DOI:
10.1039/D5RA07824E
(Paper)
RSC Adv., 2025,
15, 46425-46433
Water-soluble binuclear aquo Cu(II) complex with an amide ligand as an electrocatalyst for the OER, HER and CO2RR
Received
13th October 2025
, Accepted 4th November 2025
First published on 25th November 2025
Abstract
A dinuclear copper complex (1) with the formula Cu2L2 (where L = [3-((pyridin-2-ylmethyl)carbamoyl)isonicotinic acid]) was synthesized and characterized by single-crystal XRD and mass spectrometry. The complex showed a high order of electrocatalytic hydrogen evolution reaction (HER) and carbon dioxide reduction reaction (CO2RR) activity in acidic media, along with a moderate oxygen evolution reaction (OER) activity in the pH range of 7 to 13.5. The efficiency of 1 for the HER was calculated as follows: TOF = 1679 s−1, TON = 586 and F.E. = 83% in 56 equivalents of perchloric acid. For the CO2RR, a TOF value of 4 h−1, a TON of 18 and an F.E. of 92% were observed, with a percentage selectivity of ∼99.99% for CO2 to (COOH)2. The best water oxidation activity was accomplished at pH 13.5 with the following electroanalytical efficacy parameters: TOF = 9 s−1 (peak current method), TOFFOWA = 31 s−1, TON = 66 and F.E. = 84%. The high stability of the molecular catalyst was analyzed through CV, FESEM, EDX and DLS.
Introduction
Sustainable generation of carbon-neutral fuels is one of the major milestones mankind has been trying to achieve for decades. The conversion of solar energy to fuels with high calorific values, such as molecular hydrogen and oxygen, can be the solution to major issues like the energy crisis and global warming. The increased CO2 concentration in the atmosphere, the major cause of global warming, can be tackled by following the natural biochemical outline of photosynthesis i.e. converting CO2 into complex carbohydrates. The abundance of salty water on our planet that is unfit for drinking without treatment has inspired humans to use it for energy generation. Splitting of water through electrolysis generates hydrogen and oxygen through proton reduction and water oxidation, respectively. In this process, the half-reaction of water oxidation is considered the bottleneck since it involves 4e− transfer with several bond rearrangements through proton-coupled electron transfer (PCET). Thus, it is important to develop and employ a catalyst that can mimic PSII with high stability, efficiency and low overpotential. Several previous reports have revealed that Pt,1,2 Ir,3 Ru4 and Rh5 can achieve higher efficacy in artificial photosynthesis, but their scarce availability, difficulties in mining and high cost have compelled us to find alternatives. Thus, imitating photosynthesis by employing the natural blueprint of photosynthesis using earth-abundant metal-based molecular catalysts is highly desirable. Most of the molecular catalysts reported so far work as homogeneous catalysts upon dissolution in organic solvents.6–8 This adds both economic and environmental expenses to the process. However, developing water-soluble, first-row transition metal-based molecular catalysts that can oxidize water to molecular oxygen, reduce protons to hydrogen and reduce carbon dioxide to high end products like CH4, C2H4, CH3CH2OH, (COOH)2, CH3COOH and C3 alcohols with greater stability and efficiency is the real quest.9–12 Therefore, utilizing the abundant and universal solvent, i.e. water, for electrochemical catalysis is a favorable solution to such an issue. The unconstrained conversion of CO2 to a variety of products and low selectivity are major challenges. Therefore, extensive research and investigation of the mechanistic cycle are required for electrochemical CO2RR as well as HER and OER. The high availability of copper and its great stability make it one of the best candidates for generating hydrogen, oxygen and carbon reduced products.13–18
Single-crystal X-ray diffraction analysis
The single crystals of ligand L1 (CCDC 2388874) and metal complex 1 (CCDC 2388873) were obtained in methanol and DMSO, respectively. The crystallographic data and refinement parameters are presented in Tables S1.1–1.3 and S2.1–2.3 for L1 and Cu–complex 1, which demonstrate the selective bond angles and bond lengths, respectively. A triclinic crystal system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group was observed for L1, whereas the metal complex showed a monoclinic crystal system with the P21/n space group. The perspective ORTEP diagrams of both the ligand and metal complex are presented in Fig. 1. A Rigaku Oxford Diffraction X-Calibur CCD system, controlled using CrysalisPro, was used for crystallographic data collection at 298 K. The measurements were made on an Enraf Nonius Kappa CCD diffractometer with Mo-Kα (0.71073 Å). The crystal structure was solved with the Shelxs implemented in the Olex2 software package using Charge Flipping and refined with the SHELXL using Least Squares minimization.19
space group was observed for L1, whereas the metal complex showed a monoclinic crystal system with the P21/n space group. The perspective ORTEP diagrams of both the ligand and metal complex are presented in Fig. 1. A Rigaku Oxford Diffraction X-Calibur CCD system, controlled using CrysalisPro, was used for crystallographic data collection at 298 K. The measurements were made on an Enraf Nonius Kappa CCD diffractometer with Mo-Kα (0.71073 Å). The crystal structure was solved with the Shelxs implemented in the Olex2 software package using Charge Flipping and refined with the SHELXL using Least Squares minimization.19
 |
| | Fig. 1 ORTEP diagram of the ligand L1 and dinuclear Cu-complex: 1. | |
Result and discussion
Synthesis and characterization
This work describes a highly efficient water-soluble copper amide complex, namely the dinuclear aquo copper complex (1), which can act as a redox catalyst for water oxidation, and proton and carbon dioxide reduction. The electrocatalytic parameters for proton reduction, carbon dioxide reduction and water oxidation activity are summarized in Table S3 and Fig. S23. The ligand was obtained by the amidation of pyridine 3,4-dicarboxylic acid using 2-amino methyl pyridine. When the ligand was reacted with Cu(ClO4)2, a binuclear copper complex was formed (SI Scheme 1). Mass spectral analysis of the ligand L1 and 1 (Cu2L2·2H2O) revealed their base peaks at m/z = 258.27 and m/z = 673.39, respectively, which match the expected values (Fig. S1 and S2). The NMR spectrum (Fig. S3) of the ligand and the single-crystal XRD pattern of the ligand with the metal complex are displayed in Fig. 1.
