
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic–aromatic interactions drive fold switch of GA95 and GB95 with three residue difference†
Chen
Chen‡
ab,
Zeting
Zhang‡
*ab,
Mojie
Duan
*c,
Qiong
Wu
a,
Minghui
Yang
 ab,
Ling
Jiang
ab,
Maili
Liu
ab and
Conggang
Li
*ab
ab,
Ling
Jiang
ab,
Maili
Liu
ab and
Conggang
Li
*ab
aKey Laboratory of Magnetic Resonance in Biological Systems, State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, National Center for Magnetic Resonance in Wuhan, Wuhan National Laboratory for Optoelectronics, Wuhan Institute of Physics and Mathematics, Innovation Academy of Precision Measurement, Chinese Academy of Sciences, Wuhan 430071, China. E-mail: conggangli@apm.ac.cn; zhangzeting@wipm.ac.cn
bGraduate University of Chinese Academy of Sciences, Beijing 100049, China
cInterdisciplinary Institute of NMR and Molecular Sciences, School of Chemistry and Chemical Engineering, The State Key Laboratory of Refractories and Metallurgy, Wuhan University of Science and Technology, Wuhan 430081, China. E-mail: mduan@wust.edu.cn
First published on 18th December 2024
Abstract
Proteins typically adopt a single fold to carry out their function, but metamorphic proteins, with multiple folding states, defy this norm. Deciphering the mechanism of conformational interconversion of metamorphic proteins is challenging. Herein, we employed nuclear magnetic resonance (NMR), circular dichroism (CD), and all-atom molecular dynamics (MD) simulations to elucidate the mechanism of fold switching in proteins GA95 and GB95, which share 95% sequence homology. The results reveal that long-range interactions, especially aromatic π–π interactions involving residues F52, Y45, F30, and Y29, are critical for the protein switching from a 3α to a 4β + α fold. This study contributes to understanding how proteins with highly similar sequences fold into distinct conformations and may provide valuable insights into the protein folding code.
Introduction
Protein folding is crucial for achieving the functional conformation of proteins, and elucidating its mechanisms is vital for comprehending the rules of this complex process. Traditionally, it was widely accepted that the conformation of a protein is determined by its sequence, while the function is determined by its conformation.1,2 This traditional paradigm has been challenged by the discovery of metamorphic proteins,3,4 which perform diverse protein functions by remodeling their structures and reversibly interconvert between different states.5–7 Notably, metamorphic proteins have been found to be widely distributed and involved in a variety of biological processes,8–10 and may be more common than previously thought.11,12 Despite progress in understanding fold evolution and structure resolution, the mechanisms of interconversion between different structures remain limited.13,14 Therefore, understanding the mechanism underlying structural transition is essential for deciphering metamorphic protein folding.In recent years, Alexander et al. have designed proteins GA and GB by introducing mutations in two structural domains of the streptococcal protein G.15,16 These proteins exhibit high sequence homology but distinct folding patterns: GA folds into a 3α structure, while GB folds into a 4β + α structure. Their fold switching is accomplished through only a small number of amino acid mutations. Notably, in proteins GA95 and GB95, all three mutation sites are hydrophobic amino acids, two of which are aromatic amino acids. Aromatic residues are known to cluster in the hydrophobic core of folded proteins, where they stabilize proteins through hydrophobic effect, as well as π–π or cation–π interactions17 due to their quadrupolar electrostatic character.18,19 The configuration of the aromatic cluster is found to play crucial roles in protein stability, molecular recognition processes as well as catalytic functions of enzymes.20–24 Despite the mentioned relevance, the impact of aromatic cluster side-chain interactions on the fold switch of proteins between different structures is still poorly characterized.
Research on the mechanisms of fold switching between GA and GB has attracted widespread interest. Studies have suggested that structural differences in the denatured states play key roles in determining their folding pathway.25–28 Additionally, from thermodynamic and kinetic perspectives, fold switching of GA and GB is related to the N- and C-terminal sequences,29–32 and are supported by certain local interactions.33–35 However, the mechanism underlying the fold switch between GA and GB remains insufficiently well-defined. Current studies primarily rely on theoretical models or computer simulations due to the challenges in obtaining soluble protein samples and their inherent instability. Although AlphaFold is a powerful tool for predicting protein static structures36–39 and protein–protein interactions,40 the current default algorithms still face challenges in predicting metamorphic protein conformations and the effects of sequence variation.41,42 Importantly, AlphaFold cannot provide protein folding pathways,43,44 limiting its applications in exploring folding mechanisms for metamorphic proteins. Therefore, experimental data is essential for elucidating the exact mechanism of fold switching between GA and GB.
This study investigates how the proteins GA95 and GB95, which share 95% sequence homology, fold into different conformations by using nuclear magnetic resonance (NMR) and all-atom molecular dynamics (MD) simulations, revealing the intrinsic mechanism of their fold switching in GA and GB. The findings highlight the impact of a transition of early contacts of aromatic residues and the subsequent formation of an aromatic cluster in driving fold switching.
