
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic evolution of nitrous oxide from nitric monoxide over Pt-loaded titanium dioxide under UV irradiation†
Ryo
Asayama‡
a,
Masanori
Takemoto‡
 a,
Arata
Suzuki
a,
Ryuichi
Watanabe
a,
Fuminao
Kishimoto
a,
Kenta
Iyoki
abc,
Tatsuya
Okubo
a and
Toru
Wakihara
*ad
a,
Arata
Suzuki
a,
Ryuichi
Watanabe
a,
Fuminao
Kishimoto
a,
Kenta
Iyoki
abc,
Tatsuya
Okubo
a and
Toru
Wakihara
*ad
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: wakihara@chemsys.t.u-tokyo.ac.jp
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), Kawaguchi, Saitama 332-0012, Japan
cDepartment of Environment Systems, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa-shi, Chiba 277-8563, Japan
dInstitute of Engineering Innovation, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 7th May 2025
Abstract
This study presents a photocatalytic evolution of nitrous oxide (N2O) from nitric monoxide (NO), well known as a harmful gas contained in exhaust gas. Pt nanoparticles (NPs) were loaded on titanium dioxide (TiO2) using different methods including impregnation, photo-deposition and chemical reduction methods. Bare TiO2 (without metal loading) did not exhibit high N2O evolution under UV irradiation, but all Pt-loaded TiO2 photocatalysts did exhibit improved N2O evolution.
Despite nitrous oxide (N2O) being the “third” greenhouse gas, it has recently attracted attention in various fields. In the petrochemical industry, selective oxidation of hydrocarbons is vital for producing valuable fine chemicals,1 and there has been growing interest in utilizing green and atom-efficient oxidants such as H2O2 and N2O for catalytic oxidation.2 Both H2O2 and N2O release monoatomic oxygen, forming harmless H2O and N2, respectively. N2O, in particular, is suitable as an oxidant in liquid and gas-phase reactions due to its high solubility in nonpolar media and gaseous nature. For instance, catalytic oxidation of CH4 into methanol using N2O over zeolites has been proposed, converting two greenhouse gases into a useful chemical.3,4 In the medical field, the demand of N2O, also known as “laughing gas,” has been increasing.5,6 Considering these advantages, N2O holds significant potential for clean technologies in fine chemical synthesis and medical developments.
Plant-scale manufacturing of N2O was done at Jinhong Gas and Heng'an Gas Co., Ltd., where the decomposition of NH4NO3 is used for N2O production. According to interviews with these companies, N2O produced through the current process is available to only high-end users in practical applications because of the high cost of the process (approximately 3000–5000 $ per Mg).7 The oxidation of NH3 is considered an alternative reaction for N2O production.7,8 However, there are concerns about the current price increase of NH3, which is probably a barrier to achieving inexpensive N2O production. As is well known, anthropogenic N2O is mainly released into the atmosphere during agricultural and industrial activities,9 and N2O capture can be a promising technique for the effective utilization of N2O.10,11 However, the concentration of N2O in the atmosphere is quite low (approximately 0.3–0.4 ppm). Many types of gas molecules, such as CO2 and H2O, are also present in the atmosphere, which makes selective separation of N2O in the atmosphere further challenging. Considering the future demand of N2O, further implementation of low-cost N2O production is highly recommended.
Nitric monoxide (NO) is a well-known gaseous pollutant that mainly comes from the exhaust gases of automobiles, plants, and thermal power stations. In current de-NOx systems, NO is converted into N2 through selective catalytic reduction with NH3 over zeolites12,13 or V-based TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14,15 before being discharged into the atmosphere. Recently, the use of NO as a feed gas for valuable nitrogen compounds has been a focus of research. For example, in several studies, the catalytic conversion of NO to NH3 has been investigated.16–20 Thus, NO has great potential as a feed gas toward the low-cost production of N2O.
14,15 before being discharged into the atmosphere. Recently, the use of NO as a feed gas for valuable nitrogen compounds has been a focus of research. For example, in several studies, the catalytic conversion of NO to NH3 has been investigated.16–20 Thus, NO has great potential as a feed gas toward the low-cost production of N2O.