HER study
The electrocatalytic reduction of protons by complex 1 in water was examined by cyclic voltammetry and bulk electrolysis. In the cathodic scan of CV of 1 in an aqueous solution, two quasi-reversible peaks at −0.1 V and −0.75 V vs. Ag/AgCl (aqueous) were observed, in agreement with previous reports.20,21,33 These can be assigned to the CuIICuII/CuIICuI and CuIICuII/CuICuI redox processes, respectively (Fig. 2(A)). The addition of perchloric acid as the source of proton to the catalyst solution produced interesting results, with a continuous increase in the current intensity with incremental addition up to 56 equivalents (Fig. 2(B) and 3). Thus, an ic/ip value of 91.80 was obtained for 1 at the maximum acid concentration, where ic is the catalytic peak current with 56 equivalents of perchloric acid, and ip is the peak current without acid. This resulted in a high TOF of 1679 s−1, as calculated using eqn (A) (where n = 2 i.e. the number of electrons involved in the catalytic cycle, R = 8.314 J K−1 mol−1, T = 298 K, ν = 100 mV s−1). This implies that the 1 is a highly active catalyst for HER.
 |
| | Fig. 2 (A) CV curve and DPV of the cathodic segment of 1 in water; c = 0.5 mM, scan rate = 100 mV s−1, internal reference: Ag/AgCl (aqueous), and working electrode: glassy carbon. (B) CV curves of 0.5 mM complex 1 in water with the addition of 0–56 equivalents of perchloric acid. | |
 |
| | Fig. 3 Comparison of the CV curves of complex 1 in water without acid and with 56 equivalents of acid. | |
In the CV results, it was observed that the peak current of the CuIICuII/CuICuI couple continuously improved as the concentration of the acid in the homogeneous catalytic solution increased. To quantify the H2 generated during catalysis, CPE (controlled potential electrolysis) was carried out for 2 hours at −1.6 V using N2 to neutralize the atmospheric gases in the working electrode cell. 56 equivalents of perchloric acid are used as the source of protons during CPE. After the completion of CPE, the solution was subjected to mass spectroscopic analysis, and the gas from the headspace was evaluated by gas chromatography. GC detected H2 with high concentration during the analysis (Fig. S7). The faradaic efficiency was calculated22,23 as ∼83% using the total charge accumulated during 2 hours of CPE and the change in the volume of the catalytic solution in the cell holding the working electrode. The TON of 1 after 2 hours of CPE was calculated using the volume of hydrogen generated in the meantime, the number of moles of catalyst used and the relative catalytic loss calculated from the CV analysis done after CPE (Fig. S12(a)). Thus, a TON of 1172 was observed after the calculation.24 The mass spectrum of complex 1 after CPE clearly exhibited the addition of a proton to the complex skeleton (metal centre) (Fig. S11(b)). To investigate the effect of different acids on the catalytic activity of 1, we also used acetic acid, orthophosphoric acid and hydrochloric acid along with the same electrochemical parameters (Fig. S14) used for perchloric acid. The trend of activity of 1 was as follows: perchloric acid > hydrochloric acid > orthophosphoric acid > acetic acid, with TOF values, for the reduction of proton, of 1679, 10.15, 9.02 and 0.768 s−1, respectively.
Thus, with the electrochemical analysis and the mass spectroscopic data, we propose that the reduction of proton occurs through two consecutive one-electron reduction reactions, leading to CuIICuII/CuIICuI and CuIICuII/CuICuI. Later, a proton may be transferred from the medium to one of the metal centers, forming the CuI–H species. Then, a –H from the water molecules dangling from the copper metal center makes a H⋯H bond, which generates a dihydrogen in the last step25 (Fig. S11(b) and S25(a)).
| |
 | (A) |
OER study
The CV exhibited a pH-dependent irreversible peak at 0.96 V vs. Ag/AgCl (aqueous) (1.07 V vs. Ag/AgCl (aqueous) in DPV), attributed to the CuIICuII/CuIICuIII redox couple (Fig. 4). The same peak was found to be the catalytic peak for water oxidation when subjected to electrochemical catalysis at different pH values ranging from 7 to 13.5. Another irreversible peak was observed for the CuIICuII/CuIICuI couple of complex 1 at 0.24 V vs. Ag/AgCl (aqueous) (0.15 V vs. Ag/AgCl in DPV (aqueous)) at a scan rate of 100 mV s−1. The complex reported herein showed no change in the irreversible nature of the CuIICuII/CuIICuI couple upon changing the scan rate from 2 mV s−1 to 80 mV s−1 (Fig. 6(A)).26 1 exhibited the highest efficiency for OER at pH 13.5. The overpotential values of 1 for OER catalysis at different pH values are provided in Table 1. The electrocatalytic TOF or the Kcat of complex 1 was calculated by the peak current method using eqn (S1) and (S2). The diffusion coefficient was also calculated using the same equation. Thus, by employing eqn (B) along with the slope of the graph depicting icat/ip vs. square root of the scan rate (Fig. 6(B)), the TOF of 1 was calculated to be 9 s−1.| |
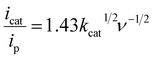 | (B) |
 |
| | Fig. 4 CV and DPV of the anodic segment of 1 in water; c = 0.25 mM, scan rate = 100 mV s−1, and internal reference: Fc/Fc+. | |
Table 1 Overpotentials of complex 1 at different pH values during catalysis (calculated using E = 1.23–0.059 pH)
| pH (buffer solution) |
Experimental potential value for CuII/CuIII |
Overpotential |
| 7 |
1.29 V |
470 mV |
| 8 |
1.30 V |
540 mV |
| 9 |
1.31 V |
610 mV |
| 10 |
1.30 V |
660 mV |
| 11 |
1.31 V |
730 mV |
| 12 |
1.32 V |
790 mV |
| 13 |
1.33 V |
868 mV |
| 13.5 |
1.33 V |
900 mV |
Electrochemical OER catalysis by 1 was also examined by changing the concentration of complex 1 in the aqueous buffer solution at pH 13.5 (Fig. S15). It was observed that the catalysis of the complex in these experimental conditions was completely independent of the concentration of the complex in the solution, delineating a WNA pathway. The pH-based electrochemical data (Fig. 5(A)) of complex 1 are available in Table 1, and using the same information, a Pourbaix diagram is constructed (Fig. 5(B)). CPE was performed at +1.45 V vs. Ag/AgCl (aqueous) with 0.5 mM complex 1. The results of CPE revealed a faradaic efficiency of ∼84% and a TON of 66 under the given experimental conditions (Fig. S12(b)). Gas chromatography results obtained after 2 hours of CPE at +1.45 V vs. Ag/AgCl are added in Fig. S8. The mass spectrum of the solution after 2 hours of CPE displayed a peak at m/z = 688.15, which might be attributed to the [Cu2L2–O–O–H]+ species, an expected intermediate in the WNA (water nucleophilic attack) pathway, along with a peak of the precursor of the WNA step at m/z = 672.16 (Fig. S11(a)). The calculation of TOF using FOWA (foot of the wave analysis) was done based on the CV results obtained at pH 13.5 (conc. = 0.5 mM, scan rate = 100 mV s−1). The ic/ip vs. 1/{1 + e[(E0,ap − E)(F/RT)]} plot was drawn to calculate the slope starting from the foot of the wave zone and, hence, to calculate the KWNA of 1 in the WNA pathway. The value of TOF obtained from the FOWA was 31 s−1 (KWNA) (Fig. 7). To prove that complex 1 is a suitable catalyst, 100 consecutive CV cycles were performed at pH 13.5 using the standard experimental conditions. During CV, it was observed that the current intensity decreased after the completion of each cycle. After the completion of 100 cycles, the working electrode was rinsed with distilled water, and a CV test was done in a fresh pH 13.5 buffer solution without the catalyst. This CV curve exhibited no sign of catalytic activity, showing that there was no accumulation of any metal oxide particles or precatalyst. The decrease in current during consecutive cycling may be due to the accumulation of oxygen gas on the working electrode. FESEM and EDX analyses of the working electrode were also done before and after the 100 CV cycles to investigate if any metal oxide traces were accumulated on the electrode. In agreement with our postulation, no trace of CuOx nanoparticles was observed in the EDX analysis; rather, only elements of the buffer solution were observed at higher concentrations. Catalysis at such high pH raises the concern of molecular disintegration into nanoparticles in solution. Thus, DLS analysis of the complex solution was also carried out after CPE for 2 hours at +1.45 V. Since a majority of the particles observed in the DLS were found to be around ∼1 micron, this negates the possibility of the generation of CuOx nanoparticles in the catalytic solution (Fig. S9). To analyze the homogeneity of complex 1, CV was repeated after CPE. An irreversible peak was observed for the CuIICuII/CuIICuI couple from −0.24 V vs. Ag/AgCl before CPE at pH 13.5; later, a catalytic loss of 17.56% was observed in the CV curve recorded. The catalytic curve of the CuIICuII/CuIICuIII couple was observed with a loss in current density by a few percent, but the other curve of the CuIICuII/CuIICuI couple remained exactly the same. This certifies that the catalyst remained in a homogeneous state in the buffer solution throughout catalysis, even at such a highly oxidative condition27 (Fig. S13). As the mass spectral data and electrochemical analysis (Fig. S15) affirmatively indicate the WNA pathway and identify the intermediates formed during the oxidation of water to molecular oxygen28–31 by 1, the parent catalyst molecule may undergo one-electron oxidation to generate the CuII/CuIII species, followed by the formation of a hydroxo intermediate (Fig. S11(a)). Later, 1 may undergo a proton coupled electron transfer (PCET) process, which possibly forms the peroxo radical. Finally, the attack on copper by one water molecules releases molecular oxygen, along with the regeneration of the catalyst. We have presented a plausible mechanism in Fig. S25(b).
 |
| | Fig. 5 (A) CV curves of complex 1 in phosphate buffer from pH 7–13.5 at a scan rate of 100 mV s−1 and 0.5 mM concentration using glassy carbon as the working electrode. (B) Pourbaix diagram of complex 1. | |
 |
| | Fig. 6 (A) CV of 0.5 mM complex 1 in phosphate buffer at pH 13.5 with increasing scan rates from 2 mV s−1 to 100 mV s−1. (B) The plot of icat/ip as a function of the inverse of the square root of the scan rates used in the experiment of complex 1. | |
 |
| | Fig. 7 The cyan line shows the CV curve of complex 1 (0.5 mM) at pH 13.5. The red line indicates the data points used for the FOWA. The i/ip vs. 1/{1 + e[(E0,ap − E)(F/RT)]} plot assuming a WNA. The fitting points for the extraction of rate constants at the foot of the wave are taken from the WNA graph. The red line indicates the points taken into consideration for the calculation of TOF. | |
CO2RR study
Homogeneous CO2RR (carbon dioxide reduction reaction)32 by complex 1 was studied using a 0.5 mM aqueous solution of the catalyst by means of cyclic voltammetry (within the potential range from 1.50 V to −2.22 V vs. Ag/AgCl) and controlled potential electrolysis of an aqueous CO2 purged solution of 1. An initial run of CV with the catalyst solution was followed by CO2 purging of the bulk solution for 60 minutes. The CV scan in the aforementioned potential range did not enhance the catalytic peak current to a significant extent. On adding the acid, the peak current increased at two places: −1.22 V and −1.85 V, corresponding to proton reduction and CO2 reduction, respectively. The increment was noticed up to 8 equivalents of acid. A 14.3-fold increment in catalytic current was recorded for the CO2RR activity (Fig. 8). The UV-visible spectra displayed a mild decrease in absorbance, with no change in the spectra after the addition of acid to the catalytic solution purged with carbon dioxide. Gas chromatography and HPLC were employed to analyze the reduction products in the gas phase and solution, respectively. CPE at −2.15 V vs. Ag/AgCl with 0.5 mM of 1 (purged with CO2 for 60 minutes) for 270 minutes resulted in 24.1 coulomb charge. During CPE,33 the reaction mixture was screened by mass spectroscopy for the identification of intermediates produced during catalysis34,35 (Fig. S11(c)). For the analysis of the gaseous product, a constant volume of the gas mixture from the headspace of the coulometry cell was injected into the GC. GC traces showed the presence of three types of reduction products, namely H2, CO and CH4. However, the amount of the gases produced was very less. The TOF, TON and FE for CO and CH4 were obtained as 0.045 h−1, 0.20, and 0.08% and 0.05 h−1, 0.22 and 0.36%, respectively. In order to identify the product in the solution, using a similar methodology, a fixed amount (20 µL) of the reaction mixture was injected into the HPLC at regular time intervals. The HPLC plot showed the presence of only oxalic acid. Therefore, in the CO2RR, oxalic acid, methane and CO were obtained. For all the products, the number of moles produced was quantified. From this data, we calculated the % selectivity of 99.9% for oxalic acid.
 |
| | Fig. 8 CV curves of 0.5 mM complex 1 in water after 60 minutes of purging with CO2 and consecutive addition of perchloric acid. Scan rate = 100 mV s−1, internal reference: Ag/AgCl (aqueous), working electrode: glassy carbon. | |
In the line with the literature on CO2RR by polynuclear copper complexes,35,36 we identified 1-*CHO (a peak at m/z = 702.23), 1-H (m/z = 647.17), 1-C2O4H2 (m/z = 763.47) in the LCMS analysis (Fig. S11(c)). However, on HRMS, oxalate ions (Fig. S24) were observed, delineating the formation of oxalic acid during the electrocatalytic reduction of carbon dioxide. The formation of hydrogen has already been discussed in the HER study. Here, the most important feature is the very high selectivity to oxalic acid, with a faradaic efficiency of 92%, a TOF of 4 h−1 and a TON of 18 (HPLC plots in Fig. S22 and S23). The almost exclusive formation of oxalic acid may be due to the binuclear copper center, which provides the required stereochemical disposition to catch two 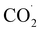 radicals (one with each metal) in the void space between the two parallel pyridine rings, stabilizing them through π–π interactions between the pyridine ring and the CO2 π electrons. This arrangement readily leads to C–C bond formation between the radicals to form an oxalato bridge.37 In an electrochemical process, two consecutive one electron reduction reactions occur to form CuI–CuI. Each CuI is coordinated by CO2. Coordinated CO2 is then converted to
radicals (one with each metal) in the void space between the two parallel pyridine rings, stabilizing them through π–π interactions between the pyridine ring and the CO2 π electrons. This arrangement readily leads to C–C bond formation between the radicals to form an oxalato bridge.37 In an electrochemical process, two consecutive one electron reduction reactions occur to form CuI–CuI. Each CuI is coordinated by CO2. Coordinated CO2 is then converted to 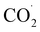 radicals. The close approach of the
radicals. The close approach of the 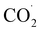 radicals then forms the C–C bond to produce oxalate. Then, two protons are added to release oxalic acid and regenerate 1. Thus, the plausible mechanism delineating CO2RR is given in SI (Fig. S25(c)).
radicals then forms the C–C bond to produce oxalate. Then, two protons are added to release oxalic acid and regenerate 1. Thus, the plausible mechanism delineating CO2RR is given in SI (Fig. S25(c)).
Theoretical calculations using DFT
DFT38 calculations using the B3LYP39,40 functional and DGTZVP basis set41 were employed to analyze and optimize complex 1 in the IEFPCM42 continuum solvation model with water as the solvent. The optimized structure (Fig. 9) yielded the minimum energy geometry of catalyst 1, along with the electronic energy and thermochemical data. Atomic ESP and Mulliken charge43,44 distribution were calculated (Fig. S17). The ESP7,45 plot (Fig. S26) exhibited that the electron density is highly localized around the carbonyl, acidic and aquo oxygen atoms. Since equal and identical kinds of oxo bonds are borne by both the metal centers, high electron density is observed around both the copper centers. The theoretical HOMO–LUMO gap41 was calculated using the data observed, which provided a particularly low energy difference of ΔE = 0.437 eV.35 This value can be attributed to high electron transport and reactivity in the catalytic solution. This can be the reason behind the high activity of the complex.
 |
| | Fig. 9 (A) Optimized structure of complex 1. (B) Theoretically calculated HOMO–LUMO gap of complex 1. | |
Materials
Pyridine-3,4-dicarboxylic acid, thionyl chloride, 2-(aminomethyl)pyridine, triethylamine, copper carbonate (for the preparation of copper perchlorate), perchloric acid, disodium hydrogen phosphate, trisodium phosphate, sodium hydroxide and all the AR-grade solvents were procured from Spectrochem and used without further purification. The HPLC-grade solvents and oxalic acid, which was used as the reference in HPLC, were procured from Sigma-Aldrich. 99.99% pure CO2 procured from Sam Air Products & Equipment, India, was used in the electrochemical reduction assays.
Characterization
All electrochemical analyses were performed using a CHI-6003E potentiostat with a 3-electrode setup (glassy carbon (GC), platinum wire and Ag/AgCl) at a 100 mV s−1 scan rate unless mentioned otherwise. UV-visible spectra were recorded on a PerkinElmer (Lambda 750) spectrophotometer. For LC-MS studies, a Thermo Scientific (USA) UltiMate 3000 was used. To analyze the gas, a trace 1110 gas chromatograph from Thermo Fisher Scientific was used. The FESEM analysis was performed on a Sigma 300 from Carl Zeiss Microscope Ltd, Germany, equipped with JSM 6390 LV from Jeol, Japan, for EDS. HPLC was performed using a Waters 2487 Dual λ Absorbance Detector. For DFT analysis, Gaussian 16 (ref. 46) was used, and GaussView 6.0.16 (ref. 47) was employed for visualizing the optimized structures.
Conclusions
This electrochemical study reveals the trifunctional nature of the water-soluble electrocatalyst 1, with high reducing ability and a moderately efficient oxidizing ability in generating H2, O2 and (COOH)2 in the aqueous medium. The identification of [Cu2L2–O–O–H]+ on the mass spectra, along with the independence of KWNA on the concentration of the catalyst, proves that 1 follows a WNA pathway for OER. The water molecule coordinated to the Cu metal center plays a crucial role in the generation of H2, because the hydrogen atom in the CuI–H bonds and the hydrogen atom of water molecules form H⋯H bonds. This probable intramolecular H⋯H bond formation can be the reason for such a high HER efficiency of 1. For CO2RR, the dinuclear nature of the complex facilitates the generation of a C–C bond between two  radicals, hence generating an oxalato bridge efficiently. Analysis of products in the gas phase using gas chromatography quantified H2, O2, CO and CH4, whereas HPLC was employed to detect and quantify the amount of oxalic acid produced after CPE in the catalytic solution with CO2 and acid. Theoretical calculations engendered a very low HOMO–LUMO gap of 0.437 eV, which facilitates such a high activity of the homogeneous catalyst. The crucial intermediates formed during the reduction of protons to dihydrogen and carbon dioxide to oxalic acid as the major products, along with oxidation of water to molecular oxygen, were identified from the LC-MS spectra recorded after CPE at respective potentials. The HRMS spectra identified the formation of oxalate ions after CPE at −2.15 V for carbon dioxide reduction. Thorough exploration of the mechanisms via computational analysis using DFT is underway.
radicals, hence generating an oxalato bridge efficiently. Analysis of products in the gas phase using gas chromatography quantified H2, O2, CO and CH4, whereas HPLC was employed to detect and quantify the amount of oxalic acid produced after CPE in the catalytic solution with CO2 and acid. Theoretical calculations engendered a very low HOMO–LUMO gap of 0.437 eV, which facilitates such a high activity of the homogeneous catalyst. The crucial intermediates formed during the reduction of protons to dihydrogen and carbon dioxide to oxalic acid as the major products, along with oxidation of water to molecular oxygen, were identified from the LC-MS spectra recorded after CPE at respective potentials. The HRMS spectra identified the formation of oxalate ions after CPE at −2.15 V for carbon dioxide reduction. Thorough exploration of the mechanisms via computational analysis using DFT is underway.
Author contributions
Md. Adnan Khan – methodology, formal analysis, data curation. Sonal Shruti – methodology. Swaraj Sengupta – formal analysis. Sahanwaj Khan – methodology. Prabhakar Bhardwaj – formal analysis. Pankaj Kumar – data curation. Subhendu Naskar – conceptualization, funding acquisition, supervision, writing, review and editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
CCDC 2388873 (metal complex Cu2L2·2H2O) and CCDC 2388874 (ligand) contain the supplementary crystallographic data for this paper.48a,b
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra07824e.
Acknowledgements
S. N. thanks SERB, New Delhi, Government of India, for funding this work through the SERB Project (“CRG/2023/001349” dated 15 March, 2024). S. N. acknowledges the NMR spectroscopic facility and computational facility in the Department of Chemistry, BIT Mesra, supported by the DST FIST Program (SR/FST/CSI-242/2012). The single-crystal X-ray diffraction facility and other analytical services availed from CIF, BIT Mesra, are greatly acknowledged. The authors also acknowledge the Department of Chemical Engineering, BIT Mesra, for gas chromatography and high-performance liquid chromatography measurements.
References
- F. Li and J. B. Baek, Nat. Catal., 2019, 2, 477–478 CrossRef CAS.
- L. M. Salonen, D. Y. Petrovykh and Y. V. Kolen'ko, Mater. Today Sustain., 2021, 11, 100060 Search PubMed.
- Z. N. Zahran, Y. Tsubonouchi, E. A. Mohamed and M. Yagi, ChemSusChem, 2021, 9, 1775–1793 Search PubMed.
- M. Sutradhar, A. J. L. Pombeiro and J. A. L. da Silva, Coord. Chem. Rev., 2021, 439, 213911 CrossRef CAS.
- A. M. R. Ramírez, S. Heidari, A. Vergara, M. Villicaña Aguilera, P. Preuss, M. B. Camarada and A. Fischer, ACS Mater. Au, 2023, 3, 177–200 CrossRef PubMed.
- M. A. Khan, S. Khan, S. Sengupta, B. N. Mongal and S. Naskar, Coord. Chem. Rev., 2024, 504, 215679 CrossRef.
- M. Raj, K. Makhal, A. Mishra, B. S. Mallik and S. K. Padhi, Inorg. Chem., 2023, 62, 10993–11008 CrossRef CAS PubMed.
- M. D. Kärkä, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed.
- J.-W. Wang, X.-Q. Zhang, H.-H. Huang and T.-B. Lu, ChemCatChem, 2016, 8, 3287–3293 CrossRef CAS.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- X. Zhong, H.-J. Peng, C. Xia and X. Liu, J. Mater. Chem. A, 2024, 12, 19663–19684 RSC.
- S. L. Jain, P. Bhattacharyya, H. L. Milton, M. Z. Slawin, J. A. Crayston and J. Derek Woollins, Dalton Trans., 2004, 6, 862–871 RSC.
- R. C. DiDomenico, K. Levine, L. Reimanis, H. D. Abruña and T. Hanrath, ACS Catal., 2023, 13, 4938–4948 CrossRef.
- T.-T. Li and Y.-Q. Zheng, Dalton Trans., 2016, 45, 12685–12690 RSC.
- G. Ruan, P. Ghosh, N. Fridman and G. Maayan, J. Am. Chem. Soc., 2021, 143(28), 10614–10623 CrossRef CAS PubMed.
- E. Ahmad, K. Majee, J. Patel, B. Das and S. K. Padhi, Eur. J. Inorg. Chem., 2017, 28, 3409–3418 CrossRef.
- P. Garrido-Barros, D. Moonshiram, M. Gil-Sepulcre, P. Pelosin, C. G. Suriñach, J. B. Buchholz and A. Llobet, J. Am. Chem. Soc., 2020, 142, 17434–17446 CrossRef CAS PubMed.
- O. V. Dplomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 59–75 CrossRef PubMed.
- T. Fang, L.-Z. Fu, L.-L. Zhou and S.-Z. Zhan, Electrochim. Acta, 2015, 161, 388–394 CrossRef CAS.
- H. A. Younus, H. Emam, N. Ahmad, M. Negm, M. Alomar, F. M. Elantabli, M. M. El-Rabiei, R. A. Hajri, S. Zhang and M. A. Abri, ChemElectroChem, 2024, 11, e202300710, DOI:10.1002/celc.202300710.
- S. Nestke, M. Kügler, J. Scholz, M. Wilken, C. Jooss and I. Siewert, Eur. J. Inorg. Chem., 2017, 28, 3376–3382 CrossRef.
- A. Z. Haddad, S. P. Cronin, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2017, 56, 11254–11265 CrossRef CAS PubMed.
- S. Karim, A. Chakraborty, D. Samanta, E. Zangrando, T. Ghosh and D. Das, Catal. Sci. Technol., 2020, 10, 2830–2837 RSC.
- M. Raj, K. Makhal, D. Raj, A. Mishra, B. S. Mallik and S. K. Padhi, Dalton Trans., 2023, 52, 17797–17809 RSC.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136(1), 273–281 CrossRef CAS PubMed.
- G. Ruan, L. Engelberg, P. Ghosh and G. A. Maayan, Chem. Commun., 2021, 57, 939–942 RSC.
- S. Khan, S. Sengupta, M. A. Khan, M. P. Sk, N. Ch. Jana and S. Naskar, Inorg. Chem., 2024, 63, 1888–1897 CrossRef CAS PubMed.
- S. Khan, S. Sengupta, M. A. Khan, M. P. Sk and S. Naskar, Dalton Trans., 2023, 52, 7590 RSC.
- M. A. Khan, S. Khan, S. Sengupta and S. Naskar, New J. Chem., 2025, 49, 6963–6974 RSC.
- X. Zhang, Y.-Y. Li, J. Jiang, R. Zhang, R.-Z. Liao and M. Wang, Inorg. Chem., 2020, 59, 5424–5432 CrossRef CAS PubMed.
- V. Boor, J. E. B. M. Frijns, E. PerezGallent, E. Giling, A. T. Laitinen, E. L. V. Goetheer, L. J. P. vandenBroeke, R. Kortlever, W. de Jong, O. A. Moultos, T. J. H. Vlugt and M. Ramdin, Ind. Eng. Chem. Res., 2022, 61, 14837–14846 CrossRef CAS PubMed.
- L.-L. Zhou, T. Fang, J.-P. Cao, Z.-H. Zhu, X.-T. Su and S.-Z. Zhan, J. Power Sources, 2015, 273, 298e304 Search PubMed.
- S. K. S. Akhter, D. Srivastava, A. Mishra, N. Patra, P. Kumar and S. K. Padhi, Chem.–Eur. J., 2024, 30, e202403321 CrossRef PubMed.
- J.-W. Liu, D. Peng, S.-J. Liu, H.-R. Wen, Z. H. Zhu, J. Zhao and J. L. Chen, ACS Sustainable Chem. Eng., 2025, 13, 2564–2573 CrossRef CAS.
- Z. Zhang, Y. Yang, J. Wang, Xu. Jing and C. Duan, Inorg. Chem. Front., 2025, 12, 5389–5396 RSC.
- S. Sengupta, S. Khan, B. N. Mongal, W. Lewis, M. Fleck, S. K. Chattopadhyay and S. Naskar, Polyhedron, 2020, 191, 114798 CrossRef CAS.
- K. Ye, Y. Li and R. Liao, RSC Adv., 2016, 6, 90035–90045 RSC.
- M. M. Ibrahim, G. A. M. Mersal, A. M. Fallatah, K. Althubeiti, H. S. El-Sheshtawy, M. F. A. Taleb, M. R. Das, R. Boukherroub, M. S. Attia and M. A. Amin, RSC Adv., 2022, 12, 8030 RSC.
- J. M. Dressel, E. N. Cook, S. L. Hooe, J. J. Moreno, D. A. Dickie and C. W. Machan, Inorg. Chem. Front., 2023, 10, 972–978 RSC.
- M. Khajehzadeh, M. Moghadam, S. Rahmaniasl and M. Rajabi, J. Mol. Struct., 2021, 1230, 129660 CrossRef CAS.
- A. Ramdass, V. Sathish, M. Velayudham, P. Thanasekaran, S. Umapathy and S. Rajagopal, RSC Adv., 2015, 5, 38479 RSC.
- F. Ma and X. Chen, ACS Appl. Nano Mater., 2023, 6, 16546–16554 CrossRef CAS.
- Z. Jiang, W. Sun, H. Shang, W. Chen, T. Sun, H. Li, J. Dong, J. Zhou, Z. Li, Y. Wang, R. Cao, R. Sarangi, Z. Yang, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2019, 12, 3508 RSC.
- E. Paul, R. Raza, S. R. Dhara, N. Baildya and K. Ghosh, RSC Adv., 2024, 14, 32759–32770 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView 6.0. 16, Semichem Inc., Shawnee Mission, 2016 Search PubMed.
-
(a) CCDC 2388873: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2l5tf1;
(b) CCDC 2388874: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2l5tg2.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a










 radicals (one with each metal) in the void space between the two parallel pyridine rings, stabilizing them through π–π interactions between the pyridine ring and the CO2 π electrons. This arrangement readily leads to C–C bond formation between the radicals to form an oxalato bridge.37 In an electrochemical process, two consecutive one electron reduction reactions occur to form CuI–CuI. Each CuI is coordinated by CO2. Coordinated CO2 is then converted to
radicals (one with each metal) in the void space between the two parallel pyridine rings, stabilizing them through π–π interactions between the pyridine ring and the CO2 π electrons. This arrangement readily leads to C–C bond formation between the radicals to form an oxalato bridge.37 In an electrochemical process, two consecutive one electron reduction reactions occur to form CuI–CuI. Each CuI is coordinated by CO2. Coordinated CO2 is then converted to  radicals. The close approach of the
radicals. The close approach of the  radicals then forms the C–C bond to produce oxalate. Then, two protons are added to release oxalic acid and regenerate 1. Thus, the plausible mechanism delineating CO2RR is given in SI (Fig. S25(c)).
radicals then forms the C–C bond to produce oxalate. Then, two protons are added to release oxalic acid and regenerate 1. Thus, the plausible mechanism delineating CO2RR is given in SI (Fig. S25(c)).
 radicals, hence generating an oxalato bridge efficiently. Analysis of products in the gas phase using gas chromatography quantified H2, O2, CO and CH4, whereas HPLC was employed to detect and quantify the amount of oxalic acid produced after CPE in the catalytic solution with CO2 and acid. Theoretical calculations engendered a very low HOMO–LUMO gap of 0.437 eV, which facilitates such a high activity of the homogeneous catalyst. The crucial intermediates formed during the reduction of protons to dihydrogen and carbon dioxide to oxalic acid as the major products, along with oxidation of water to molecular oxygen, were identified from the LC-MS spectra recorded after CPE at respective potentials. The HRMS spectra identified the formation of oxalate ions after CPE at −2.15 V for carbon dioxide reduction. Thorough exploration of the mechanisms via computational analysis using DFT is underway.
radicals, hence generating an oxalato bridge efficiently. Analysis of products in the gas phase using gas chromatography quantified H2, O2, CO and CH4, whereas HPLC was employed to detect and quantify the amount of oxalic acid produced after CPE in the catalytic solution with CO2 and acid. Theoretical calculations engendered a very low HOMO–LUMO gap of 0.437 eV, which facilitates such a high activity of the homogeneous catalyst. The crucial intermediates formed during the reduction of protons to dihydrogen and carbon dioxide to oxalic acid as the major products, along with oxidation of water to molecular oxygen, were identified from the LC-MS spectra recorded after CPE at respective potentials. The HRMS spectra identified the formation of oxalate ions after CPE at −2.15 V for carbon dioxide reduction. Thorough exploration of the mechanisms via computational analysis using DFT is underway.