Results and discussion
Effects of mutation sites on protein fold of GA95 and GB95
Soluble protein samples were fundamental for this study. Encouragingly, unlike previous specialized expression and purification systems used to produce the target protein,16 we designed a fusion protein expression construct, His-SUMO-GA95/GB95, which can successfully obtain soluble GA95 and GB95 in a routine E. coli expression system. Both GA95 and GB95 consist of 56 amino acid residues, differing at positions 20, 30, and 45 (Fig. 1). This difference causes GA95 to fold into a 3α, while GB95 folds into a 4β + α structure. To examine the short-rang effects of the mutation sites L20A, I30F, and L45Y on folding, we initially obtained NMR spectra of the truncated proteins GA95-38 and GB95-38 containing two mutation sites, as well as the truncated proteins GA95-45 and GB95-45 containing all three mutation sites (Fig. 2A, B, F and G). The 2D 1H–15N HSQC spectra revealed poorly dispersed signals, with the chemical shifts of the peaks centered at 7.5–8.5 ppm, a characteristic of disordered proteins. This indicates that neither truncated protein folds into a tertiary structure. Despite the two or three residue differences, the spectra of GA95-38/GB95-38 or GA95-45/GB95-45 are very similar, suggesting that the short-range effect on protein fold switching is minimal.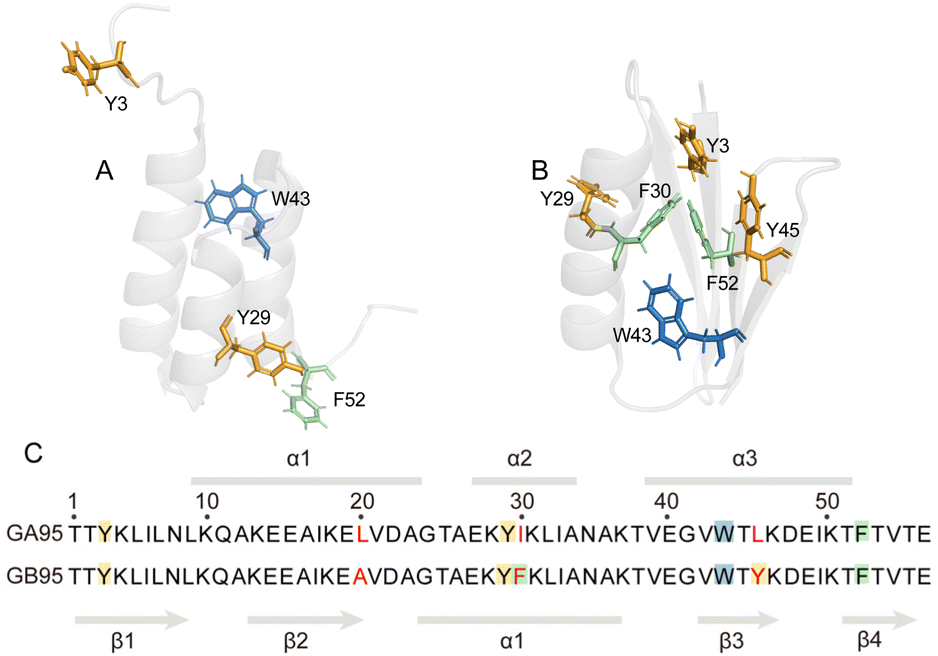 | ||
| Fig. 1 Structures of GA95 and GB95. (A) and (B) The tertiary structures of GA95 (PDB id: 2KDL) and GB95 (PDB id: 2KDM). Aromatic amino acid residues are shown in stick representations with different colors, Y is shown in orange, W is shown in blue, and F is shown in green; (C) sequence comparison and secondary structure of GA95 and GB95. Three residue differences (20, 30, 45) of GA95 and GB95 are highlighted in red. Aromatic amino acid residues are highlighted with colors consistent with the stick representations. | ||
 | ||
| Fig. 2 Amino acid nodes in GA95 and GB95 folded from secondary to tertiary structures. (A) 1H–15N HSQC spectrum of GA95-38 (red). (B) 1H–15N HSQC spectrum of GA95-45 (green). (C) 1H–15N HSQC spectrum of GA95-51 (orange). (D) 1H–15N HSQC spectrum of GA95-52 (blue). (E) 1H–15N HSQC spectrum of GA95 (black). (F) 1H–15N HSQC spectrum of GB95-38 (red). (G) 1H–15N HSQC spectrum of GB95-45 (green). (H) 1H–15N HSQC spectrum of GB95-54 (orange). (I) 1H–15N HSQC spectrum of GB95-55 (blue). (J) 1H–15N HSQC spectrum of GB95 (black). (K) Histogram of secondary structure content statistics for GA95 and its truncated proteins GA95-38, GA95-45, GA95-51 and GA95-52. (L) Histogram of secondary structure content statistics for GB95 and its truncated proteins GB95-38, GB95-45, GB95-54 and GB95-55. The content of the α-helix is labeled in black, and the content of the β-sheet is labeled in red. | ||
We further extended the truncated proteins GA95-n and GB95-n (n is the number of residues) to various lengths (Table 1) to explore the minimal sequence required for tertiary structure formation. HSQC spectra (Fig. 2A–J) were used to assess their folds; well-dispersed spectra suggest stable tertiary structure formation. We found that GA begins to be partially folded when the sequence reaches 52 residues (GA95-52), while GB needs three more residues, reaching 55 to be partially folded (GB95-55) (Fig. 2D and I). The structural transition from disordered to partial folded structures was also observed by one-dimensional 19F spectra of GA95-n and GB95-n, consistent with the HSQC results (Fig. S1 and S2†). These observations suggest that the three mutation sites affect the final folding of the tertiary structure mainly through the long-range interactions with the C-terminal residues.
| Proteins | Sequence |
|---|---|
| a Mutation sites are in red. | |
| GA95-38 |

|
| GA95-45 |

|
| GA95-51 |

|
| GA95-52 |

|
| GA95-53 |

|
| GA95-(38–56) |
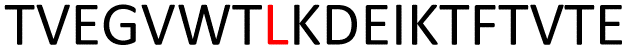
|
| GB95-38 |

|
| GB95-45 |

|
| GB95-54 |

|
| GB95-55 |

|
| GB95-(38–56) |

|
We also noticed that Gly41 had weaker signal intensity in GB95-45 compared to GA95-45 in 1H–15N HSQC spectra (Fig. S3†), and its chemical shifts changed with sequence length (Fig. S4†). This suggests that although the proteins had not yet folded into a tertiary structure, the secondary structure may have been formed, resulting in observable dynamic changes. We then utilized circular dichroism (CD) to measure the secondary structure content of the truncated proteins containing two or three mutation sites in GA95 and GB95 (Fig. 2K, L, S5 and S6†). As expected, differences in secondary structures were indeed observed. There was a small difference between GA95-45 and GB95-45, whereas an increased disparity was observed between GA95-51 and GB95-54. GA95-51 had significantly more α-helices, while GB95-54 had more β-sheets.
The NMR and CD results of these truncation variants suggest that the folding of GA95 and GB95 is closely related to the three mutation sites and potentially dependent on key interactions involved in the C-terminal region. Similarly, truncation on the N-terminal region was also performed. CD experiments showed that the C-terminal fragments GA95-(38–56) and GB95-(38–56) containing L45Y are disordered, with a negative peak appearing at wavelengths of less than 200 nm (Fig. S7†), which is different from the results in GB1,45 possibly due to sequence differences. This observation indicated that the folding of GA and GB into different conformations relies on long-range interactions between the N- and C-terminal regions. This is also demonstrated by structural changes in GA and GB that highlight the importance of these regions in folding.16
Folding pathway of GA95 and GB95 by MD simulations
Since NMR and CD experiments only provide equilibrium information of tertiary and secondary structures and cannot directly reveal the folding pathway, we utilized all-atom molecular dynamics simulations of GA95-53 and GB95 to gain insights into the effects of three mutation sites on the folding pathway. We initiated the replica exchange with solute tempering (REST2) simulations from the initial conformations, and the initial structures of different simulation replicas are depicted in Fig. S8.† The folding free energy landscapes (FELs) of GA95-53 and GB95 at room temperature (300 K) were reconstructed based on the REST2 simulations (Fig. 3A and C), and their FELs reveal a multi-step folding mechanism. The folding of GA95-53 and GB95 involves a transition from the unfolded state (state 4) to the folded intermediate states (state 3 and state 2) and finally to the folded state (state 1) (Fig. 3F and G). The data indicate that in the unfolded state of the protein (state 4), different transient secondary structures are already present, with α-helices observed in GA95-53 and short β-sheets observed in GB95 (Fig. 3F, G and S9†). Thus, the three mutation sites induce different secondary structure propensities at the early stage of folding, consistent with the CD experimental results of the truncated variants. | ||
| Fig. 3 Shift in interactions during fold switching of GA95 and GB95. (A) Free energy landscape of folding process of GA95-53. The free energy minima are colored in blue, and the saddle point (state T) between the two minima 3 and 4 is labeled by a dashed circle. (B) The occupancy of important interactions in different states of the folding process of GA95-53. (C) Free energy landscape of folding process of GB95. (D) and (E) The occupancy of important interactions in different states of the folding process of GB95. (F) and (G) The representative structures of important states in the folding process of GA95-53(left) and GB95(right). The native structures of GA95-53 or GB95 are shown in gray. The residues Y29, I30/F30, Y45, and F52, which are important in the folding pathways, are depicted by ball-and-stick. Y29, I30/F30, Y45, and F52 are colored green, yellow, pink, and blue, respectively. | ||
As indicated in Fig. 2C and D, the transition from a disordered structure to a tertiary structure occurs from residue 51 to 52 in GA95, suggesting that F52 is crucial for folding, particularly as some studies have also pointed out that F52 is involved in key interactions within protein GB.16,35,46 The interwoven aromatic interactions within proteins are well-established as pivotal for structural stability.20,24,47,48 GA95's NMR structure (PDB id: 2KDL) indicated a potential π–π interaction between F52 and Y29, and the aromatic rings of F52 can interact with that of both F30 and Y45 within spatial proximity simultaneously in GB95's NMR structure (PDB id: 2KDM). Therefore, it is reasonable to assume that the π–π interactions involving these three residues form an aromatic cluster, which contributes to folding switch of GA95 and GB95. To analysis the π–π interaction network during folding, we counted the occupancy of structural states in which residue F52 interacts either with Y29 or I30/F30 in the different folded states of the protein. In GA95, only the Y29–F52 interaction was observed (Fig. 3B). In contrast, in GB95, the F52–Y29 interaction was observed only in state 4 and state 3, while the F52–F30 interaction began to occur in state 3 and became fully dominant in the subsequent folded states (state 2 and state 1) (Fig. 3D). These findings indicate that the F52–Y29 interaction persists throughout the folding process of GA95, whereas in the mid-to-late stages (from state 3 to state 2) of GB95 folding, there is a transition in interactions, with the F52–Y29 interaction being entirely replaced by the F52–F30 interaction. This shift is possibly due to a higher density of the π-electron cloud in the F30 side chain.49,50
To understand what causes the shift in long-range π–π interactions from F52–Y29 to F52–F30 during the β3–β4 formation step, transitioning from the unfolding state (state 3) to the partially folded state (state 2) of GB95 (Fig. 3D, E, and 3G), we analyzed inter-residue interactions at different folding states of GB95. We observed that the interaction between Y45 and F52 occurs at state 2 (Fig. 3E). Combined with the previous observations that the F30–F52 interaction initiates at state 3 and fully replaces F52–Y29 at state 2 (Fig. 3D), this suggests a significant synergistic role for Y45 in rearranging F52 interactions, resulting in folding switch from 3α to 4β + α. This finding also aligns with Sikosek et al.'s report that the interaction between F52 and Y45 is critical in the protein folding switch.35
Experimental verification of the key interactions of F52 in the fold of GA95 and GB95
The MD simulations showed that Y29 and F52 contact is a folding rate-limit step (Fig. 3B) in GA95-53. By mutation at F52 to or Y29 to A in GA95-53, the narrowly dispersed 1H–15N HSQC spectra indicate the mutations disrupted the native structure (Fig. 4A and B), confirming that F52–Y29 interaction is essential in GA95 folding. | ||
| Fig. 4 Validation of F52 interactions. (A) 1H–15N HSQC spectra of GA95-53 and GA95-53 Y29A; (B) 1H–15N HSQC spectra of GA95-53 and GA95-53 F52A; (C)1H–15N HSQC spectra of GB95 and GB95 F30I; (D) 1H–15N HSQC spectra of GB95-55 and GB95-55 Y29A; the wild type is marked in blue and the mutant is marked in red. | ||
The literature and our MD simulations proposed that π–π interactions between F52 and F30 also play a key role in the folding of the 4β + α structure of GB.16,35 A former study showed that the F52A mutation would induce the unfolding of GB95.16 In this study, we found that the introduction of mutation F30I also disrupted the structure and drove the unfolding of the protein (Fig. 4C). These results demonstrate that the long-range π–π interaction between F30 and F52 is essential in GB95 folding. Additionally, Y29 also plays a significant role in GB95 folding. Introducing a mutation at Y29 resulted in partially unfolded structures of GB95, as observed in the 1H–15N HSQC spectrum (Fig. 4D). However, compared to the complete unfolding of GB95 induced by F30I, the Y29A mutation only led to partial unfolding, suggesting that F30 plays a more critical role than Y29 in GB95 folding. Based on the NMR and MD simulations result above, we propose that the shift in interaction from Y29 to F30 is crucial for the fold switching between GA95 and GB95.