Here, the photocatalytic evolution of N2O from NO as the feed gas over Pt-loaded TiO2 is reported. Pt NPs were loaded onto TiO2 (P25) using different loading methods. The photocatalysts hardly converted NO into N2O without ultraviolet (UV) irradiation, and the amount of N2O evolved increased under UV irradiation. Additionally, the Pt-loaded TiO2 photocatalyst exhibited higher N2O evolution than did the bare TiO2 photocatalyst, suggesting that Pt species can act as active sites in this case. These findings should help promote the development of N2O production from a low-cost feedstock.
Pt-loaded TiO2 photocatalysts were synthesized using different loading methods, including chemical reduction, impregnation, and photo-deposition methods, and are denoted as Pt/TiO2-cr, Pt/TiO2-imp and Pt/TiO2-pd, respectively. The details of the syntheses and characterizations of the photocatalysts are described in the ESI.†Fig. 1 shows transmission electron microscopy (TEM) images of the bare and Pt-loaded TiO2 photocatalysts. Bare P25 was composed of small TiO2 NPs with diameters of less than 20 nm, as shown in Fig. 1(a). Spherical Pt NPs were located on the surface of the TiO2 NPs, and the Pt particles were less than 10 nm in diameter, as shown in Fig. 1(b–d). X-ray diffraction confirmed that the crystalline phase and the crystalline sizes of TiO2 were not affected by Pt loading (Fig. S1, Table S1†). All of the Pt-loaded TiO2 samples became black in color upon being loaded with Pt, and UV-vis spectra of the Pt-loaded TiO2 indicated light absorption in the visible-light region, due to the surface plasmon resonance of Pt NPs (Fig. S2†). The characteristics of the Pt NPs in the Pt-loaded TiO2 photocatalysts are summarized in Table S1.† TEM images indicated that the particle sizes of Pt followed in the order Pt/TiO2-cr > Pt/TiO2-pd > Pt/TiO2-imp. Dispersion of Pt determined from CO pulse measurements followed in the order Pt/TiO2-imp > Pt/TiO2-pd > Pt/TiO2-cr. These results confirmed that the accessible surface area of the Pt NPs followed in the order Pt/TiO2-imp > Pt/TiO2-pd > Pt/TiO2-cr. X-ray fluorescence revealed that there was no significant difference in Pt content amoung the three photocatalysts (1.7–1.8 wt%). Therefore, the three Pt-loaded TiO2 photocatalysts with almost the same amount of Pt NPs and with different particle sizes were obtained.
 | ||
| Fig. 1 TEM images of (a) bare and (b–d) Pt-loaded TiO2 photocatalysts prepared using different loading methods, namely (b) impregnation, (c) chemical reduction and (d) photo-deposition. | ||
Photocatalytic conversions of NO into N2O over Pt-loaded TiO2 photocatalysts were investigated using a quartz fixed-bed reactor made in house (Fig. 2). Typically, the input gas was passed through the water tank, and UV light was irradiated onto the upper side of the photocatalysts. Prior to photocatalytic tests, samples were pre-treated to completely rid them of carbon species. The details for the photocatalytic tests are described in ESI.† N2O evolutions over different photocatalysts were compared in Fig. 3. None of the photocatalysts without UV irradiation produced any considerable amount of N2O. Upon UV irradiation, the evolved amount of N2O over bare TiO2 increased to some extent, and considerably more so did (>100 ppm) over each of the three Pt-loaded TiO2 photocatalysts. These results were indicative of N2O evolution having been accelerated by light irradiation and of Pt NPs playing a key role in enhancing N2O evolution. Reaction mechanism is hereafter discussed using Pt/TiO2-pd as the photocatalyst.
 | ||
| Fig. 2 Schematic of the fixed-bed reactor made in house. | ||
 | ||
| Fig. 3 N2O evolutions over different photocatalysts. Photocatalytic tests were carried out at an SV of 3500 h−1. | ||
Fig. 4 shows representative gas chromatography (GC) profiles of the outlet gases in the photocatalytic tests under UV irradiation. No considerable peak was observed under He flow (in the absence of NO). When NO/He was introduced as the feedstock gas, there was a peak at 1.36 min, which could be assigned to N2O. This result indicated that N2O evolved from the introduced NO. A decreased N2O evolution was observed when water was not introduced into the reaction system, suggestive of water also playing a part in the photocatalytic reaction.
 | ||
| Fig. 4 GC profiles of the gases produced from different feed stocks. The catalytic tests were carried out using Pt/TiO2-pd under UV irradiation at an SV of 3500 h−1. | ||
Fig. 5 shows GC profiles in the region of 6.5–7.5 min at various gas hourly space velocity (GHSV) values. No peak was observed at 7 min for a GHSV of 3500 h−1 (black line), but peaks at 7 min became obvious at lower GHSV values (red and blue lines). These peaks can be assigned to O2. Furthermore, the peaks originating from O2 evolution were not observed at 7 min without UV irradiation (dashed, blue line). In summary, O2 evolution was found to be triggered by UV irradiation together with N2O evolution from NO feed stock. Fig. 6 shows a schematic illustration explaining the proposed mechanism for photocatalytic evolution of N2O over TiO2 with Pt loading. The reductive/oxidative and overall reactions of this system were estimated, and each potential versus a normal hydrogen electrode (NHE) can be presented as follows.
 | ||
| Fig. 5 GC profiles of the product gas over Pt/TiO2-pd under UV irradiation at different SV conditions. | ||
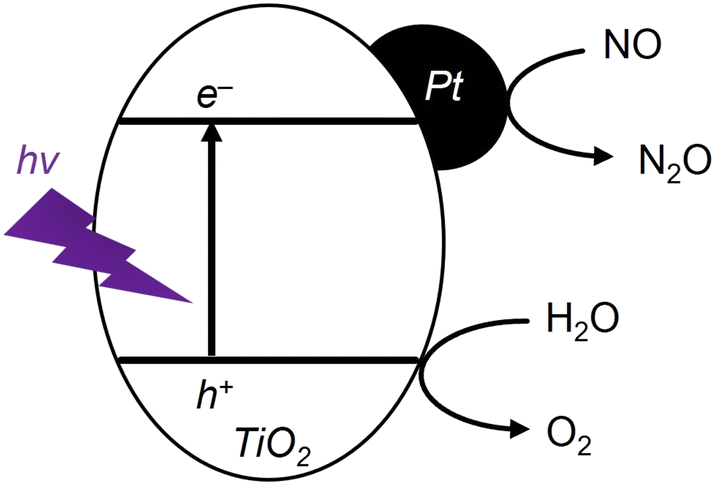 | ||
| Fig. 6 Proposed mechanism for N2O evolution over Pt-loaded TiO2 under UV irradiation. | ||
Reductive reaction:
| 4NO + 4H+ + 4e− → 2N2O + 2H2O (1.54 V vs. NHE) | (1) |
Oxidative reaction:
| 2H2O + 4h+ → O2 + 4H+ (1.23 V vs. NHE) | (2) |
Overall reaction:
| 4NO → 2N2O + O2 | (3) |
| 2NO + O2 → 2NO2 | (4) |
In summary, low-cost N2O manufacturing approach using NO as feed was proposed. The photocatalysts tested hardly converted NO into N2O without UV irradiation, and the amount of N2O that evolved increased under UV irradiation. In particular, the Pt-loaded TiO2 photocatalyst exhibited higher N2O evolution than did the bare TiO2 photocatalyst, suggesting that Pt enhanced the efficiency of photogenerated charges. Furthermore, decreased evolution of N2O was observed without H2O, indicative of the involvement of H2O in this photocatalytic reaction. To the best of our knowledge, this was the first study to demonstrate the photocatalytic evolution of N2O from NO. Although further improvements (e.g., suppression of oxidation of NO and increase of conversion rate) is still required, these findings should be considered to help promote the development of N2O production from a low-cost feedstock.
Data availability
The data that support the findings of this study are openly available.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the New Energy and Industrial Technology Development Organization (NEDO) under the Moonshot Project, the Japan Society for the Promotion of Science (JSPS), a KAKENHI Grant-in-Aid for Transformative Research Areas (A) JP20A206/20H05880, a Grant-in-Aid for Scientific Research (S) JP23H05454, a Grant-in-Aid for Early-Career Scientists JP24K17585 and the Materials Processing Science project (“Materealize”) of MEXT, Grant Number JPMXP0219192801.References
- V. N. Parmon, G. I. Panov, A. Uriarte and A. S. Noskov, Catal. Today, 2005, 100, 115–131 CrossRef CAS.
- K. Severin, Chem. Soc. Rev., 2015, 44, 6375–6386 RSC.
- P. Xiao, Y. Wang, Y. Lu, T. De Baerdemaeker, A.-N. Parvulescu, U. Müller, D. De Vos, X. Meng, F.-S. Xiao, W. Zhang, B. Marler, U. Kolb, H. Gies and T. Yokoi, Appl. Catal., B, 2023, 325, 122395 CrossRef CAS.
- P. Xiao, Y. Wang, K. Nakamura, Y. Lu, T. De Baerdemaeker, A.-N. Parvulescu, U. Müller, D. De Vos, X. Meng, F.-S. Xiao, W. Zhang, B. Marler, U. Kolb, R. Osuga, M. Nishibori, H. Gies and T. Yokoi, ACS Catal., 2023, 13, 11057–11068 CrossRef CAS.
- A. C. Huizink, Curr. Opin. Psychol., 2022, 45, 101312 CrossRef PubMed.
- M. A. Gillman, Curr. Drug Res. Rev., 2019, 11, 12–20 CrossRef CAS.
- Z. Tang, I. Surin, A. Rasmussen, F. Krumeich, E. V. Kondratenko, V. A. Kondratenko and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2022, 61, e202200772 CrossRef CAS.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- C. D. Dorich, R. T. Conant, F. Albanito, K. Butterbach-Bahl, P. Grace, C. Scheer, V. O. Snow, I. Vogeler and T. J. van der Weerden, Curr. Opin. Environ. Sustain., 2020, 47, 13–20 CrossRef.
- K. Yamashita, Z. Liu, K. Iyoki, C. T. Chen, S. Miyagi, Y. Yanaba, Y. Yamauchi, T. Okubo and T. Wakihara, Chem. Commun., 2021, 57, 1312–1315 RSC.
- S. Yamaguchi, P. Hu, H. Ya, P. Xiao, A. Nakata, T. Miyazaki, Y. Morikawa, J. N. Kondo, K. Tange, K. Tonokura, M. Takemoto, Y. Yonezawa, T. Yokoi, K. Iyoki, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2025, 388, 113550 CrossRef CAS.
- M. Jablonska, RSC Adv., 2022, 12, 25240–25261 RSC.
- S. Mohan, P. Dinesha and S. Kumar, Chem. Eng. J., 2020, 384, 123253–123262 CrossRef CAS.
- H. Li, X. Yi, J. Miao, Y. Chen, J. Chen and J. Wang, Catalysts, 2021, 11, 906–920 CrossRef CAS.
- Y. Inomata, S. Hata, M. Mino, E. Kiyonaga, K. Morita, K. Hikino, K. Yoshida, H. Kubota, T. Toyao, K.-i. Shimizu, M. Haruta and T. Murayama, ACS Catal., 2019, 9, 9327–9331 CrossRef CAS.
- B. Suzumura, K. Tanaka, K. Kitazume, S. Hioki, A. Kubo and M. Iwamoto, Catal. Sci. Technol., 2023, 13, 4131 RSC.
- K. Tanaka, B. Suzumura, K. Kitazume, H. Suzuki, H. Teduka and M. Iwamoto, Catal. Lett., 2024, 154, 2090 CrossRef CAS.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- A. G. Ramu, R. Renukadevi, P. Silambarasan, I.-S. Moon, M. Govindarasu and D. Choi, J. Environ. Chem. Eng., 2023, 11, 110751 CrossRef CAS.
- D. Wang, Z. W. Chen, K. Gu, C. Chen, Y. Liu, X. Wei, C. V. Singh and S. Wang, J. Am. Chem. Soc., 2023, 145, 6899 CrossRef CAS PubMed.
- S. B. Rawal, S. Bera and W. I. Lee, Catal. Lett., 2012, 142, 1482–1488 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- T. Suguro, F. Kishimoto, N. Kariya, T. Fukui, M. Nakabayashi, N. Shibata, T. Takata, K. Domen and K. Takanabe, Nat. Commun., 2022, 13, 5698 CrossRef CAS.
- T. Suguro, F. Kishimoto and K. Takanabe, Energy Fuels, 2022, 36, 8978–8994 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5re00119f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |