 2003, 24, 2287–2293 CrossRef CAS PubMed.
- M. A. Elsawy, K.-H. Kim, J.-W. Park and A. Deep, Hydrolytic degradation of polylactic acid (PLA) and its composites, Renewable Sustainable Energy Rev., 2017, 79, 1346–1352 CrossRef CAS.
- P. E. Haers, R. Suuronen, C. Lindqvist and H. Sailer, Biodegradable polylactide plates and screws in orthognathic surgery: technical note, J. Craniomaxillofac. Surg., 1998, 26, 87–91 CrossRef CAS.
- T. A. Swetha, A. Bora, K. Mohanrasu, P. Balaji, R. Raja, K. Ponnuchamy, G. Muthusamy and A. Arun, A comprehensive review on polylactic acid (PLA) – synthesis, processing and application in food packaging, Int. J. Biol. Macromol., 2023, 234, 123715 CrossRef CAS PubMed.
- A. Morão and F. de Bie, Life Cycle Impact Assessment of Polylactic Acid (PLA) Produced from Sugarcane in Thailand, J. Polym. Environ., 2019, 27, 2523–2539 CrossRef.
- M. van der Zee and K. Molenveld, The fate of (compostable) plastic products in a full scale industrial organic waste treatment facility, Wageningen Food & Biobased Research, 2020 Search PubMed.
- M. F. Cosate de Andrade, P. M. S. Souza, O. Cavalett and A. R. Morales, Life Cycle Assessment of Poly(Lactic Acid) (PLA): Comparison Between Chemical Recycling, Mechanical Recycling and Composting, J. Polym. Environ., 2016, 24, 372–384 CrossRef CAS.
- Y. Tokiwa and B. P. Calabia, Biodegradability and biodegradation of poly(lactide), Appl. Microbiol. Biotechnol., 2006, 72, 244–251 CrossRef CAS.
- Y.-C. Chien, C. Liang and S.-h. Yang, Exploratory study on the pyrolysis and PAH emissions of polylactic acid, Atmos. Environ., 2011, 45, 123–127 CrossRef CAS.
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a circular economy, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- U. Suwanmanee, V. Varabuntoonvit, P. Chaiwutthinan, M. Tajan, T. Mungcharoen and T. Leejarkpai, Life cycle assessment of single use thermoform boxes made from polystyrene (PS), polylactic acid, (PLA), and PLA/starch: cradle to consumer gate, Int. J. Life Cycle Assess., 2013, 18, 401–417 CrossRef CAS.
- V. Piemonte, S. Sabatini and F. Gironi, Chemical Recycling of PLA: A Great error Opportunity Towards the Sustainable Development?, J. Polym. Environ., 2013, 21, 640–647 CrossRef CAS.
- F. Iñiguez-Franco, R. Auras, K. Dolan, S. Selke, D. Holmes, M. Rubino and H. Soto-Valdez, Chemical recycling of poly(lactic acid) by water-ethanol solutions, Polym. Degrad. Stab., 2018, 149, 28–38 CrossRef.
- R. Petrus, D. Bykowski and P. Sobota, Solvothermal Alcoholysis Routes for Recycling Polylactide Waste as Lactic Acid Esters, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- V. Piemonte and F. Gironi, Kinetics of Hydrolytic Degradation of PLA, J. Polym. Environ., 2013, 21, 313–318 CrossRef CAS.
- G. W. Coates and Y. D. Y. L. Getzler, Chemical recycling to monomer for an ideal, circular polymer economy, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- A. Duda and A. Kowalski, in Handbook of Ring-Opening Polymerization, 2009, pp. 1–51, DOI:10.1002/9783527628407.ch1.
- F. D. Kopinke, M. Remmler, K. Mackenzie, M. Möder and O. Wachsen, Thermal decomposition of biodegradable polyesters—II. Poly(lactic acid), Polym. Degrad. Stab., 1996, 53, 329–342 CrossRef CAS.
- L. Zhou, Z. Zhang, C. Shi, M. Scoti, D. K. Barange, R. R. Gowda and E. Y. X. Chen, Chemically circular, mechanically tough, and melt-processable polyhydroxyalkanoates, Science, 2023, 380, 64–69 CrossRef CAS PubMed.
- B. A. Abel, R. L. Snyder and G. W. Coates, Chemically recyclable thermoplastics from reversible-deactivation polymerization of cyclic acetals, Science, 2021, 373, 783–789 CrossRef CAS PubMed.
- P. Olsén, K. Odelius and A.-C. Albertsson, Ring-Closing Depolymerization: A Powerful Tool for Synthesizing the Allyloxy-Functionalized Six-Membered Aliphatic Carbonate Monomer 2-Allyloxymethyl-2-ethyltrimethylene Carbonate, Macromolecules, 2014, 47, 6189–6195 CrossRef.
- D. K. Schneiderman, M. E. Vanderlaan, A. M. Mannion, T. R. Panthani, D. C. Batiste, J. Z. Wang, F. S. Bates, C. W. Macosko and M. A. Hillmyer, Chemically Recyclable Biobased Polyurethanes, ACS Macro Lett., 2016, 5, 515–518 CrossRef CAS PubMed.
- A. Duda and S. Penczek, Thermodynamics of L-lactide polymerization. Equilibrium monomer concentration, Macromolecules, 1990, 23, 1636–1639 CrossRef CAS.
- D. R. Witzke, R. Narayan and J. J. Kolstad, Reversible Kinetics and Thermodynamics of the Homopolymerization of l-Lactide with 2-Ethylhexanoic Acid Tin(II) Salt, Macromolecules, 1997, 30, 7075–7085 CrossRef CAS.
- C. Alberti and S. Enthaler, Depolymerization of End-of-Life Poly(lactide) to Lactide via Zinc-Catalysis, ChemistrySelect, 2020, 5, 14759–14763 CrossRef CAS.
- C. F. Gallin, W.-W. Lee, J. A. Byers and A. Simple, Selective, and General Catalyst for Ring Closing Depolymerization of Polyesters and Polycarbonates for Chemical Recycling, Angew. Chem., Int. Ed., 2023, 62, e202303762 CrossRef CAS PubMed.
- D. Cam and M. Marucci, Influence of residual monomers and metals on poly (l-lactide) thermal stability, Polymer, 1997, 38, 1879–1884 CrossRef CAS.
- T. M. McGuire, A. Buchard and C. Williams, Chemical Recycling of Commercial Poly(l-lactic acid) to l-Lactide Using a High-Performance Sn(II)/Alcohol Catalyst System, J. Am. Chem. Soc., 2023, 145, 19840–19848 CrossRef CAS.
- L. Cederholm, J. Wohlert, P. Olsén, M. Hakkarainen and K. Odelius, “Like Recycles Like”: Selective Ring-Closing Depolymerization of Poly(L-Lactic Acid) to L-Lactide, Angew. Chem., Int. Ed., 2022, 61, e202204531 CrossRef CAS.
- J. P. Macdonald and M. P. Shaver, An aromatic/aliphatic polyester prepared via ring-opening polymerisation and its remarkably selective and cyclable depolymerisation to monomer, Polym. Chem., 2016, 7, 553–559 RSC.
- E. Lizundia, V. A. Makwana, A. Larrañaga, J. L. Vilas and M. P. Shaver, Thermal, structural and degradation properties of an aromatic–aliphatic polyester built through ring-opening polymerisation, Polym. Chem., 2017, 8, 3530–3538 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry parallel, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- O. Y. Abdelaziz, K. Li, P. Tunå and C. P. Hulteberg, Continuous catalytic depolymerisation and conversion of industrial kraft lignin into low-molecular-weight aromatics, Biomass Convers. Biorefin., 2018, 8, 455–470 CrossRef CAS.
- M. B. Figueirêdo, F. W. Keij, A. Hommes, P. J. Deuss, R. H. Venderbosch, J. Yue and H. J. Heeres, Efficient Depolymerization of Lignin to Biobased Chemicals Using a Two-Step Approach Involving Ozonation in a Continuous Flow Microreactor Followed by Catalytic Hydrotreatment, ACS Sutainable Chem. Eng., 2019, 7, 18384–18394 CrossRef.
- P. Lian, S. Liu, Z. Ma, Y. Wang, Y. Han, G. Sun and X. Wang, Continuous-Flow Microreactor Accelerates Molecular Collisions for Lignin Depolymerization to Phenolic Monomers and Oligomers, Biomacromolecules, 2023, 24, 5152–5161 CrossRef CAS PubMed.
- B. Braun, J. R. Dorgan and S. F. Dec, Infrared Spectroscopic Determination of Lactide Concentration in Polylactide: An Improved Methodology, Macromolecules, 2006, 39, 9302–9310 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Tools and techniques for solvent selection: green solvent selection guides, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- H. Nishida, T. Mori, S. Hoshihara, Y. Fan, Y. Shirai and T. Endo, Effect of tin on poly(l-lactic acid) pyrolysis, Polym. Degrad. Stab., 2003, 81, 515–523 CrossRef CAS.
- Y. Fan, H. Nishida, Y. Shirai and T. Endo, Control of racemization for feedstock recycling of PLLA, Green Chem., 2003, 5, 575–579 RSC.
- Y. Fan, H. Nishida, S. Hoshihara, Y. Shirai, Y. Tokiwa and T. Endo, Pyrolysis kinetics of poly(l-lactide) with carboxyl and calcium salt end structures, Polym. Degrad. Stab., 2003, 79, 547–562 CrossRef CAS.
- H. Tsuji, H. Daimon and K. Fujie, A new strategy for recycling and preparation of poly (L-lactic acid): hydrolysis in the melt, Biomacromolecules, 2003, 4, 835–840 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR spectroscopy, SEC traces and inline kinetic data. See DOI: https://doi.org/10.1039/d4sc05891g |
| This journal is © The Royal Society of Chemistry 2025 |