Y45 facilitates the full shift of F52 interactions from Y29 to F30 to form an aromatic cluster in GB
We have found that F30 and F52 are critical for the 4β + α fold of GB95. However, despite the presence of F30, GA98, which evolved from the I30F mutation in GA95 mainly maintains a 3α fold with F52 still interacting with Y29 (Fig. S10†). This illustrates that the long-range π–π interaction between F52 and F30 does not occur naturally in GA. Therefore, we reasonably assume that the promotion of the F30–F52 interaction is associated with Y45 in GB, supported by the fact that L45Y is the only mutation site driving the fold switch between GA98 and GB98.16 To elucidate the role of Y45 in GB95 folding, we introduced mutations at this site. The Y45I mutation disrupted the protein structure (Fig. 5A), while the Y45F mutation restored the protein structure by reintroducing the benzene ring (Fig. 5B). Analysis of the GB95 NMR structure (PDB id: 2KDM) suggests that, in addition to the main chain hydrogen bonds involved in β34 folding, π–π interactions also occur between the side chains of F52 and Y45. Furthermore, the introduction of the L45I mutation in GA95 did not alter its protein structure (Fig. 5C), indicating that L45 is not essential in GA95. Combined with the necessity of Y45 and its side chain π-bonding in 4β + α folding, we suggest that the reason for the predominance of the interaction between F30 and F52 in GB95 is closely related to the presence of Y45. | ||
| Fig. 5 Validation of the role of Y45 in GB95 to form an aromatic cluster. (A) 1H–15N HSQC spectra of GB95 and GB95 Y45I; (B) 1H–15N HSQC spectra of GB95 and GB95 Y45F; (C)1H–15N HSQC spectra of GA95 and GA95 L45I; (D) 1H–15N HSQC spectra of GB95 and GB95 L32P. The disappearing residues are marked with arrows; the wild type is marked in blue and the mutant is marked in red. | ||
Based on the MD simulations, we found that the F30–F52 interaction facilitates the formation of main-chain hydrogen bonds of the central α-helix, ultimately triggering the folding of the 4β + α structure. However, it remains uncertain whether the F30–F52 interaction solely influences the formation of the final α-helix or plays a key role in the early stages of GB95 folding. To address this, we introduced a leucine-to-proline mutation at residue 32, known to disrupt the central α-helix.51 In GB95, this mutation only causes the disappearance of signals from some residues in the central α-helix in the 1H–15N HSQC spectrum, with new signals emerging in the center of the spectrum (Fig. 5D), indicating disruption of the central α-helix, but not the overall GB95 structure by L32P mutation. Given the fact that F30I disrupts the structure of GB95, as mentioned earlier, we conclude that the long-range π–π interaction between F30 and F52 is crucial for the overall structure formation of GB95 during early folding stages. Furthermore, the side-chain π–π interaction involving Y45 and F52, as well as the folding of β34, is heavily reliant on F30.
Based on these results, we concluded that Y45–F52 π–π interaction stabilizes β34 and forms an aromatic cluster with F30, facilitating the complete shift of F52's interaction from Y29 to F30 and ultimately stabilizing the structure of GB95. It follows that the long-range interactions, particularly the preferences for self-association among hydrophobic residues52–54 and the formation of aromatic clusters exert a significant influence on protein structures.24,47,48 More importantly, it is plausible to propose that there may be more than one potential intramolecular interaction pattern in metamorphic proteins, where a specific interaction plays a decisive role in conformational transitions, termed folding switches. These folding switches may be the fundamental regulatory mechanism in metamorphic protein systems, capable of being formed or disrupted in response to external factors such as mutation, ligand binding, or solution conditions,55,56 which facilitate changes in the population of conformational ensembles57 and affect structural transitions.58,59
The long-range interactions of the C-terminal sequences additionally stabilize the protein structure
The results obtained from truncated proteins indicate a progression from partially folded to well-folded structures as the sequences were extended, underscoring the crucial role of the C-terminal sequence in stabilizing protein structure. Specifically, T53 stabilized the 3α fold of GA95 (Fig. 4A), while T55 and E56 stabilized the 4β + α fold of GB95 (Fig. 6A and D). Simulation results further indicate that in the transition state of GA95 (state T), there is an interaction between T53 and Y29 (Fig. S11†). This finding aligns with the experimental observations showing that GA95-53 folded well and GA95-53 Y29A completely lost its native structure (Fig. 4A and S12†), verifying that T53 and Y29 stabilized the structure of GA95.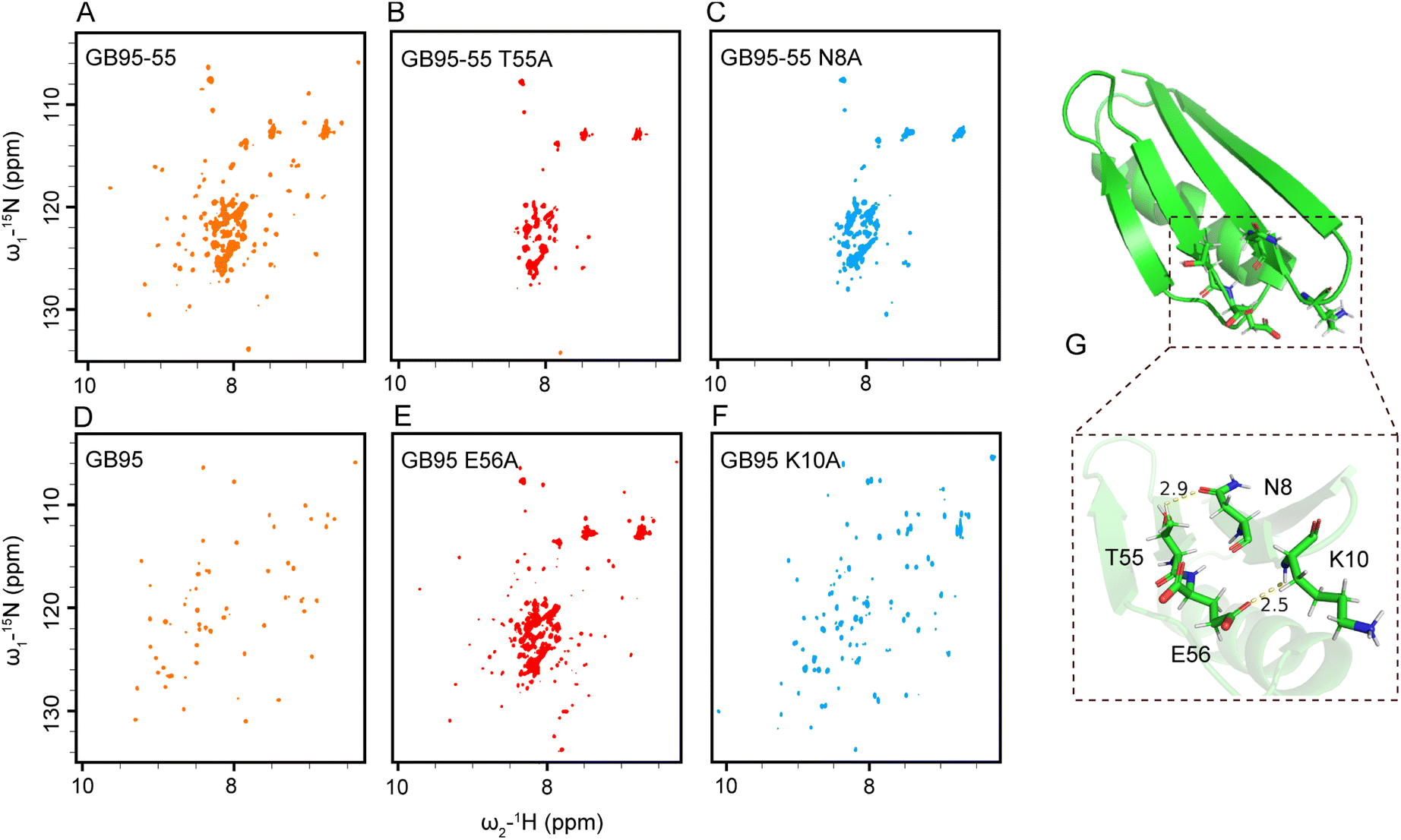 | ||
| Fig. 6 The interactions between the N- and C-terminal amino acids stabilize the tertiary structure of GB95. (A) 1H–15N HSQC spectrum of GB95-55 (orange). (B) 1H–15N HSQC spectrum of GB95-55 T55A (red). (C) 1H–15N HSQC spectrum of GB95-55 N8A (blue). (D) 1H–15N HSQC spectrum of GB95 (orange). (E) 1H–15N HSQC spectrum of GB95 E56A (red). (F) 1H–15N HSQC spectrum of GB95 K10A (blue). (G) Hydrogen bonds and bond distance formed in T55–N8 and E56–K10. | ||
To investigate the roles of T55 and E56 in GB95 folding, we analyzed the NMR structure of GB95 (PDB id: 2KDM) and found that the side chain OH group of T55 lies within hydrogen bond distance to the side chain carbonyl group of N8, and the side chain carbonyl group of E56 lies within hydrogen bond distance to the main chain NH group of K10 (Fig. 6G). It is reasonable to hypothesize that the hydrogen bonds between T55–N8 and E56–K10 contribute significantly to the folding of GB95.60 To test this hypothesis, we introduced T55S, T55A, or N8A mutations into GB95-55, respectively. As expected, the 1H–15N HSQC spectrum of GB95-55 T55S closely matched that of GB95-55 (Fig. S13†). In contrast, the spectra of GB95-55 T55A and GB95-55 N8A exhibited poor signal dispersion (Fig. 6B and C), indicating that the absence of the T55 and N8 side chains significantly destabilized the native structure. These results confirm that the hydrogen bonds involving the side chain OH group of T55 and the side chain carbonyl group of N8 play essential roles in stabilizing the 4β + α fold of GB95.
Furthermore, we introduced E56A, K10A, and K10E mutations into GB95. Comparing the 1H–15N HSQC spectra of these mutants with that of GB95 revealed that the E56A mutation results in a significant loss of the native structure (Fig. 6E), implying that the side chain of E56 plays a crucial role in the 4β + α fold in GB95. However, the K10A mutation did not affect the protein structure (Fig. 6F), nor did the K10E mutation (Fig. S14†), indicating that the side chain of K10 is not involved in key interactions. Therefore, we conclude that hydrogen bonds involving the side chain carbonyl group of E56 and the main chain NH group of K10 contribute significantly to the stabilization of the tertiary structure of GB95.
Surprisingly, the T55A or N8A mutation did not affect the tertiary structure of GB95 when the E56–K10 hydrogen bond was preserved (Fig. S15 and S16†). This suggests that the hydrogen bonds involving T55–N8 and E56–K10 act synergistically, akin to a pair of “double-breasted buckles.” When the N- and C-termini of proteins are bridged by the E56–K10 interaction, the T55–N8 hydrogen bond has little effect on protein conformation. However, if E56 is disrupted or absent, the hydrogen bond involving T55 and N8 becomes essential for maintaining conformational stability in GB95. In summary, additional long-range interactions involving the C-terminal residues are essential for the ultimate stabilization of the protein structure. In GA95, the 3α fold is stabilized by T53–Y29 interaction, whereas in GB95, the N- and C-termini of the protein are connected by T55–N8 and E56–K10 interactions, further stabilizing the 4β + α fold.
Conclusions
GA and GB have been designed as engineered proteins with highly similar sequences but distinct structures.16 This study examined the proteins GA95 and GB95 with 95% sequence homology. By combining experimental data and simulations, we elucidate how the proteins GA95 and GB95 fold into different conformations through three mutation sites, thereby elucidating the mechanism of fold switching between these two conformations (Fig. 7). Our findings indicate that the mutation sites L20A, I30F, and L45Y influence secondary structure propensities and early contacts of aromatic residues in both GA95 and GB95. Particularly, during the mid-to-late stage of GB95 folding, the aromatic–aromatic interaction of F52 is shifted, leading to the formation of an aromatic cluster involving residue F30 and Y45. This rearrangement of inter-residue interactions facilitates protein fold switching. Moreover, extensive mutational studies revealed that long-range interactions of C-terminal residues can stabilize different structures. However, AlphaFold2 (AF2) fails to predict structures for many GA and GB truncations and mutants constructed in our study or previously published work16 (Table S1 and S2†), indicating its current limitations in accurately recognizing dynamic changes in long-range interactions during protein folding.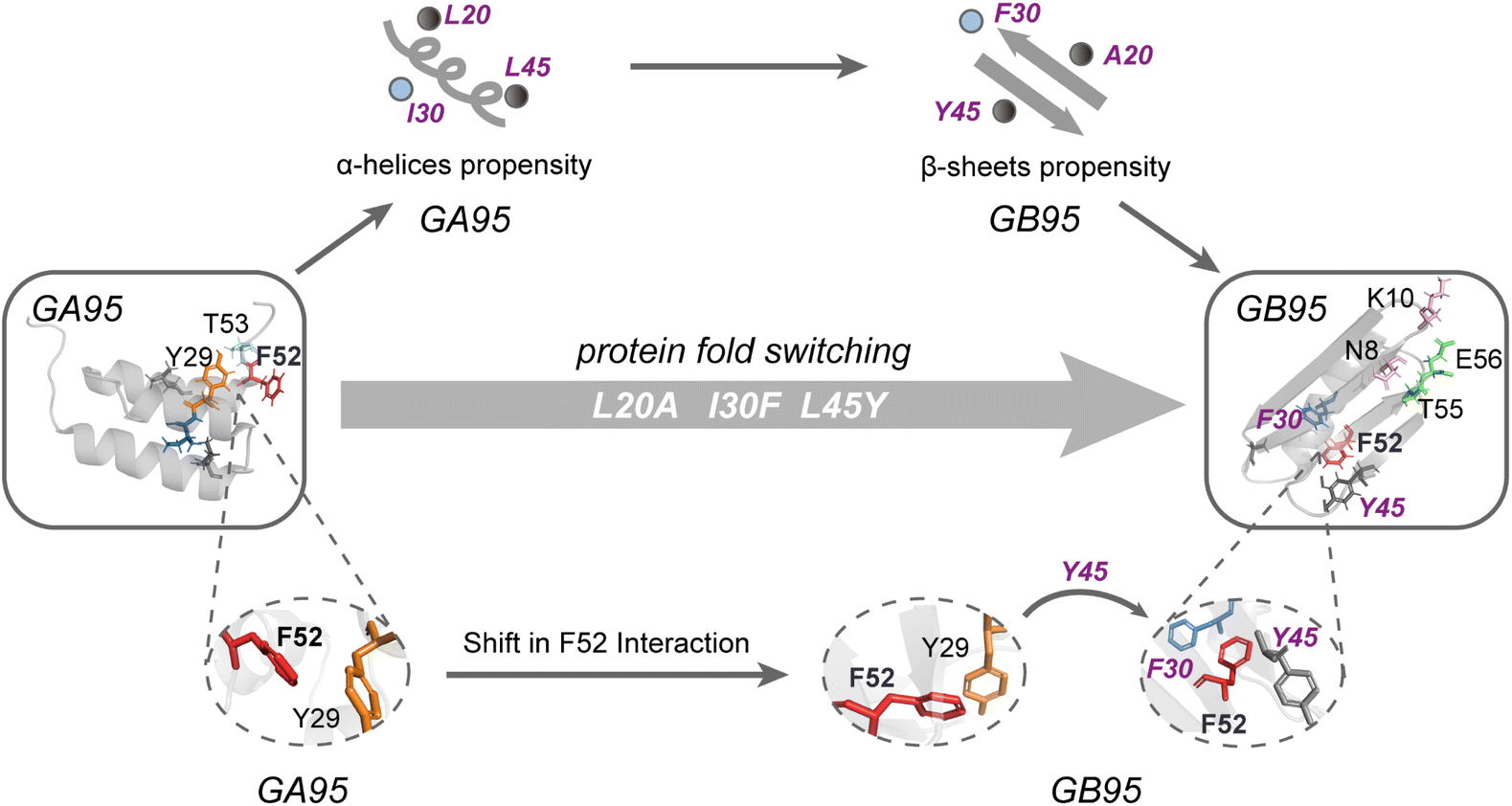 | ||
| Fig. 7 The mechanism of fold switching in GA95 and GB95. GA95 and GB95 undergo fold switching through mutations at three specific sites, altering their secondary structure preferences and residue interactions. The folded state (left: GA95 and right: GB95) is depicted in gray. In the tertiary structures of GA95 and GB95, the three mutation sites and other crucial amino acids involved in long-range interactions are shown in stick representation: L20A and L45Y in dark grey, I30F in blue, Y29 in orange, F52 in red, T53 in light blue, E56 and T55 in green, N8 and K10 in light pink. The secondary structures of proteins in the unfolded state are related to L20A, I30F, and L45Y, with GA95 preferring α-helices while GB95 prefers β-sheets. The interactions involving F52 during the folding process in GA95 and GB95 are represented as local magnification. In GA95, F52 interacts with Y29, whereas in GB95, the interaction of F52 shifts from Y29 to F30, with Y45 being the key element contributing to the complete shift of this interaction. | ||
In summary, our work provides empirical validation for studying the dynamic mechanisms underlying fold switching between different structures of proteins with highly similar sequences. It emphasizes how reshuffling aromatic residue side-chain interactions within molecules can lead to the remodeling of secondary structures—offering insights into solving future challenges in protein folding by understanding these critical inter-residue interactions.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
C. C., Z. Z., and C. L. designed research; C. C., Z. Z., and M. D. performed research; C. C., M. D., Q. W., M. Y., Z. Z., L. J., M. L., and C. L. analyzed data; C. C., Z. Z., M. D., and C. L. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by grants from the National Natural Science Foundation of China (22274161, 21925406, 21991080) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0540000).References
- C. B. Anfinsen, Science, 1973, 181, 223–230 CrossRef CAS.
- C. B. Anfinsen and H. A. Scheraga, Adv. Protein Chem., 1975, 29, 205–300 CrossRef CAS PubMed.
- P. N. Bryan and J. Orban, Curr. Opin. Struct. Biol., 2010, 20, 482–488 CrossRef CAS PubMed.
- A. F. Dishman and B. F. Volkman, Curr. Opin. Struct. Biol., 2022, 74, 102380 CrossRef CAS PubMed.
- L. L. Porte and L. L. Looger, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5968–5973 CrossRef.
- D. Chakravarty, S. Sreenivasan, L. Swint-Kruse and L. L. Porter, Nat. Commun., 2023, 14, 3177 CrossRef CAS.
- M. López-Pelegrín, N. Cerdà-Costa, A. Cintas-Pedrola, F. Herranz-Trillo, P. Bernadó, J. R. Peinado, J. L. Arolas and F. X. Gomis-Rüth, Angew Chem. Int. Ed. Engl., 2014, 53, 10624–10630 CrossRef.
- B. M. Burmann, S. H. Knauer, A. Sevostyanova, K. Schweimer, R. A. Mooney, R. Landick, I. Artsimovitch and P. Rösch, Cell, 2012, 150, 291–303 CrossRef CAS PubMed.
- X. Luo, Z. Tang, G. Xia, K. Wassmann, T. Matsumoto, J. Rizo and H. Yu, Nat. Struct. Mol. Biol., 2004, 11, 338–345 CrossRef CAS.
- A. F. Dishman and B. F. Volkman, ACS Chem. Biol., 2018, 13, 1438–1446 CrossRef CAS.
- L. L. Porter, Bioessays, 2023, 45, e2300057 CrossRef PubMed.
- M. Mezei, Proteins, 2020, 89, 3–5 CrossRef.
- L. L. Porter, I. Artsimovitch and C. A. Ramírez-Sarmiento, Curr. Opin. Struct. Biol., 2024, 86, 102807 CrossRef CAS PubMed.
- K. Madhurima, B. Nandi and A. Sekhar, Open Biol. J., 2021, 11, 210012 CrossRef CAS.
- P. A. Alexander, Y. He, Y. Chen, J. Orban and P. N. Bryan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11963–11968 CrossRef CAS.
- P. A. Alexander, Y. H, Y. Chen, J. Orban and P. N. Bryan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21149–21154 CrossRef CAS PubMed.
- G. A. Dalkas, F. Teheux, J. M. Kwasigroch and M. Rooman, Proteins, 2014, 82, 1734–1746 CrossRef CAS.
- K. Carter-Fenk and J. M. Herbert, Chem. Sci., 2020, 11, 6758–6765 RSC.
- J. H. Williams, Acc. Chem. Res., 1993, 26, 593–598 CrossRef CAS.
- S. K. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CrossRef CAS PubMed.
- C. D. Tatko and M. L. Waters, J. Am. Chem. Soc., 2002, 124, 9372–9373 CrossRef CAS.
- W. Zhang, X. Wang, G. Zhu, B. Zhu, K. Peng, T. Hsiang, L. Zhang and X. Liu, Angew Chem. Int. Ed. Engl., 2024, e202406246 CAS.
- E. Lanzarotti, L. A. Defelipe, M. A. Marti and A. G. Turjanski, ChemInform, 2020, 12, 30 CrossRef CAS.
- T. V. Tran, T. Hoang, S.-H. Jang and C. Lee, PLoS One, 2023, 18, e0290686 CrossRef CAS PubMed.
- A. Morrone, M. E. McCully, P. N. Bryan, M. Brunori, V. Daggett, S. Gianni and C. Travaglini-Allocatelli, J. Biol. Chem., 2011, 286, 3863–3872 CrossRef CAS.
- R. Giri, A. Morrone, C. Travaglini-Allocatelli, P. Jemth, M. Brunori and S. Gianni, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17772–17776 CrossRef CAS.
- L. Sutto and C. Camilloni, J. Chem. Phys., 2012, 136, 185101 CrossRef PubMed.
- S. Gianni, M. E. McCully, F. Malagrinò, D. Bonetti, A. D. Simone, M. Brunor and V. Daggett, Angew Chem. Int. Ed. Engl., 2018, 57, 12795–12798 CrossRef CAS PubMed.
- S. K. Mani, H. Balasubramanian, S. Nallusamy and S. Samuel, Protein Eng. Des. Sel., 2010, 23, 911–918 CrossRef PubMed.
- N. Hansen, J. R. Allison, F. H. Hodel and W. F. v. Gunsteren, Biochemistry, 2013, 52, 4962–4970 CrossRef CAS PubMed.
- P. Tian and R. B. Best, PLoS Comput. Biol., 2020, 16, e1008285 CrossRef CAS PubMed.
- S.-H. Chen, J. Meller and R. Elber, Protein Sci., 2015, 25, 135–146 CrossRef.
- Y. He, Y. Chen, P. A. Alexander, P. N. Bryan and J. Orban, Structure, 2012, 20, 283–291 CrossRef CAS.
- C. Song, Q. Wang, T. Xue, Y. Wang and G. Chen, J. Mol. Recognit., 2016, 29, 580–595 CrossRef CAS.
- T. Sikosek, H. Krobath and H. S. Chan, PLoS Comput. Biol., 2016, 12, e1004960 CrossRef PubMed.
- K. Tunyasuvunakool, J. Adler, Z. Wu, T. Green, M. Zielinski, A. Žídek, A. Bridgland, A. Cowie, C. Meye, A. Laydon, S. Velankar, G. J. Kleywegt, A. Bateman, R. Evans, A. Pritze, M. Figurnov, O. Ronneberger, R. Bates, S. A. A. Kohl, A. Potapenko, A. J. Ballard, B. Romera-Paredes, S. Nikolov, R. Jain and D. Hassabis, Nature, 2021, 596, 590–596 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- G. M. D. Silva, G. P. Lisi, J. Y. Cui, D. C. Dalgarno, G. P. Lis and B. M. Rubenstein, Nat. Commun., 2024, 15, 2464 CrossRef.
- H. K. Wayment-Steele, A. Ojoawo, R. Otten, J. M. Apitz, W. Pitsawong, M. Hömberger, S. Ovchinnikov, L. Colwell and D. Kern, Nature, 2024, 625, 832–839 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O’Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS.
- M. A. Pak, K. A. Markhieva, M. S. Novikova, D. S. Petrov, I. S. Vorobyev, E. S. Maksimova, F. A. Kondrashov and D. N. Ivankov, PLoS One, 2023, 18, e0282689 CrossRef CAS.
- D. Chakravarty and L. L. Porter, Protein Sci., 2022, 31, e4353 CrossRef CAS PubMed.
- J. Skolnick, M. Gao, H. Zhou and S. Singh, J. Chem. Inf. Model., 2021, 61, 4827–4831 CrossRef CAS.
- C. Outeiral, D. A. Nissley and C. M. Deane, Bioinformatics, 2021, 38, 1881–1887 CrossRef PubMed.
- F. J. Blanco and L. Serrano, Eur. J. Biochem., 1995, 230, 634–649 CrossRef CAS PubMed.
- F. J. Blanco, G. Rivas and L. Serrano, Nat. Struct. Biol., 1994, 1, 584–590 CrossRef CAS PubMed.
- M. Biancalana, K. Makabe, S. Yan and S. Koide, Protein Sci., 2015, 24, 841–849 CrossRef CAS PubMed.
- H.-Y. Cai, Z.-J. Xu, J. Tang, Y. Sun, K.-X. Chen, H.-Y. Wang and W.-L. Zhu, Acta Pharmacol. Sin., 2012, 33, 1062–1068 CrossRef CAS PubMed.
- V. Sivasakthi, A. Anbarasu and S. Ramaiah, Cell Biochem. Biophys., 2013, 67, 853–863 CrossRef CAS PubMed.
- Y. Zhao, J. Li, H. Gu, D. Wei, Y.-C. Xu, W. Fu and Z. Yu, Interdiscipl. Sci., 2015, 7, 211–220 CrossRef CAS PubMed.
- C.-W. Lam, Y.-P. Yuen, W.-F. Cheng, Y.-W. Chan and S.-F. Tong, Clin. Chim. Acta, 2006, 364, 256–259 CrossRef CAS PubMed.
- M. M. Gromiha and S. Selvaraj, J. Mol. Biol., 2001, 310, 27–32 CrossRef CAS PubMed.
- S. Basaka, R. P. Nobregaa, D. Tavellaa, L. M. Deveaua, N. Kogab, R. Tatsumi-Kogab, D. Baker, F. Massia and C. R. Matthewsa, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6806–6811 CrossRef PubMed.
- D. Sengupta and S. Kundu, BMC Bioinf., 2012, 13, 142 CrossRef CAS PubMed.
- J.-H. Ha and S. N. Loh, Chemistry, 2012, 18, 7984–7999 CrossRef CAS PubMed.
- X. Zhang, T. Perica and S. A. Teichmann, Curr. Opin. Struct. Biol., 2013, 23, 954–963 CrossRef CAS PubMed.
- C.-J. Tsai, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9970–9972 CrossRef CAS PubMed.
- Z. Hu, D. Bowen, W. M. Southerland, A. d. Sol, Y. Pan, R. Nussinov and B. Ma, PLoS Comput. Biol., 2007, 3, e117 CrossRef PubMed.
- J. Kniazeff, L. Shi, C. J. Loland, J. A. Javitch, H. Weinstein and U. Gether, J. Biol. Chem., 2008, 283, 17691–17701 CrossRef CAS PubMed.
- J. Orban, P. Alexander, P. Bryan and D. Khare, Biochemistry, 1995, 34, 15291–15300 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04951a |
| ‡ C. Chen and Z. Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |